
Abstract
First principle calculations were performed to investigate the structural, mechanical, electronic properties, and thermodynamic properties of three binary Mg–B compounds under pressure, by using the first principle method. The results implied that the structural parameters and the mechanical properties of the Mg–B compounds without pressure are well matched with the obtainable theoretically simulated values and experimental data. The obtained pressure–volume and energy–volume revealed that the three Mg–B compounds were mechanically stable, and the volume variation decreases with an increase in the boron content. The shear and volume deformation resistance indicated that the elastic constant Cij and bulk modulus B increased when the pressure increased up to 40 GPa, and that MgB7 had the strongest capacity to resist shear and volume deformation at zero pressure, which indicated the highest hardness. Meanwhile, MgB4 exhibited a ductility transformation behaviour at 30 GPa, and MgB2 and MgB7 displayed a brittle nature under all the considered pressure conditions. The anisotropy of the three Mg–B compounds under pressure were arranged as follows: MgB4 > MgB2 > MgB7. Moreover, the total density of states varied slightly and decreased with an increase in the pressure. The Debye temperature ΘD of the Mg–B compounds gradually increased with an increase in the pressure and the boron content. The temperature and pressure dependence of the heat capacity and the thermal expansion coefficient α were both obtained on the basis of Debye model under increased pressure from 0 to 40 GPa and increased temperatures. This paper brings a convenient understanding of the magnesium–boron alloys.
Similar content being viewed by others
Introduction
Magnesium boride alloys (MgB2, MgB4, and MgB7) as desirable compounds play an important role in many fields due to their remarkable conductivity, excellent ductility, and high hardness1,2,3. Usually, boron-rich magnesium alloys have excellent material characteristics such as mechanical properties and stability4,5. Moreover, MgB2 has been widely introduced into magnesium alloys for the reinforcement and grain refinement6,7, because of the chemical substitution and the crystal growth of substituted MgB28,9,10,11. Therefore, increasing attention has been paid to investigate the magnesium boride alloys in many academic fields.
Superconductors of magnesium diboride were reported first by Akimitsu12 in 2001. Since then, magnesium boride systems have been extensively studied through theoretical simulations and experimental analyses13,14,15. The intermediate phases of Mg–B alloys, which include MgB2, MgB4, and MgB7, were found through the continued investigation of the Mg–B binary phase diagram using the CALPHAD method based on experimental data16,17. Furthermore, Brutti et al.18 studied the vaporisation behaviour of MgB2 and MgB4 by the Knudsen effusion-mass spectrometry technique. Wenzel et al.19 predicted the crystal system and the lattice parameters of Mg–B compounds by using the electron probe micro analysis (EPMA) and X-ray diffraction (XRD) analytical approaches. Moreover, Alapati et al.4 calculated the lattice parameters of Mg–B compounds using the first principle based on the density functional theory (DFT). The elastic constants, mechanical properties, bond structure, and electronic properties of MgB7 at 0 GPa were investigated by Ozisik20. Furthermore, the heat capacity and the thermal expansion of MgB2 at 0 GPa was predicted by Saengdeejing21. The thermodynamic properties of Mg–B compounds and Al–Mg–B films were also investigated by using ab initio calculations and CALPHAD methods22,23. So far, the most effective method to obtain the hexagonal phase MgB2 is the high-pressure and high-temperature growth by using different kinds of solvents, and the external pressures and higher temperature may promote the reaction of Mg–B compounds22. Moreover, the crystal structure, electronic properties, thermodynamic properties, and mechanical properties of Mg–B compounds at different pressure and temperature have not been studied.
Assuredly, the above mentioned experimental studies have evidenced that the properties of Mg–B compounds can be calculated using DFT for establishing the trends of stability through the cohesive energies and the trends of charge transfers onto boron. Therefore, in the current article, the structural, mechanism, electronic, and anisotropic properties of MgB2, MgB4, and MgB7 under pressure from 0 to 40 GPa were investigated by using DFT calculation. The thermal expansion coefficient, Debye temperature, heat capacity, and other thermodynamic properties were theoretically studied for determining the pressure and temperature dependence of Mg–B compounds.
Computational methodology
In this study, all the calculated results were obtained by using the first-principle method through the Vienna ab initio simulation package (VASP)24 codes. PBE (Perdew–Burke–Ernzerhof)25 in GGA (generalized gradient approximation) was performed to expound the exchange-correction function26 and calculate the self-consistent electronic density. All the calculations in the current study were considered Mg 3p63s2 and B 2s22p1 as the valence electrons. To obtain an accurate calculated results, the cut-off energy Ecut was set to 500 eV. Moreover, the Brillouin-zone sampling mesh for the Monkhorst–Pack27 k-point for MgB2, MgB4, and MgB7 was set to 19 × 19 × 14, 9 × 11 × 7, and 8 × 8 × 8, respectively, due to the k-mesh was forced to be centred on the gamma point. Besides, the σ value of the first-order Methfessel–Paxton smearing was set to 0.2 eV, the convergence threshold of the self-consistent field was set to 1.0 × 10−5 eV/atom.
Results and discussions
Structural stability
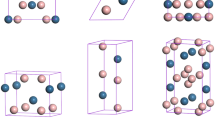
The optimised crystal texture of Mg–B compounds is shown in Fig. 1, and the corresponding calculated crystal parameters of the Mg–B compounds at 0 GPa are tabulated in Table 128,29,30,31,32. As listed in Table 1, the simulated crystal structure parameters considered in this study matched well with the reported literature data from the experimental and theoretical calculations, which verified the reasonability of the Mg–B compound models.

Optimised crystal structures of Mg–B compounds: (a) MgB2; (b) MgB4; (c) MgB7.
The energy–volume E(V) relation curves at zero absolute temperature were obtained using the first-principle method, as shown in Fig. 2. All the E(V) data were fitted to the Birch–Murnaghan model as follows33:
where B0 is the bulk modulus, B0′ is the first pressure derivative of the bulk modulus, and V0 is the equilibrium volume.

Variation between energy and volume of Mg–B compounds.
The functional pressure–volume P(V) data were obtained after the fitting of the E(V) curves to the Birch–Murnaghan model. Therefore, the P(V) curves displayed the relationship between the structural change and the pressure increase with a step of 10 GPa, as shown in Fig. 3. Moreover, the P(V) curves were calculated by using the equilibrium thermodynamic relation as follows34:

Volume ratio and pressure relation of Mg–B compounds with an interval of 10 GPa.
The volume ratio V/V0 of the three Mg–B compounds decreased with an increase in the pressure, as shown in Fig. 3, which was in agreement with the general rules. Moreover, the value of V/V0 of the Mg–B compounds under the same pressure from 10 to 40 GPa ranged in the following order: MgB2 < MgB4 < MgB7. That is, MgB7 was harder to compress under the same applied pressure, as it had the highest value of V/V0 among the three Mg–B compounds. Furthermore, MgB2 was the most sensitive to the pressure–volume relationship.
Mechanical properties
The elastic constants (Cij) of the crystal as an indispensable parameter played an important role in characterising the mechanical behaviours, because it contains a significant mechanical information under various pressures. There were nine (C11, C12, C13, C22, C23, C33, C44, C55, and C66) elastic constants for the orthorhombic crystals of MgB4 and MgB7, and six elastic constants (C11, C12, C13, C33, C44, and C66) for the hexagonal crystal of MgB2. Table 2 shown the simulated elastic constants and other elastic parameters of the Mg–B compounds; they were in agreement with the reference data. Moreover, the mechanical stability criterion of the hexagonal and orthorhombic structures of the Mg–B compounds is listed below:
For the hexagonal (MgB2) crystal35:
For the orthorhombic (MgB4 and MgB7) crystal36:
The influence of the applied pressure from 0 to 40 GPa on the calculated elastic constants for the Mg–B compounds is displayed in Fig. 4. As shown in Fig. 4, the entries in the elastic tensor Cij increased with an increase in pressure in the range from 0 to 40 GPa, and when the pressure reached to 40 GPa, all the elastic constants are well matched with the criterion of mechanical stability. Moreover, the deformation resistance of MgB2 and MgB7 is higher in the x-axis direction than in that of the other axes; this might be attributed to the fact that the largest elastic constant of C11 was observed for MgB2 and MgB7. Similarly, the C22 of MgB4 with the largest elastic constant indicated that the y-axis had the highest deformation resistance for MgB4. According to the existing literature37, the values of C44, C55, and C66 are always used to represent the ability of compounds to resist shear deformation. Therefore, MgB7 had higher values of C44 and C66 than the other two Mg–B compounds, according to Fig. 4, which implied that MgB7 had the highest resistance ability for the shear deformation.

Pressure dependence of the elastic constants of the three Mg–B compounds.
Generally, the elastic modulus contained B, G, and E could be subsequently obtained by using the VRH (Voigt–Reuss–Hill) approximation38, after the elastic constants Cij were obtained. The calculation equations are given by Ref.39,40:
For the hexagonal crystal:
For an orthorhombic crystal:
As displayed in Fig. 5, the elastic moduli consisting of bulk modulus B, shear modulus G, and Young’s modulus E increased linearly with an increase in the pressure, which indicated that the resistance to the volume deformation increased. From the reports, we inferred that the higher the bulk modulus was, the better the resistance to deformation was Ref.41. Simultaneously, we found that MgB7 had a stronger capacity to resist volume deformation and had higher hardness, as it had the highest values of the elastic moduli than others at a constant pressure. From Fig. 5, we inferred that the volume deformation resistance ability under a pressure from 0 to 30 GPa deferred to the following increased order: MgB2 < MgB4 < MgB7. Nevertheless, the ability resist to the volume change of MgB2 was stronger than others under pressures of 30–40 GPa, and it’s G and E were larger than that of MgB4’s. Therefore, it would be inaccurate to predict the hardness of MgB2 and MgB4.

Elastic modulus (B, G, and E) of Mg–B compounds various from different pressure.
In general, the B/G ratio and Poisson’s ratio ʋ can be used to explain the ductility and brittleness of Mg–B compounds42,43. Figure 6 presents the relation between the B/G ratio and Poisson’s ratio of Mg–B compounds under changed pressure. Poisson’s ratio ʋ can be calculated as follows:

Pressures dependence of the B/G ratio and Poisson’s ratio ʋ.
The B/G ratio and ʋ were proposed to describe the brittle or ductile of materials, when the values were 1.75 and 0.26, respectively. As shown in Fig. 6, the Poisson’s ratio ʋ and B/G ratio of MgB4 were 0.267 and 1.80 at 30 GPa, and 0.277 and 1.91 at 40 GPa, respectively. Thus, MgB4 showed ductile behaviour under pressures of 30–40 GPa, but brittle behaviour at 0–30 GPa, illustrating that the ductile transition for MgB4 occurred when the pressure increased to 30 GPa. Nevertheless, MgB2 and MgB7 displayed a brittle nature, and the brittleness of Mg–B compounds could be ranked in the following order: MgB4 < MgB2 < MgB7. Moreover, all the B/G ratios and ʋ values increased with an increase in the pressure, which indicates that the ductility could be improved by increasing the applied pressure on the Mg–B compounds.
The hardness H is an important parameter to measure the structure and mechanical properties, which can be calculated by using the following semi-empirical law44:
As shown in Fig. 7, the hardness of Mg–B compounds increased with an increase in the external pressure, and the values of hardness could be rowed as the following order: MgB7 > MgB2 > MgB4, which implied that MgB7 had the highest hardness; this finding matched well with the above mentioned results. On the basis of a comparison with Fig. 5, we can summarized that the effects of G and E on the hardness was greater than B.

Variation of micro-hardness at various pressures.
Elastic anisotropy plays an important role in crack behaviour and phase transformations, and its formula is defined as follows45:
As listed in Table 3, all the predicted values of AU were greater than zero, which implied that all the three Mg–B compounds were anisotropic materials. Moreover, the values of AU of MgB4 increased with an increase in the applied pressure, indicating that the anisotropy of MgB4 was enhanced by the increase in the applied pressure. Moreover, the elastic anisotropy of MgB4 was more sensitive to pressure according to the increase in the values of AU, and the anisotropy from low to high could be ranked in the following order: MgB7 < MgB2 < MgB4.
Electronic properties
To determine the effects of pressure on the mechanical properties and gain in-depth knowledge of the electronic structure of Mg–B compounds, the partial density of states (PDOS) and the total density of states (TDOS) under various pressures are shown in Fig. 8. Figure 8a,b,c shows that the MgB2 presented many peak point near the Fermi level, this indicated that MgB2 exhibited its special electrical conductivity, but the Fermi level of MgB4 and MgB7 are both in the range of zero DOS value, which implied that MgB4 and MgB7 may present semiconductor or insulator characteristics. Moreover, the primary bond peaks near the Fermi level were mainly occupied by the B 2p states and Mg 3p states for MgB2, MgB4, and MgB7. It can be seen from the Fig. 8, the DOS values of MgB4 and MgB7 at the Fermi level are all above 0, which implies that both of MgB4 and MgB7 also present metallic properties. However, for the MgB7, the valence band from 2.0 to 5.0 eV, Mg-p band contributes less than the Mg-s band near the Fermi level, The s–p hybridization between the B and Mg atoms forms covalent bonding for MgB7. Figure 8d,e,f only depict the TDOS of the Mg–B compounds at 0 GPa, 20 GPa, and 40 GPa, to demonstrate the regularity of TDOS for Mg–B compounds under various pressures. They show that there was a slight decrease in TDOS with an increase in the external pressure, which indicated that there was no structural phase transformation and small interaction potentials changed because of the decrease in the atomic distance under pressure. These figures also display the structural stability and the various electronic characteristics of Mg–B compounds under applied pressure.

Density of states of Mg–B compounds, (a, b, c) are partial density of states for MgB2, MgB4, and MgB7, respectively; (d, e, f) are total density of states under various pressures for MgB2, MgB4, and MgB7, respectively.
Thermodynamic properties
The quasi-harmonic Debye model of the phonon density of states was implemented in this part of the study to investigate the thermodynamic behaviours of the Mg–B compounds under pressure, namely the heat capacity Cv, Cp, the linear thermal expansion coefficient, and the Debye temperature ΘD of the Mg–B compounds. The above-obtained E(V) curves, as important input data for numerical minimisation programs in this model, were used to obtain more thermodynamical information of the Mg–B compounds46,47. Moreover, the vibrational thermodynamic properties were obtained at a designated temperature in the quasi-harmonic Debye model; this might be attributed to the consideration of the vibrational contribution for the internal energy. To improve the calculated precision of the thermodynamic behaviours, the 21 volume points from 0.80 a to 1.20 a of the calculated energy–volume were implied.
The Debye temperature of the Mg–B compounds was calculated from the average sound velocity by using the following formula48:
where νm, νs, and νl represent the average wave velocity and the shear and longitudinal sound velocities, respectively; h is Planck’s constant; kB is Boltzmann’s constant; n is the total number of atoms; NA is Avogadro’s number; ρ is the density; and M is the molecular weight. As shown in Fig. 9, the ΘD of the Mg–B compounds increased with an increase in the pressure and remained almost constant from 0 to 200 K but linearly decreased after 200 K. Simultaneously, the ΘD of the Mg–B compounds from low to high could be rowed as the following order: MgB2 < MgB4 < MgB7, when all the compounds were under the same temperature and pressure conditions.

Debye temperature of Mg–B compounds under various pressure and temperature.
Figure 10 shows the temperature and pressure dependence of the volumetric thermal expansion coefficient α of the Mg–B compounds. The thermal expansion coefficient is defined as \(\alpha = \frac{1}{{\text{V}}}\frac{{\partial {\text{V}}}}{{\partial {\text{T}}}}\), and the thermal expansion coefficient α increased with an increase in the pressure and the temperature. Although α was linear with T3 in the range from 0 to 300 K, α presented a gradual growth rate and changed gently when the temperature exceeded 300 K, which implied that the main thermal expansion of the Mg–B compounds occurred in the low-temperature region. In addition, α presented a decreasing tendency when the pressure increased to 40 GPa at a constant temperature. Meanwhile, the impact strength of pressure on the thermal expansion coefficient increased when the pressure was above 20 GPa.

Thermal expansion coefficient of Mg–B compounds as function of pressure and temperature.
The heat capacity are estimated by using Debye temperature and electronic structures of Mg–B compounds, which defined as follows:
where γ and β are the electronic and phonon contributions to the specific heat respectively. The temperature and the pressure dependence of the isochoric heat capacity (CV) and the isobaric heat capacity (CP) of the Mg–B compounds are displayed in Fig. 11. When the temperature was below 300 K, the variation of CV and CP exhibited an obvious and sharp rise; this subordinated Debye’s law. However, CP and CV were likely to continue to increase and remained constant after 300 K, respectively, due to the CV abided by the Dulong–Petit limit under high temperature conditions. Moreover, both the isochoric heat capacity (CV) and the isobaric heat capacity (CP) decreased with an increase in the pressure. Thus, from Fig. 11, we inferred that the heat capacity of MgB7 was higher than that of MgB4 and MgB2, indicating the stronger ability of release and absorption energy of MgB7.

Pressure and temperature dependence of heat capacity CV, CP of Mg–B compounds.
Conclusion
In this investigation, the structural, mechanical, electronic, and thermodynamic properties of Mg–B compounds were studied by using density functional theory; the conclusions of this paper can be summarized as follows:
-
(1)
The simulated elastic constants and elastic modulus through first-principle method are well coincided with the experimental values and theoretical calculations. The ratio of V/V0 decreased with an increase in the external pressure and increased with an increase in the boron content.
-
(2)
The three Mg–B compounds are mechanically stable from 0 to 40 GPa. The additional pressure on MgB2, MgB4, and MgB7 can improve it’s B, G, and E, which rowed as the following order: MgB2 < MgB4 < MgB7, but these properties of MgB2 is more excellent than that of MgB4 when the pressure reach to 30 GPa. Besides, a ductile conversion behavior at 30GPa is found in the process of increasing the pressure for MgB4.
-
(3)
The hardness of three Mg–B compounds enhanced with the increased pressure, the hardness of Mg–B compounds could be rowed as following order: MgB7 > MgB2 > MgB4. Conversely, the calculated elastic anisotropy could be ranked as following order: MgB7 < MgB2 < MgB4.
-
(4)
From the results the total density of states (TDOS) and the partial density of states (PDOS), the main orbital hybridizations of Mg–B compounds are B p and Mg d orbitals, B p and Mg s orbitals and B p and Mg p orbitals for MgB2, MgB4 and MgB7, respectively. There has no phase transformation under the rising external pressure. From the band structure, the MgB7 and the MgB4 shows semiconductor properties, but MgB2 presents excellent conductivity characteristic.
-
(5)
The Debye temperature of all Mg–B compounds reduce with an increase temperature from 0 to 1400 K but increase with an increase pressure from 0 to 40 GPa. The linear thermal expansion coefficient α increase linearly with an increase temperature and pressure, while it present a sharp increase when the pressure is rising up to 40GPa. The results of the isochoric heat capacity (CV) and the isobaric heat capacity (CP) increase gradually with an increase temperature, while the CV remain unchanged at higher temperature due to followed the Dulong–Petit limit.
Data availability
Some or all data, models, or code generated or used during the study are proprietary or confidential in nature and may only be provided with restrictions.
References
Yang, L. et al. Ultrafine nanocrystalline microstructure in Mg–B alloy for ultrahigh hardness and good ductility. Appl. Surf. Sci. 486, 102–107. https://doi.org/10.1016/j.apsusc.2019.05.006 (2019).
Chepulskii, R. V. & Curtarolo, S. First-principles solubilities of alkali and alkaline-earth metals in Mg–B alloys. Phys. Rev. Ser. B 79, 134203. https://doi.org/10.1103/PhysRevB.79.134203 (2001).
Serquis, A. et al. Microstructure and high critical current of powder in tube MgB2. Appl. Phys. Lett. 82, 1754–1756. https://doi.org/10.1063/1.1561572 (2002).
Alapati, S. V., Johnson, J. K. & Sholl, D. S. Identification of destabilized metal hydrides for hydrogen storage using first principles calculations. J. Phys. Chem. B 110, 8769–8776. https://doi.org/10.1021/jp060482m (2006).
Pediaditakis, A., Schroeder, M., Sagawe, V., Ludwig, T. & Hillebrecht, H. Binary boron-rich borides of magnesium: single-crystal investigations and properties of MgB7 and the new boride Mg similar to B-5(44). Inorg. Chem. 49, 10882–10893. https://doi.org/10.1021/ic1012389 (2010).
Lee, S. Recent advances in crystal growth of pure and chemically substituted MgB2. Phys. Sect. C 456, 14–21. https://doi.org/10.1016/j.physc.2007.01.018 (2007).
Xu, S., Moritomo, Y., Oikawa, K., Kamiyama, T. & Nakamura, A. Lattice structural change at ferromagnetic transition in Nd2Mo2O7. J. Phys. Soc. Jpn. 70, 2239–2241. https://doi.org/10.1143/jpsj.70.2239 (2001).
Varghese, N., Vinod, K., Kumar, R. G. A., Syamaprasad, U. & Sundaresan, A. Influence of reactivity of sheath materials with Mg/B on superconducting properties of MgB2. J. Appl. Phys. 4, 43914(1–4). https://doi.org/10.1063/1.2773696 (2007).
Slusky, J. S. et al. Loss of superconductivity with the addition of Al to MgB2 and a structural transition in Mg1−xAlxB2. Nature 410, 343–345. https://doi.org/10.1038/35066528 (2001).
Avdeev, M., Jorgensen, J. D., Ribeiro, R. A., Bud’ko, S. L. & Canfield, P. C. Crystal chemistry of carbon-substituted MgB2. Physica C 387, 301–306. https://doi.org/10.1016/S0921-4534(03)00722-6 (2015).
Maurin, I. et al. Carbon miscibility in the boron layers of the MgB2 superconductor. Chem. Mater. 14, 3894–3897. https://doi.org/10.1021/cm020308k (2002).
Nagamatsu, J., Nakagawa, N., Muranka, T., Zeniranim, Y. & Akimitsu, J. Superconductivity at 39 K in magnesium diboride. Nature 410, 63–64. https://doi.org/10.1002/chin.200121009 (2001).
Bohnenstiehl, S. D. et al. Experimental determination of the peritectic transition temperature of MgB2 in the Mg–B phase diagram. Thermochim. Acta 576, 27–35. https://doi.org/10.1016/j.tca.2013.11.027 (2014).
Balducci, G. et al. Thermodynamics of the intermediate phases in the Mg–B system. J. Phys. Chem. Solids 66, 292–297. https://doi.org/10.1016/j.jpcs.2004.06.063 (2005).
Bu, L.-P., Shen, Q.-T. & Wu, P. Microstructure and mechanical properties of Mg–RE–B alloys. Adv. Mater. Res. 311–313, 2251–2254. https://doi.org/10.4028/www.scientific.net/AMR.311-313.2251 (2011).
Liu, Z. K., Schlom, D. G. & Li, Q. Computational thermodynamic modeling of the Mg–B system. Calphad 25, 299–303. https://doi.org/10.1016/S0364-5916(01)00050-5 (2001).
Liu, Z. K., Schlom, D. G., Li, Q. & Xi, X. Thermodynamics of the Mg–B system: implications for the deposition of MgB2 thin films. Appl. Phys. Lett. 78, 3678–3680. https://doi.org/10.1063/1.1376145 (2001).
Brutti, S., Ciccioli, A., Balducci, G. & Gigli, G. Vaporization thermodynamics of MgB2 and MgB4. Appl. Phys. Lett. 80, 2892–2894. https://doi.org/10.1063/1.1471382 (2002).
Wenzel, T. et al. Electron probe microanalysis of Mg–B compounds: stoichiometry and heterogeneity of superconductors. Phys. Status Solidi C 198, 374–386. https://doi.org/10.1002/pssa.200306625 (2003).
Ozisik, H., Deligoz, E., Colakoglu, K. & Ateser, E. The first principles studies of the MgB7 compound: hard material. Intermetallics 39, 84–88. https://doi.org/10.1016/j.intermet.2013.03.016 (2013).
Saengdeejing, A., Wang, Y. & Liu, Z.-K. Structural and thermodynamic properties of compounds in the Mg–B–C system from first-principles calculations. Intermetallics 18, 803–808. https://doi.org/10.1016/j.intermet.2009.12.015 (2010).
Kim, S. et al. Phase stability determination of the Mg–B binary system using the CALPHAD method and ab initio calculations. J. Alloys Compd. 470, 85–89. https://doi.org/10.1016/j.jallcom.2008.02.099 (2009).
Ivashchenko, V. I. et al. Structural and mechanical properties of Al–Mg–B films: experimental study and first-principles calculations. Thin Solid Films 599, 72–77. https://doi.org/10.1016/j.tsf.2015.12.059 (2016).
Kresse, G. & Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B Condens. Matter 54, 11169–11186. https://doi.org/10.1103/PhysRevB.54.11169 (1996).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865. https://doi.org/10.1103/PhysRevLett.77.3865 (1996).
Perdew, J. P. et al. Atoms, molecules, solids, and surfaces: applications of the generalized gradient approximation for exchange and correlation. Phys. Rev. B Condens. Matter. 46, 6671–6687. https://doi.org/10.1103/PhysRevB.46.6671 (1996).
Monkhorst, H. J. Special points for Brillouin-zone integrations. Phys. Rev. B Condens. Matter 16, 1748–1749. https://doi.org/10.1103/PhysRevB.13.5188 (1976).
Chen, X. R., Wang, H. Y., Cheng, Y. & Hao, Y. J. First-principles calculations for structure and equation of state of MgB2 at high pressure. Physica B 370, 281–286. https://doi.org/10.1016/j.physb.2005.09.025 (2005).
Ravindran, P., Vajeeston, P., Vidya, R., Kjekshus, A. & Fjellvåg, H. Detailed electronic structure studies on superconducting MgB2 and related compounds. Phys. Rev. B 64, 224509. https://doi.org/10.1103/PhysRevB.64.224509 (2001).
Romaka, V. V., Prikhna, T. A. & Eisterer, M. Structure and properties of MgB2 bulks: ab initio simulations compared to experiment. IOP Conf. Ser. Mater. Sci. Eng. 756, 012020. https://doi.org/10.1088/1757-899X/756/1/012020 (2020).
Naslain, R., Guette, A. & Barret, M. Magnesium diboride and magnesium tetraboride crystal chemistry of tetraborides. J. Solid State Chem. 8, 68–85. https://doi.org/10.1021/ja01571a007 (1973).
Yakıncı, M. E., Balcı, Y., Aksan, M. A., Adigüzel, H. İ & Gencer, A. Degradation of superconducting properties in MgB2 by formation of the MgB4 phase. J. Supercond. Inc. Novel Magn. 15, 607–611. https://doi.org/10.1023/A:1021215728989 (2002).
Birch, F. Finite elastic strain of cubic crystals. Phys. Rev. 71, 809–824. https://doi.org/10.1103/PhysRev.71.809 (1947).
Birch, F. Finite strain isotherm and velocities for single-crystal and polycrystalline NaCl at high pressures and 300 K. J. Geophys. Res. Solid Earth 83(B3), 1257–1268. https://doi.org/10.1029/JB083iB03p01257 (1978).
Mouhat, F. & Coudert, F. X. Necessary and sufficient elastic stability conditions in various crystal systems. Phys. Rev. B. 90, 224104. https://doi.org/10.1103/PhysRevB.90.224104 (2014).
Yang, Q., Liu, X. J. & Bu, F. Q. First-principles phase stability and elastic properties of Al–La binary system intermetallic compounds. Intermetallics 60, 92–97. https://doi.org/10.1016/j.intermet.2015.02.007 (2015).
Tian, J. Z., Zhao, Y. H., Wang, B., Hou, H. & Zhang, Y. M. The structural, mechanical and thermodynamic properties of Ti–B compounds under the influence of temperature and pressure: first-principles study. Mater. Chem. Phys. 209, 200–207. https://doi.org/10.1016/j.matchemphys.2018.01.067 (2018).
Hill, R. The elastic behavior of crystalline aggregate. Proc. R. Soc. A Math. Phys. Eng. Sci. 65, 349–354. https://doi.org/10.1088/0370-1298/65/5/307 (1952).
Watt, J. P. & Peselnick, L. Clarification of the Hashin–Shtrikman bounds on the effective elastic moduli of polycrystals with hexagonal, trigonal, and tetragonal symmetries. J. Appl. Phys. 51, 1525. https://doi.org/10.1063/1.327804 (1980).
Qi, L. et al. The structural, elastic, electronic properties and Debye temperature of Ni3Mo under pressure from first-principles. J. Alloys Compd. 621, 383–388. https://doi.org/10.1016/j.jallcom.2014.10.015 (2015).
Tian, J. Z., Zhao, Y. H., Hou, H. & Wang, B. The effect of alloying elements on the structural stability, mechanical properties, and debye temperature of Al3Li: a first-principles study. Materials 11, 1471. https://doi.org/10.3390/ma11081471 (2018).
Liu, Z. J. et al. The melting curve of CaSiO3 perovskite under lower mantle pressures. Solid State Commun. 150, 590–593. https://doi.org/10.1016/j.ssc.2009.12.038 (2010).
Wang, P. et al. Structural, mechanical, and electronic properties of Zr–Te compounds from first-principles calculations. Chin. Phys. B Philos. Mag. Lett. 29, 076201. https://doi.org/10.1088/1674-1056/ab8da7 (2020).
Wen, Z. Q., Zhao, Y. H., Hou, H., Tian, J. Z. & Han, P. D. First-principles study of Ni–Al intermetallic compounds under various temperature and pressure. Superlattices Microstruct. 103, 9–18. https://doi.org/10.1016/j.spmi.2017.01.010 (2017).
Ranganathan, S. I. & Ostoja-Starzewski, M. Universal elastic anisotropy index. Phys. Rev. Lett. 101, 055504. https://doi.org/10.1103/PhysRevLett.101.055504 (2008).
OterodelaRoza, A., Abbasi-Perez, D. & Luana, V. Gibbs2: a new version of the quasiharmonic model code. II. Models fr solid-state thermodynamics, features and implementation. Comput. Phys. Commun. 182, 2232–2248. https://doi.org/10.1016/j.cpc.2011.05.009 (2011).
Blanco, M. A., Francisco, E. & Luana, V. GIBBS: isothermal isobaric thermodynamics of solids from energy curves using a quasiharmonic Debye model. Comput. Phys. Commun. 158, 57–72. https://doi.org/10.1016/j.comphy.2003.12.001 (2004).
Ravindran, P. et al. Density functional theory for calculation of elastic properties of orthorhombic crystals: application to TiSi2. J. Appl. Phys. 84, 4891–4904. https://doi.org/10.1063/1.368733 (1998).
Funding
This work is supported by the Natural Science Foundation of Shanxi Province (No. 201801D121111); The Natural Science Foundation of Shanxi Province (No. 201801D121108).
Author information
Authors and Affiliations
Contributions
G.Z. and C.X.: conceptualization, methodology, software; H.X. and Y.Z.: visualization and supervision. Y.D.: editing. F.S. and X.R.: investigation. M.W.: writing—reviewing, investigation and editing.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhang, G., Xu, C., Wang, M. et al. Pressure effect of the mechanical, electronics and thermodynamic properties of Mg–B compounds A first-principles investigations. Sci Rep 11, 6096 (2021). https://doi.org/10.1038/s41598-021-85654-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-021-85654-z
This article is cited by
-
High pressure studies of sc based ternary system: a DFT study
Optical and Quantum Electronics (2023)
-
Hydrostatic Pressure Effect on Lattice Thermal Conductivity in Si Nanofilms
Silicon (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
