
Abstract
Preterm birth remains the leading identifiable risk factor for cerebral palsy (CP), a devastating form of motor impairment due to developmental brain injury occurring around the time of birth. We performed genome wide methylation and whole transcriptome analyses to elucidate the early pathogenesis of CP in extremely low gestational age neonates (ELGANs). We evaluated peripheral blood cell specimens collected during a randomized trial of erythropoietin for neuroprotection in the ELGAN (PENUT Trial, NCT# 01378273). DNA methylation data were generated from 94 PENUT subjects (n = 47 CP vs. n = 47 Control) on day 1 and 14 of life. Gene expression data were generated from a subset of 56 subjects. Only one differentially methylated region was identified for the day 1 to 14 change between CP versus no CP, without evidence for differential gene expression of the associated gene RNA Pseudouridine Synthase Domain Containing 2. iPathwayGuide meta-analyses identified a relevant upregulation of JAK1 expression in the setting of decreased methylation that was observed in control subjects but not CP subjects. Evaluation of whole transcriptome data identified several top pathways of potential clinical relevance including thermogenesis, ferroptossis, ribosomal activity and other neurodegenerative conditions that differentiated CP from controls.
Similar content being viewed by others


Introduction
Despite advances in neonatal intensive care, survivors of preterm birth continue to suffer high rates of long term intellectual and/or physical impairment. Babies born prior to 28 weeks gestational age (Extremely Low Gestational Age Neonates—ELGANs) are at particularly high risk, with severe neurodevelopmental impairment reported in almost half of survivors and cerebral palsy (CP) in up to 10%1,2,3. Preterm birth remains the leading identifiable risk factor for CP, estimated to account for more than 50% of cases from population-based studies4. While promising neuroprotective therapies for ELGANs are under investigation, advancing neuroprotective care in the neonatal intensive care unit (NICU) is limited by the absence of biomarkers of brain injury that can identify infants early in the course of injury progression and help elucidate specific causal pathways to injury.
Prematurity-related neurologic injury is multifactorial and has been associated with various perinatal stressors including inflammation, intermittent hypoxia/hyperoxia, ischemia, pain, and nutritional deficiencies5. Any and all of these environmental triggers can lead to differential gene expression leading to risk for adverse outcomes. Modification of gene expression can occur by various epigenetic mechanisms, including DNA methylation6. Studies in preterm infants have demonstrated multiple differentially methylated regions (DMRs) compared to term controls7,8,9,10,11,12,13,14. Differences in DNA methylation, and associated differences in gene expression, may contribute to the risk for neurological sequelae in this high risk population.
We evaluated biological samples collected as a part of an NIH-funded Phase III Randomized Controlled Trial assessing the efficacy of erythropoietin (Epo) for neuroprotection in preterm infants (PENUT Trial, NCT01378273)15,16. Neonatal peripheral blood cell (PBC) samples from selected PENUT subjects were analyzed to determine whether examination of DMR and gene expression profiles in ELGAN survivors will lead to early neonatal biomarkers of CP. We hypothesized that early stressors of extrauterine life result in DMR and/or differentially expressed gene signatures that can distinguish infants with and without CP.
Results
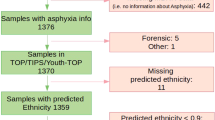
We identified total of 76 CP cases after completion of PENUT follow-up. High quality DNA was isolated from 94 PENUT subjects (n = 47 CP vs. n = 47 Control) at both timepoints of interest. The CP cohort consisted of babies with mild (n = 24), moderate (n = 16) and severe (n = 7) levels of motor impairment. Characteristics of the study population are summarized in Table 1. The majority of patients with CP had diplegia (n = 33, 70%), followed by spastic quadriplegia (n = 10, 21%) and hemiplegia (n = 4, 9%). While most CP subjects had a Gross Motor Function Classification Scale (GMFCS) level of 0 (n = 19, 40%), various levels of functional impairment were represented in the cohort (Table 1). Gene expression data were generated from a subset of subjects with adequate RNA (RIN > 5); 56 subjects (n = 29 CP vs n = 27 Control) on day 1 and 23 subjects (n = 12 CP vs n = 11 Control) on day 14.
DNA methylation analyses
Only 22 CpGs for the CP versus control day 1 comparison and 5 CpGs on day 14 were identified spread across the genome, so there was no evidence for any DMRs that differentiated CP cases from controls. In testing the day 1 and 14 interaction, we identified only one DMR on chromosome 15 (chr15: 40861240–40861791; mean proportional change 0.015, p = 8.62 e−0.01) that included 11 CpGs and showed decreased methylation between day 1 and 14 in the CP cases, whereas stable methylation was observed over time in controls (Fig. 1). The single gene associated with this DMR, RNA Pseudouridine Synthase Domain Containing 2 (RPUSD2), is a protein coding gene without reported functional correlates to neurological disease. No additional DMRs were identified when the analyses were restricted to the cases with moderate or severe CP. Additionally, there was no evidence for differential gene expression for RPUSD2 in the subset of patients with available transcriptome data (log2 fold change −0.016; p = 0.994). For the between day comparisons, there were multiple DMRs identified within the CP (n = 302, Supplemental Table S1) and control groups (n = 339, Supplemental Table S2).

Significant DMR on chromosome 15 for the interaction (day 1 to 14 change) in CP vs Control. Methylation decreases in CP babies on day 14 (pink) compared to day 1 (blue), whereas appears stable in Control babies (red and green). Individual CpG numbers (denoted by green vertical bars) are listed as follows: cg09167084; cg18988510; cg12754238; cg20139049; cg17356999; cg15454857; cg21874278; cg14760714; cg12307764; cg18013830; cg00066468. Figure generated with the Bioconductor Gviz package (version 1.28.3).
Whole transcriptome analyses
There was no evidence for differentially expressed genes between CP and control infants at day 1, day 14 or the interaction (day 1 to 14 change). For the between day comparisons, numerous genes that were significantly upregulated or downregulated over time were identified in both CP (n = 579, Supplemental Table S3) and control infants (n = 841, Supplemental Table S4). In order to evaluate genes with differential expression between day 1 and 14 that were unique to CP versus control, we created a Venn diagram to demonstrate the overlap in significant genes between the two conditions (Fig. 2). iPathwayGuide analysis of differentially expressed genes between day 1 and 14 that were unique to each diagnosis identified several top pathways of potential clinical relevance (Table 2).

Venn diagram of differentially expressed genes between day 1 and 14 that are unique in CP (yellow) compared to control (blue) ELGANs. Figure generated using the CRAN package gplots (version 3.0.1.1).

iPathwayGuide meta-analysis of DNA methylation and gene expression data identified 2 genes (highlighted in white) unique to CP, and 4 genes (black box) unique to Controls, that demonstrated evidence of differential methylation and gene expression. Figure generated using Advaita Bio’s iPathway Guide (https://www.advaitabio.com/ipathwayguide).
Meta-analyses of DNA methylation and transcriptome analyses
Secondary iPathwayGuide meta-analyses were performed to identify genes and/or pathways that were unique to CP subjects or controls across both methylation and gene expression datasets. We extracted the DMRs that were significant for the day 1 to 14 comparisons that were unique to each diagnosis and output the set of associated genes for each DMR. We generated a Venn diagram to evaluate the overlap between genes associated with a DMR and genes that demonstrated differential expression (Fig. 3). We identified only 2 genes, and no relevant pathways, that were differentially expressed between day 1 and 14 only in CP cases, and 4 genes only in Controls, observed consistently across both DNA methylation and gene expression datasets (Fig. 3). These genes and functional information are summarized in Table 3.
Discussion
DNA methylation is known to play an important role in brain development17,18,19,20,21, with alterations described in infants born preterm and those with postnatal stress14,22. While epigenetic changes are tissue-specific and it is recognized that between-tissue variation in DNA methylation exceeds between-individual differences, some inter-individual variation is reflected across brain and blood indicating that peripheral tissues may have some utility in identifying biomarkers of disease phenotypes that manifest in the brain23,24,25. Given this biological context and limited plausibility for identifying PBC DMRs as epigenetic biomarkers of prematurity-related developmental brain injury, it is not surprising that our genome wide DNA methylation analyses demonstrated limited evidence for early epigenetic factors in this tissue type that relate to later CP in ELGANs. Meanwhile, whole transcriptome analyses identified a profile of genes that are uniquely differentially expressed between day 1 and 14 in patients who later developed CP. Further investigation of these pathways may elucidate important insights into the early pathogenesis of CP.
DNA methylation and cerebral palsy
We identified one DMR in the interaction (day 1 to 14 change) comparison between CP and controls, without associated evidence of differential gene expression. In our secondary analyses, we identified 2 DMR-associated differentially expressed genes in CP cases (CSRP1, USP44) and 4 in Controls (CRELB2, DCAF11, FEM1A, JAK1). Apart from JAK1, these genes are widely expressed and generally involved in cell cycle, growth or differentiation without direct correlates to neurological disease. JAK1, however, is involved in regulating cytokine signaling, mediating innate immune response, and has been linked to the growth, differentiation and aging process of nerve cells. The JAK/STAT pathway has been implicated in the pathogenesis of CP and may serve as a mediator of Epo neuroprotection26. The role of methylation in regulating JAK1 expression may warrant further investigation.
Prior studies have suggested epigenetic mechanisms for CP pathogenesis in preterm infants. Two studies reported evidence for differential methylation in young children and adolescents with CP born preterm compared to term born controls27,28. These studies utilized sequencing data and evaluated individual CpG probes and/or evaluated differences among each gene, which may suffer from noise compared to DMRs which pool information across genomically adjacent probes in order to boost true signal29,30,31. These two studies also used fewer subjects (four pairs of discordant twins, and 22 CP and 21 controls, respectively). In addition to the methodological differences, our results may differ from these studies given our evaluation of methylation differences in the first 2 weeks of life. Thus, we cannot exclude the possibility of epigenetic changes that occur outside of the newborn period that can lead to CP risk in infants born preterm.
Two recent studies analyzed archived blood spots to evaluate neonatal DNA methylation differences between CP cases and controls. Bahado-Singh and colleagues reported a case–control study (n = 23 CP and n = 21 controls) in which they identified 230 differentially methylated CpG probes that were linked to canonical pathways involved in neuronal function and were found to be overall predictive of CP32. This study utilized a 450 K array which investigates half the CpGs as the EPIC array used in our study, requiring a less stringent correction for multiplicity. Their analyses, however, were based on comparisons using beta values (% methylation estimates) rather than the more customary M-values (used in the current study), which satisfy the assumptions of normality for statistical testing. While recent reports suggest that M-value preprocessing may not impact results in large scale datasets33, other reports suggest that M-value methods may allow more reliable identification of true positives, particularly in studies with smaller sample sizes34,35. More consistent with our results, Mohandas and colleagues reported a study evaluating neonatal blood spots from 15 CP-discordant monozygotic twin pairs (12 born preterm) and described 33 CpG probes and 2 DMRs36, although these did not meet significance threshold after adjustment for multiplicity. To our knowledge, this report represents the largest study to date evaluating DNA methylation in neonatal blood samples from preterm infants and the risk for later CP. Our data suggest that epigenetic changes reflected in PBCs in the first 2 weeks of life have a limited role in the pathogenesis of CP.
Transcriptome profiles and cerebral palsy
Although we did not identify significant differentially expressed genes in our direct comparisons between CP and control groups, evaluation of genes with differential expression from day 1 to 14 that were unique to each group provided insights into pathways with potential relevance to neurological disease. Two of the top canonical pathways identified included Parkinson’s and Huntington’s disease, both neurodegenerative conditions affecting the basal ganglia with associated motor dysfunction. The link between thermogenesis and CP is less clear, although this process is regulated by dopaminergic signaling in the CNS37. Of interest is the identification of ferroptosis, which has recently emerged as an important oxidative stress-induced cell death pathway and has been implicated in the pathogenesis of neurodegenerative diseases such as Alzheimer’s, traumatic brain injury, and stroke38. As this pathway is uniquely upregulated in babies with CP, it warrants further investigation. Similarly, the upregulation of ribosomal genes observed only in control infants suggests that this developmental change may be an important mediator of CP risk and that impaired upregulation of genes encoding ribosomal machinery may contribute to the development of CP. This finding is consistent with prior reports that have linked ribosomal activity to CP and other neurological diseases39,40,41,42. While these results do not provide specific causal pathways to CP, further exploration of relevant pathways may provide novel insights and gene targets for future studies.
Prior studies have evaluated neonatal transcriptome data in mixed cohorts of preterm and term born children with CP. Ho and colleagues evaluated neonatal peripheral blood from 20 preterm-born (< 37 weeks gestational age) children with CP and 1:1 gender and gestational age matched controls43. They reported that inflammatory, hypoxic and thyroidal gene sets were upregulated in preterm-born CP cases compared to controls. Of note, RNA was extracted from archived neonatal blood spots and significant degradation was noted (average RIN 2.3 ± 0.7) which may have impacted reproducibility of these findings. Van Eyk and colleagues demonstrated transcriptional dysregulation of trophic signaling pathways in patient-derived immortalized B-cell lines of a large cohort of CP cases (n = 182) and network analyses demonstrated significant overlap with genes observed in autism44. Cell lines used in these analyses were derived from multiple separate biorepositories which may have contributed technical variability. While methodological differences may explain variable results across studies, these studies and ours suggests that investigating transcriptome profiles in babies with CP can provide insights into pathogenic mechanisms and potentially identify early therapeutic targets.
Technical notes and limitations
We evaluated mixed PBC specimens to generate DNA methylation and RNA transcriptome data. It is acknowledged that several factors including age, prematurity45 and erythropoietin treatment46 may impact cell proportions. We chose to use the surrogate variable analysis approach, which ensures orthogonality of our covariates and has been shown to be a reasonable approach for cell-type mixture adjustment under most scenarios47. Another important technical note is to acknowledge that these PBC pellets were not primarily stored for RNA preservation. Thus, we faced significant RNA degradation which limited our sample size and introduced technical variability into our analyses. We limited the impact of this on our analyses by requiring a minimum RIN (> 5) and using robust analytic and QC approaches to increase confidence that differences observed were biological and not due to differential degradation or other technical confounders. Given the stringent significance criteria and multiplicity correction used for our primary analyses, we performed secondary pathway analyses to identify potentially relevant DMRs or differentially expressed genes that were uniquely changing within CP versus controls. These findings should be considered hypothesis generating and warrant further investigation in future studies. Likewise, future studies will need to investigate the impact of important clinical confounders that may impact methylation profiles. While our principle components analyses demonstrated that the selected clinical factors explored did not correlate with methylation changes in our dataset (supplemental figures), our study was not designed to investigate methylation in these co-morbidities. Finally, we included all severity levels and classifications of CP in order to increase sample size and power for our analyses. It is acknowledged that the pathogenesis of the more severe forms or subtypes of CP may differ and future investigations focusing on specific phenotypes may yield different results.
Conclusion
While the role of methylation in regulating JAK1 expression warrants further investigation, we found limited evidence of distinguishing methylation changes in peripheral blood cells that related to the development of CP in this selected population of ELGANs. Whole transcriptome analyses may implicate ferroptosis and ribosomal activity as potential canonical pathways leading to CP risk in the preterm ELGAN which warrant further investigation.
Methods
Study population
This is an ancillary study to the NIH-funded PENUT Trial15,16, a randomized, placebo-controlled study of Epo treatment in 941 preterm infants 24-0/7 to 27–6/7 weeks of gestation. Infants were enrolled between December 2013 and September 2016 from 19 U.S. sites. Written informed consent was obtained from the parent of each participant and the study was approved by the institutional review boards at each participating study site. Infants were randomized to Epo treatment or placebo within 24 h of birth, with study drug continued until 32–6/7 weeks corrected age. Infants with major life-threatening or chromosomal anomalies hematopoietic crises such as DIC or hemolysis, polycythemia, congenital infection or prior use of Epo were excluded. Detailed characteristics of the study population have been previously reported16. Developmental assessment at 24 ± 2 month corrected age included diagnosis and classification of CP by standardized neurological examination based on ELGAN Neurological Exam Study protocol48,49 and the Gross Motor Function Classification System (GMFCS)50,51. The GMFCS is a 5-level system to aid in classification of CP on the basis of voluntary gross motor skills, with a level of 5 the most severe. For this study, the subset of infants with any diagnosis of CP by neurological exam were identified and included if they had adequate biological specimens available at both timepoints of interest. Severity of motor impairment was classified as mild, moderate or severe based on CP subtype and GMFCS level as prescribed by the parent study15,16. This CP cohort was distributionally matched 1:1 to a cohort of control infants without CP by gestational age (within 3 days), treatment allocation and sex. This study is reported in accordance with the Strengthening the Reporting of Observational Studies in Epidemiology (STROBE) guidelines and all methods were carried out in accordance with relevant guidelines and regulations52.
Specimen processing, DNA and RNA isolation
Serial blood samples were obtained at baseline (day 1), day 7, 9 and 14 to measure circulating inflammatory and brain-specific biomarkers. Samples were spun for 8 min at 2000 G, plasma separated from PBC pellet and each stored at -70 °C. Stored day 1 and 14 PBC specimens from selected PENUT subjects were divided into 2 aliquots for DNA and RNA isolation. DNA isolation was performed using the commercially available QIAamp Blood DNA isolation kit (Qiagen, Inc., Valencia, CA) according to the manufacturer’s protocol. DNA purity was assessed by measuring OD260/280 and OD260/230 ratios. Samples with OD260/280 ratios and OD260/230 ratios ≥ 1.8 were deemed of good quality DNA. RNA isolation was performed using a commercially available RNA isolation kit (Macherey–Nagel's NucleoSpin RNA Blood kit, cat#740200.50; Macherey–Nagel, Bethlehem, PA) using the manufacturer's “RNA isolation from 200 uL blood” protocol. RNA quantities was determined by spectrometry using the NanoDrop 8000 Spectrophotometer (ThermoFisher Scientific, Waltham, MA) and quality assessment using an Agilent 2100 Bioanalyzer (Agilent, Santa Clara, CA). As the PBC pellets were not collected primarily for RNA analyses, we used experimental approaches designed to address issues with storage-associated RNA degradation (described below). We restricted analyses to samples with RNA integrity number (RIN) > 5.
DNA methylation
Whole genome DNA methylation was determined using commercially available Illumina Infinium MethylationEPIC BeadChip Assay (Illumina, San Diego, CA). 500 ng of DNA is first treated with sodium bisulfite, using the EZ DNA Methylation Kit (Zymo Research) and following the Illumina-specified instructions. Converted DNAs are then processed on the Illumina Infinium Methylation EPIC 8-Sample Beadchip following the Infinium HD Methylation 15019521v01 protocol. DNA from the conversion step is denatured and neutralized to prepare it for amplification. The denatured DNA is amplified overnight at 37 °C. Next, amplified bisulfite-converted DNA is enzymatically fragmented for 60 min at 37 °C, precipitated with isopropanol and air dried. DNA is then re-suspended in hybridization buffer. Eight samples are applied to each beadchip, kept separate with an IntelliHyb seal. The prepared beadchip is incubated overnight in a hybridization oven at 48 °C with rocking. The amplified and fragmented DNA samples anneal to locus-specific 50mers during hybridization. Following hybridization, unhybridized and non-specifically hybridized DNA is washed away, and the chip is prepared for staining and extension. The chip undergoes staining and extension in capillary flow-through chambers. Allele-specific single-base extension of the oligos on the beadchip, using the captured DNA as template, incorporates detectable labels on the beadchip and determines the methylation profile for the sample. After staining, beadchips are scanned using the Illumina iScan + with ICS v3.3.28, and intensity data is extracted with Illumina GenomeStudio software (GenomeStudio v2011.1 with Methylation Analysis Module v1.9.0).
Gene transcription analyses
Whole transcriptome analyses were performed using the Human Clariom S Array (Affymetrix, ThermoFisher Scientific, Waltham, MA), following the GeneChip Pico protocol, which is optimized for degraded sample types including FFPE samples, frozen tissues and PBCs. This assay can generate robust expression profiles from as little as 100 pg of total RNA. Briefly, amplified double-stranded cDNAs are synthesized and converted to cRNAs via in vitro transcription. After clean-up, double-stranded, antisense cDNAs are synthesized using 20 µg cRNAs. Using 5.5 µg of cDNA samples are then fragmented and labeled, and then hybridized onto the Clariom S Human Arrays for 16 h at 45 °C. Arrays are then washed, stained and scanned using an Affymetrix GeneChip Scanner 3000.
Bioinformatics and data analysis
For methylation data, raw data from Illumina Human Methylation EPIC arrays (IDAT files) were imported into R (r-project.org) using the Bioconductor minfi package53. These arrays have extensive quality control (QC) probes used to determine the quality of each processing step. Any arrays that failed QC were either re-processed or discarded. We also discarded any probes that have binding indistinguishable from background for > 5% of the subjects. We then background adjusted the raw probe intensities using ‘out of bounds’ probes, which are non-complementary probes that are not expected to hybridize and, thus, provide a measure of non-specific probe binding54, followed by a functional quantile normalization that uses control probes to eliminate technical differences between arrays55. Following normalization, we generated a principal components analysis (PCA) plot to evaluate the relative contribution of clinical variables (e.g. treatment allocation, baseline Epo level, gestational age, sex, severe bronchopulmonary dysplasia, necrotizing enterocolitis Bell’s stage 2b or 356, intraventricular hemorrhage grade 3 or 457, severe retinopathy of prematurity (requiring laser or bevacizumab), culture-proven sepsis, maternal race/ethnicity, CP status48,49 and GMFCS50,51) and cell components to the variability within each principal component (Supplemental Figures S1-13). Based on these plots, we included sex and race/ethnicity as confounders in our models. In addition, to control for other unobserved variability (for example, differences in cell composition from each sample), we used the Bioconductor sva package to estimate surrogate variables which we included in the models58. We made various inter-group comparisons using the Bioconductor limma package59 and DMRcate package29 to detect DMRs. This software accounts for the fact that individual CpG probes may be less reliable and suffer from both false positives and false negatives. Conversely, CpGs that are closely spaced tend to be highly correlated, with the correlation between CpGs dropping off as the genomic distance increases. Therefore, by grouping CpGs within genomic regions (within < 1000bps) and ‘smoothing’ the statistics used to detect differential methylation from adjacent CpGs, we increased power to detect DMRs. To reduce false positives, we required that any DMR consist of at least 10 CpGs and selected individual CpGs based on a false discovery rate (FDR) < 0.05. The group comparisons performed included: (1) Day 1 CP vs no CP, (2) Day 14 CP vs no CP, (3) the interaction (Day 1 to Day 14 change) CP vs no CP and (4) the Day 1 to 14 change within each diagnosis. Sensitivity analyses were also performed with parallel comparisons between controls and the CP cohort limited to the moderate and severely affected babies (GMFCS level ≥ 1). Given our study design, we fit different models depending on the comparison. For the first two comparisons, we fit a conventional analysis of variance (ANOVA) model, adjusting for sex, race, ethnicity and seven surrogate variables. For the remaining models, since we had repeated measures for each subject, we fit an ANOVA model that included patient-level blocking factors, an interaction between CP status and sampling day, and eight surrogate variables. To ensure goodness of fit for the individual CpG comparisons, we evaluated histograms of the resulting p-values, after which we combined into DMRs and tested for differential methylation.
For transcriptome data, we summarized and normalized transcript expression data using the RMA algorithm60, as implemented in the Bioconductor oligo package61. Analogous to the DNA methylation analyses, we used the Bioconductor sva package62 to estimate surrogate variables to adjust for known and unknown sources of technical and clinical variability. We then fit a linear mixed model that fits the patient as a random effect and includes these variables as well as the variable of interest (CP or control), and made comparisons using empirical Bayes adjusted contrasts59. We performed group comparisons as described above for the DNA methylation analyses. Genes were considered significant based on a FDR < 0.0563. We defined genes that may be affected by changes in methylation as those genes that fulfilled our significance criteria (FDR < 0.05) in a given contrast that were also within a 1 Mb region centered on a DMR. We performed several QC plots including normalized unscaled standard errors (NUSE), relative log expression (RLE), and MA plots using the RMA algorithm52 to evaluate the quality of the normalization and adequacy of surrogate variables to control for excess technical variability.
Secondarily, we evaluated the profile of genes that demonstrated significant up or downregulation between day 1 and 14 that was unique to CP cases as compared to controls. Impact analyses (iPathwayGuide) were performed to identify relevant pathways that were unique to each diagnosis. Functional information for candidate genes was derived from the GeneCards database (genecards.org).
Data availability
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
Stoll, B. J. et al. Neonatal outcomes of extremely preterm infants from the NICHD Neonatal Research Network. Pediatrics 126, 443–456. https://doi.org/10.1542/peds.2009-2959 (2010).
O’Shea, T. M. et al. The ELGAN study of the brain and related disorders in extremely low gestational age newborns. Early Human Dev. 85, 719–725. https://doi.org/10.1016/j.earlhumdev.2009.08.060 (2009).
Horbar, J. D. et al. Trends in mortality and morbidity for very low birth weight infants, 1991–1999. Pediatrics 110, 143–151 (2002).
Schieve, L. A. et al. Population impact of preterm birth and low birth weight on developmental disabilities in US children. Ann. Epidemiol. 26, 267–274. https://doi.org/10.1016/j.annepidem.2016.02.012 (2016).
Back, S. A. White matter injury in the preterm infant: pathology and mechanisms. Acta Neuropathol. https://doi.org/10.1007/s00401-017-1718-6 (2017).
Feinberg, A. P. Phenotypic plasticity and the epigenetics of human disease. Nature 447, 433–440. https://doi.org/10.1038/nature05919 (2007).
Schroeder, J. W. et al. Neonatal DNA methylation patterns associate with gestational age. Epigenetics 6, 1498–1504. https://doi.org/10.4161/epi.6.12.18296 (2011).
Menon, R., Conneely, K. N. & Smith, A. K. DNA methylation: an epigenetic risk factor in preterm birth. Reprod. Sci 19, 6–13. https://doi.org/10.1177/1933719111424446 (2012).
Fernando, F. et al. The idiopathic preterm delivery methylation profile in umbilical cord blood DNA. BMC Genomics 16, 736. https://doi.org/10.1186/s12864-015-1915-4 (2015).
Burris, H. H. et al. Associations of LINE-1 DNA Methylation with Preterm Birth in a Prospective Cohort Study. J. Dev. Origins Health Dis. 3, 173–181 (2012).
Cruickshank, M. N. et al. Analysis of epigenetic changes in survivors of preterm birth reveals the effect of gestational age and evidence for a long term legacy. Genome Med. 5, 96. https://doi.org/10.1186/gm500 (2013).
Parets, S. E. et al. Fetal DNA methylation associates with early spontaneous preterm birth and gestational age. PLoS ONE 8, e67489. https://doi.org/10.1371/journal.pone.0067489 (2013).
Piyasena, C. et al. Dynamics of DNA methylation at IGF2 in preterm and term infants during the first year of life: an observational study. Lancet 385 Suppl 1, S81, https://doi.org/10.1016/S0140-6736(15)60396-8 (2015).
Sparrow, S. et al. Epigenomic profiling of preterm infants reveals DNA methylation differences at sites associated with neural function. Transl. Psychiatry 6, e716. https://doi.org/10.1038/tp.2015.210 (2016).
Juul, S. E., Mayock, D. E., Comstock, B. A. & Heagerty, P. J. Neuroprotective potential of erythropoietin in neonates; design of a randomized trial. Maternal Health Neonatology Perinatol. 1, 27. https://doi.org/10.1186/s40748-015-0028-z (2015).
Juul, S. E. et al. A randomized trial of erythropoietin for neuroprotection in preterm infants. N. Engl. J. Med. 382, 233–243. https://doi.org/10.1056/NEJMoa1907423 (2020).
Teter, B., Finch, C. E. & Condorelli, D. F. DNA methylation in the glial fibrillary acidic protein gene: map of CpG methylation sites and summary of analysis by restriction enzymes and by LMPCR. J. Neurosci. Res. 39, 708–709. https://doi.org/10.1002/jnr.490390611 (1994).
Fan, G. et al. DNA hypomethylation perturbs the function and survival of CNS neurons in postnatal animals. J. Neurosci. 21, 788–797 (2001).
Takizawa, T. et al. DNA methylation is a critical cell-intrinsic determinant of astrocyte differentiation in the fetal brain. Dev. Cell 1, 749–758 (2001).
Hutnick, L. K. et al. DNA hypomethylation restricted to the murine forebrain induces cortical degeneration and impairs postnatal neuronal maturation. Hum. Mol. Genet. 18, 2875–2888. https://doi.org/10.1093/hmg/ddp222 (2009).
Sauvageot, C. M. & Stiles, C. D. Molecular mechanisms controlling cortical gliogenesis. Curr. Opin. Neurobiol. 12, 244–249 (2002).
Mehta, D. et al. Childhood maltreatment is associated with distinct genomic and epigenetic profiles in posttraumatic stress disorder. Proc. Natl. Acad. Sci. USA 110, 8302–8307. https://doi.org/10.1073/pnas.1217750110 (2013).
Davies, M. N. et al. Functional annotation of the human brain methylome identifies tissue-specific epigenetic variation across brain and blood. Genome Biol. 13, R43. https://doi.org/10.1186/gb-2012-13-6-r43 (2012).
Hannon, E., Lunnon, K., Schalkwyk, L. & Mill, J. Interindividual methylomic variation across blood, cortex, and cerebellum: implications for epigenetic studies of neurological and neuropsychiatric phenotypes. Epigenetics 10, 1024–1032. https://doi.org/10.1080/15592294.2015.1100786 (2015).
Edgar, R. D., Jones, M. J., Meaney, M. J., Turecki, G. & Kobor, M. S. BECon: a tool for interpreting DNA methylation findings from blood in the context of brain. Transl. Psychiatry 7, e1187. https://doi.org/10.1038/tp.2017.171 (2017).
Tao, W., Wen, F., Zhang, H. & Liu, G. The signal transduction mediated by erythropoietin and proinflammatory cytokines in the JAK/STAT pathway in the children with cerebral palsy. Brain Develop. 31, 200–207. https://doi.org/10.1016/j.braindev.2008.06.011 (2009).
Jiao, Z. et al. Wholegenome scale identification of methylation markers specific for cerebral palsy in monozygotic discordant twins. Mol. Med. Reports 16, 9423–9430. https://doi.org/10.3892/mmr.2017.7800 (2017).
Crowgey, E. L., Marsh, A. G., Robinson, K. G., Yeager, S. K. & Akins, R. E. Epigenetic machine learning: utilizing DNA methylation patterns to predict spastic cerebral palsy. BMC Bioinf. 19, 225. https://doi.org/10.1186/s12859-018-2224-0 (2018).
Peters, T. J. et al. De novo identification of differentially methylated regions in the human genome. Epigenet. Chromatin 8, 6. https://doi.org/10.1186/1756-8935-8-6 (2015).
Jaffe, A. E. et al. Bump hunting to identify differentially methylated regions in epigenetic epidemiology studies. Int. J. Epidemiol. 41, 200–209. https://doi.org/10.1093/ije/dyr238 (2012).
Jaenisch, R. & Bird, A. Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nat. Genet. 33(Suppl), 245–254. https://doi.org/10.1038/ng1089 (2003).
32Bahado-Singh, R. O. et al. Deep Learning/Artificial Intelligence and Blood-Based DNA Epigenomic Prediction of Cerebral Palsy. International journal of molecular sciences 20, https://doi.org/10.3390/ijms20092075 (2019).
33van Rooij, J. et al. Evaluation of commonly used analysis strategies for epigenome- and transcriptome-wide association studies through replication of large-scale population studies. Genome biology 20, 235, https://doi.org/10.1186/s13059-019-1878-x (2019).
Zhuang, J., Widschwendter, M. & Teschendorff, A. E. A comparison of feature selection and classification methods in DNA methylation studies using the Illumina Infinium platform. BMC Bioinf. 13, 59. https://doi.org/10.1186/1471-2105-13-59 (2012).
Du, P. et al. Comparison of Beta-value and M-value methods for quantifying methylation levels by microarray analysis. BMC Bioinf. 11, 587. https://doi.org/10.1186/1471-2105-11-587 (2010).
Mohandas, N. et al. Epigenome-wide analysis in newborn blood spots from monozygotic twins discordant for cerebral palsy reveals consistent regional differences in DNA methylation. Clin. Epigenet. 10, 25. https://doi.org/10.1186/s13148-018-0457-4 (2018).
Folgueira, C. et al. Hypothalamic dopamine signaling regulates brown fat thermogenesis. Nat. Metabol. 1, 811–829. https://doi.org/10.1038/s42255-019-0099-7 (2019).
Ratan, R. R. The Chemical Biology of Ferroptosis in the Central Nervous System. Cell Chem. Biol. https://doi.org/10.1016/j.chembiol.2020.03.007 (2020).
39Zomzely, C. E., Roberts, S., Gruber, C. P. & Brown, D. M. Cerebral protein synthesis. II. Instability of cerebral messenger ribonucleic acid-ribosome complexes. J Biol. Chem. 243, 5396–5409 (1968).
Von Walden, F. et al. Muscle contractures in patients with cerebral palsy and acquired brain injury are associated with extracellular matrix expansion, pro-inflammatory gene expression, and reduced rRNA synthesis. Muscle Nerve 58, 277–285. https://doi.org/10.1002/mus.26130 (2018).
Hetman, M. & Slomnicki, L. P. Ribosomal biogenesis as an emerging target of neurodevelopmental pathologies. J. Neurochem. 148, 325–347. https://doi.org/10.1111/jnc.14576 (2019).
Koren, S. A. et al. Tau drives translational selectivity by interacting with ribosomal proteins. Acta Neuropathol. 137, 571–583. https://doi.org/10.1007/s00401-019-01970-9 (2019).
Ho, N. T. et al. Gene expression in archived newborn blood spots distinguishes infants who will later develop cerebral palsy from matched controls. Pediatr. Res. 73, 450–456. https://doi.org/10.1038/pr.2012.200 (2013).
van Eyk, C. L. et al. Analysis of 182 cerebral palsy transcriptomes points to dysregulation of trophic signalling pathways and overlap with autism. Translational psychiatry 8, 88. https://doi.org/10.1038/s41398-018-0136-4 (2018).
Vatansever, U. et al. Nucleated red blood cell counts and erythropoietin levels in high-risk neonates. Pediatr Int 44, 590–595. https://doi.org/10.1046/j.1442-200x.2002.01630.x (2002).
Ferber, A., Fridel, Z., Weissmann-Brenner, A., Minior, V. K. & Divon, M. Y. Are elevated fetal nucleated red blood cell counts an indirect reflection of enhanced erythropoietin activity?. Am. J. Obstet. Gynecol. 190, 1473–1475. https://doi.org/10.1016/j.ajog.2004.02.033 (2004).
McGregor, K. et al. An evaluation of methods correcting for cell-type heterogeneity in DNA methylation studies. Genome Biol. 17, 84. https://doi.org/10.1186/s13059-016-0935-y (2016).
Kuban, K. C. et al. An algorithm for identifying and classifying cerebral palsy in young children. J. Pediatr. 153, 466–472
Kuban, K. C. et al. Video and CD-ROM as a training tool for performing neurologic examinations of 1-year-old children in a multicenter epidemiologic study. J. Child Neurol. 20, 829–831 (2005).
Palisano, R. J. et al. Validation of a model of gross motor function for children with cerebral palsy. Phys Ther 80, 974–985 (2000).
Palisano, R. J., Rosenbaum, P., Bartlett, D. & Livingston, M. H. Content validity of the expanded and revised Gross Motor Function Classification System. Dev. Med. Child Neurol. 50, 744–750. https://doi.org/10.1111/j.1469-8749.2008.03089.x (2008).
guidelines for reporting observational studies. 52von Elm, E. et al. The Strengthening the Reporting of Observational Studies in Epidemiology (STROBE) statement. Lancet 370, 1453–1457. https://doi.org/10.1016/S0140-6736(07)61602-X (2007).
Fortin, J. P., Triche, T. J. Jr. & Hansen, K. D. Preprocessing, normalization and integration of the Illumina HumanMethylationEPIC array with minfi. Bioinformatics 33, 558–560. https://doi.org/10.1093/bioinformatics/btw691 (2017).
Triche, T. J. Jr., Weisenberger, D. J., Van Den Berg, D., Laird, P. W. & Siegmund, K. D. Low-level processing of Illumina Infinium DNA Methylation BeadArrays. Nucleic Acids Res. 41, e90. https://doi.org/10.1093/nar/gkt090 (2013).
Fortin, J. P. et al. Functional normalization of 450k methylation array data improves replication in large cancer studies. Genome Biol. 15, 503. https://doi.org/10.1186/s13059-014-0503-2 (2014).
Bell, M. J. Neonatal necrotizing enterocolitis. N. Engl. J. Med. 298, 281–282 (1978).
Papile, L. A., Burstein, J., Burstein, R. & Koffler, H. Incidence and evolution of subependymal and intraventricular hemorrhage: a study of infants with birth weights less than 1,500 gm. J. Pediatr. 92, 529–534 (1978).
Leek, J. T. & Storey, J. D. Capturing heterogeneity in gene expression studies by surrogate variable analysis. PLoS Genet. 3, 1724–1735. https://doi.org/10.1371/journal.pgen.0030161 (2007).
59Smyth, G. K. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Statistical applications in genetics and molecular biology 3, Article3, https://doi.org/10.2202/1544-6115.1027 (2004).
Irizarry, R. A. et al. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics 4, 249–264. https://doi.org/10.1093/biostatistics/4.2.249 (2003).
Carvalho, B. S. & Irizarry, R. A. A framework for oligonucleotide microarray preprocessing. Bioinformatics 26, 2363–2367. https://doi.org/10.1093/bioinformatics/btq431 (2010).
Leek, J. T., Johnson, W. E., Parker, H. S., Jaffe, A. E. & Storey, J. D. The sva package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinformatics 28, 882–883. https://doi.org/10.1093/bioinformatics/bts034 (2012).
Reiner, A., Yekutieli, D. & Benjamini, Y. Identifying differentially expressed genes using false discovery rate controlling procedures. Bioinformatics 19, 368–375 (2003).
Acknowledgements
The PENUT Trial (NCT01378273) was supported by the National Institutes of Health National Institute of Neurological Disorders and Stroke (1U01NS077953). We are indebted to the PENUT investigators, coordinators and the PENUT families. This ancillary study was supported by the Cerebral Palsy Alliance Research Foundation (PG10617).
Funding
This study was funded by the Cerebral Palsy Alliance Research Foundation.
Author information
Authors and Affiliations
Contributions
A.N.M. made substantial contributions to the conception or design of the work; interpretation of data; and drafted the work for publication. T.K.B. made substantial contributions to the conception or design of the work; analysis and interpretation of data; and substantively revised the work for publication. J.M. made substantial contributions to the conception or design of the work; analysis and interpretation of data; and substantively revised the work for publication. K.M.P. made substantial contributions to the interpretation of data; and substantively revised the work for publication. B.C. made substantial contributions to the conception or design of the work; the acquisition and analysis of data; and substantively revised the work for publication. S.E.J. made substantial contributions to the conception or design of the work; the acquisition, analysis, and interpretation of data; and substantively revised the work for publication. All authors have approved the submitted version (and any substantially modified version that involves the author's contribution to the study); and have agreed both to be personally accountable for the author's own contributions and to ensure that questions related to the accuracy or integrity of any part of the work, even ones in which the author was not personally involved, are appropriately investigated, resolved, and the resolution documented in the literature.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Massaro, A.N., Bammler, T.K., MacDonald, J.W. et al. Whole genome methylation and transcriptome analyses to identify risk for cerebral palsy (CP) in extremely low gestational age neonates (ELGAN). Sci Rep 11, 5305 (2021). https://doi.org/10.1038/s41598-021-84214-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-021-84214-9
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
