
Abstract
Several DNA-binding proteins show the affinities for their specific DNA sites that positively depend on the length of DNA harboring the sites, i. e. antenna effect. DNA looping can cause the effect for proteins with two or more DNA binding sites, i. e. the looping mechanism. One-dimensional diffusion also has been suggested to cause the effect for proteins with single DNA sites, the diffusion mechanism, which could violate detailed balance. We addressed which mechanism is possible for E. coli TrpR showing 104-fold antenna effect with a single DNA binding site. When a trpO-harboring DNA fragment was connected to a nonspecific DNA with biotin-avidin connection, the otherwise sevenfold antenna effect disappeared. This result denies the looping mechanism with an unknown second DNA binding site. The 3.5-fold repression by TrpR in vivo disappeared when a tight LexA binding site was introduced at various sites near the trpO, suggesting that the binding of LexA blocks one-dimensional diffusion causing the antenna effect. These results are consistent with the chemical ratchet recently proposed for TrpR-trpO binding to solve the deviation from detailed balance, and evidence that the antenna effect due to one-dimensional diffusion exists in cells.
Similar content being viewed by others


Introduction
In the chromosomes, there are a limited number of DNA sites where a protein binds with high affinities for its complex to perform the genetic commands, specific sites. These proteins are also known to bind to most parts of DNA, nonspecific sites, in much weaker affinities. The complexes with these sites are generically called specific and nonspecific complexes. In this view, DNA can be considered as a sequence of specific and nonspecific sites with nonspecific sites occupying most parts of DNA. If the high affinity for a specific site is determined solely by its local sequence or structure, nonspecific sites should have no function other than competing against specific sites for such a protein.
Contrary to this simple competitive view, the affinities of several proteins are reported to increase when their specific sites exist on longer DNA. This length effect was compiled under the name of “antenna effect” irrespective of the mechanisms1. The earliest report was for LacI and the enhancement of the affinity was 10–1,000 fold2,3. Since a LacI molecule is homotetrameric and can bind to two specific sites on DNA simultaneously, the change in the apparent affinity has been attributed to the additional stabilization by the second intramolecular binding to later found operators on the same DNA molecule4. The longer the DNA, the more frequent formation of the intramolecular DNA loop stabilizing the complex. Thus, this looping mechanism provides a clear explanation of the antenna effect for a protein molecule with two or more DNA-binding sites like LacI5,6,7,8,9. In contrast, the application of the mechanism for the proteins with single DNA-binding sites is difficult. In the case of the homodimeric bacterial repressor, only a single binding site is formed from two helix-turn-helix motifs of two subunits. Thus, the putative second DNA-binding site must be attributed to an unfound site or arbitrary surface of the protein, but such sites have not so far became evidenced. Moreover, there are accumulating examples in structural biology where a significant stabilization is attributed to only specific molecular interactions but not ones with an arbitrary surface. Thus, the looping mechanism for a protein with a single DNA-binding site is still questioned.
The antenna effect for a protein with single binding site, E. coli EcoRI methyltransferase, was found by Surby and Reich10 as the first systematic study of the effect. By using gel-shift assay, they showed that the affinity for whole DNA fragments increased 20-fold as the length increased from 14 to 775 bp, while the observed dissociation rate constant was independent of the length. From the results, they proposed a mechanism based on the accelerated association by one-dimensional diffusion along DNA with a declared reservation of the “violation of the thermodynamic rule”, without further comment.
This rule is usually called detailed balance and prohibits the existence of net circulation flow among reaction components at equilibrium, and thus claims that acceleration of a rate must be accompanied by the acceleration of its reverse rate of every step in the equilibrated reaction, maintaining the binding affinity for the specific site unchanged. This rule has been strictly established in the timescale at equilibrium in statistical mechanics11. Since the affinity is also determined at equilibrium, the rule seemingly denies the antenna effect caused by one-dimensional diffusion.
We found that the “equilibrium” required for detailed balance is different than the one required for determining an affinity. The former is stricter and must be established on the level of molecular systems. In contrast, the latter is a macroscopic steady state of the ensemble average of the molecular systems and does not necessarily demand the establishment of the former. As discussed later in more detail, if some of the reaction component molecules alter their intrinsic affinity/stability according to oscillating conformational changes with random phases, the latter is possible to be established to show a macroscopic affinity but the rule does not hold in the non-equilibrium state. We named the mechanism “chemical ratchet”, which is consistent with the 10,000-fold antenna effect of TrpR-trpO binding12.
Since there is neither evidence for nor counterevidence against the looping mechanism for TrpR-trpO binding, we designed a connection of DNA segments with avidin–biotin, which allows DNA looping but hampers one-dimensional diffusion by disrupting B-DNA helical structure. When a trpO-harboring DNA segment was connected to a nonspecific DNA segment with the intact phosphodiester bonds, the antenna effect was seven fold. In contrast, when they are connected with avidin–biotin, no antenna effect was observed (one-fold). We thus concluded that one-dimensional diffusion, but not DNA looping, is the cause of the length dependence of the affinity of TrpR for trpO.
The existence of one-dimensional diffusion in cells has been suggested for EcoRV by the observed correlation between the first-order cleaving rates in vitro and the titer values of bacteriophage lambda in vivo for the wild-type and mutant enzymes13. We examined the existence of antenna effect caused by one-dimensional diffusion in vivo by our block method. We inserted a LexA binding site with various affinities at various distances from the trpO regulating the expression of lacZ monitor. The measured expression of lacZ showed a good correlation to the LexA affinity for the sites, providing evidence for the existence of one-dimensional diffusion as well as antenna effect due to the diffusion in vivo, suggesting the cross-talk between two proteins at a distance on DNA.
Results
Elimination of DNA looping from the major mechanism of antenna effect
The dissociation equilibrium constant Kd of TrpR-trpO binding is defined as
All the symbols and their definitions are listed in Table S1 (Supplementary). Since the hydroxyl radical footprinting14 can directly quantify the amount of TrpR protein complexed at trpO site, we first determined the ratio of [TrpR-trpO complex] to [trpO]total at equilibrium at various [trpO]total and then determined the Kd value according to Eq. (2) by the least square fit as described in our preceding paper12. In our experimental condition the amount of TrpR was in excess over trpO, and thus [free TrpR] was approximated by [TrpR]total.
When the 36 bp DNA harboring trpO at its center is connected to 232 bp nonspecific DNA fragment, the affinity is enhanced, and 7.4-fold antenna effect was observed (Table 1). We tested the looping mechanism by connecting the two fragments with biotin-avidin binding. Since this connection preserves DNA looping, the antenna effect by the looping mechanism should be preserved. The looping may even enhance the effect because tetrameric avidin molecule can connect the two DNA fragments at an angle much smaller than 180°. Moreover, the flexible (–CH2–)9 residue in the biotin linker may facilitate DNA looping. In fact, a similar avidin connection proved the enhancer action mediated by DNA looping4,15. In contrast, this connection is expected to hinder one-dimensional diffusion of a protein along DNA at the joint, because of the diameter of avidin being much larger than that of DNA and because of the positively charged avidin surface opposite to the DNA surface. The sliding, a mode of one-dimensional diffusion in which a protein molecule tracks the DNA groove, especially, should be blocked by the disruption of the DNA grooves.
The obtained results clearly showed that the 7.4-fold antenna effect is caused by one-dimensional diffusion but not by DNA looping: the disappearance of the effect by biotin-avidin connection (Table 1). Moreover, we confirmed that the addition of biotin-avidin at the end of 36 bp trpO DNA did not change its affinity for TrpR (Table 1).
Effect of the binding of LexA protein near the operator in vivo
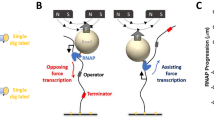
It is not easy to get evidence for the existence of one-dimensional diffusion in vivo. The existence of antenna effect in vivo may be more difficult. In vitro, we have hindered one-dimensional diffusion by introducing avidin between two DNA fragments as shown above. We then used in vivo a similar method to hinder one-dimensional diffusion by introducing LexA protein and LexA binding sites near trpO in a low copy plasmid DNA. If the antenna effect exists also in vivo, the formation of the tight LexA-DNA complex near trpO is expected to decrease the affinity of TrpR for trpO, and then suppresses the repression by TrpR. Therefore, disappearance of the antenna effect due to blocked one-dimensional diffusion, similar to the in vitro experiment described above, can be detected. The block should consequently decrease the affinity of TrpR for trpO, enhancing the expression of the monitor gene lacZ cloned downstream of trpO. In this way, we examine the existence of one-dimensional diffusion as well as the antenna effect due to the diffusion.
A series of low-copy mini-F plasmids16 harboring a LexA binding site17 was constructed. The part upstream from the intact trpO-trpR promoter was remain intact and was followed by a lacZ reporter gene (Fig. 1). The bound LexA is expected to block the sliding of TrpR into the trpO to diminish the antenna effect, activating lacZ transcription from the trpO-trpR promoter. But a similar block also could be induced by direct contact between the bound LexA and/or the initiating RNA polymerase and/or TrpR. Such protein–protein contacts are known to require the presence of the protein molecules on the same face of the double-stranded DNA as shown for λ repressor cI6 and RNA polymerase7,18, resulting in an iterative pattern of the presence and the absence of the effect at every 5–6 bp, a half pitch of DNA helix. Therefore, the spacing was changed between the LexA site and the promoter among the 5 constructs (Fig. 1).

Structures of the trpO site with the LexA binding sites upstream (a). The position of a LexA binding site inserted and the position of a spacer are indicated with the trpO ordinate, and the functional elements are indicated as “-35 box”, “-10 box”, and “trpO”. Transcription starts at + 1. (b). The sequences of the LexA binding sites and the spacers shown in Panel A and Table 2.
The control strain harboring the plasmid with no LexA site showed a repression by TrpR of about fourfold (data lines 1 and 2 in Table 2) as previously reported on a λ lysogen harboring the trpO19, whereas the strains harboring plasmids carrying the consensus LexA site showed the expected disappearance of repression irrespective of the spacings (lines 3–7). The independence of the iterative pattern at every 5 bp (lines 3–7) indicated that the loss of repression was not due to the direct contact of the bound LexA to RNA polymerase or TrpR7. Furthermore, a weakened LexA target allowed intermediate repression (lines 8 and 9), and overproduction of TrpR recovered the repression depending on the affinities of LexA site (lines 10–13). These results indicate that the tight binding of LexA decreased the affinity of TrpR for the trpO at a distance. The recovery of repression at a higher level of TrpR in vivo is consistent with the mechanism that the affinity was decreased enough by the partial blocking of sliding only from upstream. These results are consistent with the model that the one-dimensional diffusion exerts antenna effect.
Discussion
The DNA looping mechanism has a characteristic dependence on DNA length. It becomes difficult for the DNA length shorter than the persistent length, ca. 50 nm or 150 bp, since a looping of shorter DNA costs energy because of rigidity of double-stranded DNA20,21,22. This length limitation may be mitigated in two cases. In the first case, a flexible peptide links two DNA-binding sites as in the case of E. coli AraC5. In the second, relevant energy is supplied by two specific interactions in the case of the λ repressor cI6 and E. coli LacI4 or by a specific binding of another protein in the case of E. coli gal repressor9. However, TrpR molecule, 2–3 nm for a homodimer, is too small and has neither a flexible peptide linker nor extra binding surfaces generating the relevant energy on the tested DNA fragments. Moreover, when the specific site is cloned at the center of DNA as in our experiments, looping DNA should be facilitated for DNA longer than twice the persistent length, 300 bp. However, the major increase in the affinity of TrpR, 720 fold, was observed as increasing DNA length from 18 to 200 bp12, for which DNA looping is difficult to occur. Thus, consideration of DNA rigidity contradicts the looping mechanism of TrpR.
Since the one-dimensional diffusion and the looping mechanisms predict different dependences on DNA length of association and dissociation rates, we tried to measure their dependences. However, the TrpR-trpO binding cannot be fluorescently monitored23, and the rates were too fast to be determined with other available methods.
The largest difficulty of the diffusion mechanism as a cause of antenna effect is its deviation from detailed balance. This rule holds in the timescale where all the degrees of freedom of the reaction molecules become stationary11, conventionally expressed as “at equilibrium”, which might cause misunderstanding if the concept of timescale is not taken into account24. We here defined the timescale of a reaction, the summed average times cost in its forward and reverse steps. If a reactant is in excess over the other, the timescale of the binding can be defined as \(\left\{ {k_{ + } \left[ {{\text{excess}}\,{\text{reactant}}} \right] + k_{ - } } \right\}^{ - 1}\), which is the inverse of decay time of the binding reaction shown in Fig. 2a, indicating how fast the reaction reaches its stationary state.

Comparison of chemical ratchet mechanism with a conventional description of a binding reaction. The binding reaction composed of two reactants and a product complex (free DNA, free protein, and the complex) is here considered. (a) The simplest binding reaction and its potential of mean force along the reaction coordinate in conventional description. (b) The product complex exists in two forms, the more stable complexA and a less stable complexB in conventional description. (c) Chemical ratchet in which two or more potentials of mean force alternate. During reaction A, the association is dominant to form more stable complexA which is more stable, while during reaction B, the dissociation becomes dominant to dissociate unstable complexB, generating an alternating reaction flow. In the dissociation reaction of reaction B, a rate constant, \(k_{ - }^{B}\), may not be able to be defined because the complexB may not in the local minimum of the potential.
In Fig. 2, we schematically show three mechanisms of bimolecular binding. Among them, the simplest basic protein-DNA binding reaction is shown in Panel (a), while the reaction with additional forms of complex is shown in Panel (b). The potential mean force along the reaction coordinate shows local minima of the number of states of free reactants and complexes and the potential is time independent. In panels (a) and (b), the slowest timescale is that of the binding reactions and others of internal degrees of freedom are faster. Therefore, detailed balance holds on the slowest timescale14, binding in Panels (a) and (b).
In chemical ratchet, the potential of mean force changes in time: reaction A and reaction B alternate with more stable complexA and with less stable conplexB, respectively, in the example shown in Panel (c). Although the schematic illustrations in Panels (b) and (c) may look similar, there is a critical difference. Two complexes are converted to each other via the direct \(k_{ \pm }^{AB}\) pathway and/or the stepwise \(k_{ \pm }^{A}\) and \(k_{ \pm }^{B}\) pathways via the free state in Panel (b). The conversion is described with rate equations. In contrast, in Panel (c), chemical ratchet, two complexes are alternative. ComplexA cannot exist in reaction B, and complexB cannot exist in reaction A, making the switching unable to be described with a single set of rate equations where all steps are stochastic. During reaction A, the equilibrium is inclined toward complexA, while during reaction B, it is inclined toward the free state because of unstable complexB. Therefore, in a microscopic view, the dominant reaction during reaction A is association, while that during reaction B is dissociation, making alternating reaction flow.
The phases of switching are random for each DNA molecule and, and thus the ensemble average essentially becomes time independent due to the cancellation among the microscopic differences, resulting in a stationary state at non-equilibrium. Since detailed balance requires perfect equilibrated stationary state, this non-equilibrium state of chemical ratchet is indifferent to the detailed balance of the binding reaction.
We are now ready to explain the length dependence of Kd in Eq. (1), the antenna effect of TrpR. One-dimensional diffusion of TrpR along DNA can accelerate its association to trpO site as well as its dissociation from the site. If reaction A is more dependent on one-dimensional diffusion, while reaction B is less, the association is more accelerated for longer DNA, while dissociation not as much. Therefore, the longer the DNA, the smaller the value of Kd.
The length dependence of Kd can be kinetically derived under the condition that DNA length is short enough for one-dimensional diffusion to be equilibrated. Furthermore, we also assume that the isomerization between the specific and nonspecific complexes is also equilibrated in the timescale of the binding. The kinetic changes of the complex and free components are calculated and averaged over a cycle of the switching to obtain Kd (Supplementary). The calculated length dependence well agreed with the theoretical curve obtained as a stationary solution of a stricter differential equation 12. As shown in the red line in Fig. 3a, the calculation is only significant for DNA length shorter than the sliding distance (ca. 600 bp) previously determined12 because of the assumption of rapid diffusion..

(a) The theoretical length dependence (red line) with the parameters fitted to the experimental results agreed with that obtained from the stationary solution of the reaction–diffusion differential equation (blue line). The experimentally determined values of Kd (filled circles) and the theoretical curve in blue were taken from Ref12. The size of the trpO site is a common parameter and showed essentially the same value in the two analyses to be 18 bp. (b) One of the possible molecular models for the chemical ratchet of TrpR (gray homodimer) binding to trpO. DNA is illustrated as a thick brown bar and trpO site is illustrated as double stranded DNA to emphasize the specific interaction (red box) in the major DNA groove. ComplexA is stable, while complexB is unstable due to the specific interaction damaged by an infrequent DNA bending at trpO (see the text).
To support the reality of the chemical ratchet model, we should propose at least one possible molecular model based on the knowledge on protein-DNA binding (Fig. 3b). ComplexA and complexB have straight and bent DNAs at trpO, respectively. ComplexA is a stable complex, while complexB is an unstable intermediate. ComplexB tends to dissociate in concerted manner with the bending because DNA bending at trpO in the complex distorts the DNA grooves and disrupts the specific interactions between TrpR and trpO DNA. The reactions from complexB, its dissociation or straightening DNA, cannot be described with rate equations because complexB is not at a significant local minimum on the potential of mean force. The DNA bending as well as straightening in the complex are expected to occur much less frequently than those of naked DNA due to the specific protein-DNA interactions, providing the degree of freedom slower than that of binding. Therefore, bending DNA and straightening DNA can be the switching of chemical ratchet. There are, however, a lot of other possible models for a chemical ratchet.
The research on TrpR and trpO has a long successful history25 but there remains a problem on the specificity and the level of TrpR in cells. In E. coli cells, TrpR must significantly saturate the trpO site to show its function, and thus the intercellular concentration of TrpR must be close to values of Kd. If we suppose the concentration is equal to Kd, and suppose the affinity for nonspecific site is S-fold weaker than that of the trpO, the amount of the specific complex at trpO per genome and that of the complex at a nonspecific site are respectively,
When N is the number of exposed nonspecific sites, there should be at least \(\frac{1}{2} + \frac{2N}{{S + 1}} \sim \frac{2N}{S}\) molecules of TrpR per genome. The value of N is at least 106, because about a half of the DNA in E.coli cells is shown to be exposed by the quantitative footprinting assay of IHF in vivo26. In the gel-retardation assay using 90 bp trpO DNA fragment in vitro, the value of S has been determined to be ca. 300 because half of DNA molecules retain one nonspecific complex per specific complex at [TrpR] = 300 Kd23. From the values of N and S, there should be at least 3,000 TrpR molecules per genome existing in cells.
However, this estimate is contradictory to the measured level of TrpR, ca. 300 molecules per genome27, by an order smaller than those required to occupy trpO. The antenna effect of TrpR can solve this problem, if the sliding distance in vivo is 500 bp or longer, because the value of S in the cell is expected to be an order of magnitude larger than that for 90 bp (Fig. 3a).
Antenna effect, which can be driven by chemical ratchet, may provide a novel interpretation to experimental observation of DNA sequence dependency of protein binding affinity. Lukatsky and his colleague found that several proteins enhance the affinity of the specific complex depending on the sequence of DNA segments flanking the specific site in vivo and in vitro28,29,30,31. The enhancing sequences were repetitive or homopolymeric sequence, which had been known to facilitate one-dimensional diffusion32, as the authors estimated the contribution of the diffusion.
The studies on one-dimensional diffusion have been developed with the focus on best combination of different diffusion modes33, the overlooking of the specific site34, speed-selectivity paradox35,36, and so on. Since the acceleration is a kinetic effect, the contribution of the acceleration is temporally limited to the phenomena with timescales similar to the accelerated association rates, say seconds or less, as long as detailed balance holds. Chemical ratchet can link the rapid kinetic effect to the physiological phenomena with much slower timescales through antenna effect. In this way, one-dimensional diffusion may contribute to gene expression and its regulation in more general way than the kinetic effect.
Materials and methods
Protein and DNA
E. coli TrpR protein was provided by Dr. Jeannette Carey. The 36 bp and 268 bp DNAs were prepared with PCR and purified by electrophoresis in an 8% polyacrylamide gel, followed by simple diffusion from the crushed gel slices. The biotin-attached 36 bp and 232 bp DNA fragments were prepared using 5′-end biotinylated primers. The 36 bp biotinylated DNA was next preincubated with a two-fold excess of avidin (avidin DN from Upstate Biotechnology) for 1 h, then mixed with a two-fold excess of 232 bp biotinylated DNA. Avidin from some other commercial sources could not be used because of nonspecific binding of DNA. The 36 + 232 bp DNA connected by biotin-avidin was isolated by using 8% polyacrylamide gel.
A series of DNA fragments harboring a LexA binding site were prepared by PCR using different primers and inserted at a XhoI site of pFF6 plasmid, which carries lacZ with dual origins of pBR101 and the miniF18. Single-copy plasmids were then prepared by replacing the pBR origin with a trpO fragment at the Hind III site of the plasmid. The plasmids were used to transform E. coli MC4100 strain and the transformants were grown in M9 medium with or without 0.25 mM L-tryptophan. Growth was rapidly halted at OD600 = 0.5–0.7 by immersing an aliquot in liquid nitrogen. The samples were then kept frozen until the standard β-galactosidase assay was performed.
Hydroxyl radical footprinting
All the binding and footprinting experiments were performed as already described15. The Fenton reagent was freshly prepared from concentrated solutions and all the measurements for a DNA were finished before the aging period that had been determined in preliminary experiments. Reaction was stopped by an addition of glycerol to 20%. To satisfy the single-cutting condition, the period of cleavage reaction was limited so that less than 20% of the full-length DNA fragment had disappeared according to the results of preliminary experiments. Fitting the data to Eqs. (1) and (2) was carried out by the least-squares method with MacCurveFit 1.5 and the standard deviations were obtained from the sum of the squared errors.
Measurements of TrpR-trpO binding in vivo
In the measurement in vivo, determination of the Mirror unit, required many cautions. The observed value was dependent on the lot of culture media and the recovery procedure of E. coli cells from its stock solution. Thus we prepared a large volume of the media and kept using the same lot. The cells were recovered three times in the fresh lot with the same dilutions into the fresh medium and the same shaking process taking three days. Growth was rapidly halted at OD600 = 0.5–0.7 by immersing an aliquot in liquid nitrogen. The samples were then kept frozen until the standard β-galactosidase assay was performed. This whole process was repeated three times as listed in Table 2.
References
Shimamoto, N. One-dimensional diffusion of proteins along DNA. Its biological and chemical significance revealed by single-molecule measurements. J. Biol. Chem. 274, 15293–15296 (1999).
Winter, R. B. & von Hippel, P. H. Diffusion-driven mechanisms of protein translocation on nucleic acids. 2. The Escherichia coli repressor-operator interaction: equilibrium measurements. Biochemistry 20, 6948–6960 (1981).
Khoury, A. M., Lee, H. J., Lillis, M. & Lu, P. Lac repressor-operator interaction: DNA length dependence. Biochem. Biophys. Acta. 1087, 55–60 (1990).
Borowiec, J. A., Zhang, L., Sasse-Dwight, S. & Gralla, J. D. DNA supercoiling promotes formation of a bent repression loop in lac DNA. J. MoL Biol. 196, 101–111 (1987).
Harmer, T., Wu, M. & Schleif, R. The role of rigidity in DNA looping-unlooping by AraC. Proc Natl. Acad. Sci. USA 98, 427–431 (2001).
Hochschild, A. & Ptashne, M. Cooperative binding of λ repressors to sites separated by integral the DNA helix. Cell 44, 681–687 (1986).
Ushida, C. & Aiba, H. Helical phase dependent action of CRP: effect of the distance between the CRP site and the -35 region on promoter activity. Nucleic Acids Res. 18, 6325–6330 (1990).
Vanzi, F., Broggio, C., Sacconi, L. & Pavone, F. S. Lac repressor hinge flexibility and DNA looping: single molecule kinetics by tethered particle motion. Nucl. Acids Res. 34, 3409–3420 (2006).
Aki, T. & Adhya, S. Repressor induced site-specific binding of HU for transcriptional regulation. EMBO J. 16, 3666–3674 (1997).
Surby, M. A. & Reich, N. O. Facilitated Diffusion of the EcoRI DNA Methyltransferase Is Described by a Novel Mechanism. Biochemistry 35, 2209–2217 (1996).
van Kampen, N. G. Stochastic Processes in Physics and Chemistry 3rd edn, 114–117 (Elsevier, New York, 2007).
Shimamoto, N. et al. Dependence of DNA length on binding affinity between TrpR and trpO of DNA. Sci. Rep-UK 10, 15624 (2020).
Jeltsch, A., Wenz, C., Stahl, F. & Pingoud, A. Linear diffusion of the restriction endonuclease EcoRV on DNA is essential for the in vivo function of the enzyme. EMBO J. 15, 5104–5111 (1996).
Shicker, P. & Heumann, H. Hydroxyl radical footprinting. In Methods of molecular Biology DNA-Protein Interactions (ed. Kneale, G. G.) 21–32 (Humana Press, Totowa, 1994).
Mueller-Storm, H. P., Sogo, J. M. & Schaffner, W. An enhancer stimulates transcription in trans when attached to the promoter via a protein bridge. Cell 59, 767–777 (1989).
Kitagawa, M., Wada, C., Yoshioka, S. & Yura, T. Expression of ClpB, an analog of the ATP-dependent protease regulatory subunit in Escherichia coli, is controlled by a heat shock σ factor (σ32). J. Bacteriol. 173, 4247–4253 (1991).
Lewis, L. K., Harlow, G. R., Gregg-Jolly, L. A. & Mount, D. W. Identification of high affinity binding sites for LexA which define new DNA damage-inducible genes in Escherichia coli. J. Mol. Biol. 241, 507–523 (1994).
Gaston, K., Bell, A., Kolb, A., Buc, H. & Busby, S. Stringent spacing requirements for transcription activation by CRP. Cell 62, 733–743 (1990).
Kelley, R. L. & Yanofsky, C. Trp aporepressor production is controlled by autogenous regulation and inefficient translation. Proc. Natl. Acad. Sci. USA 79, 3120–3124 (1982).
Hagerman, P. J. Investigation of the flexibility of DNA using transient electric birefringence. Biopolymers 20, 1503–1535 (1981).
Taylor, W. H. & Hagerman, P. J. Application of the method of phage T4 DNA ligase-catalyzed ring-closure to the study of DNA structure: II. NaCl-dependence of DNA flexibility and helical repeat. J. Mol. Biol. 212, 363–376 (1990).
Smith, S. B., Finzi, L. & Bustamante, C. Direct mechanical measurements of the elasticity of single DNA molecules by using magnetic beads. Science 258, 1122–1126 (1992).
Carey, J. Gel retardation at low pH resolves trp repressor-DNA complexes for quantitative study. Proc. Natl. Acad Sci. USA 85, 975–979 (1988).
van Kampen, N. G. Stochastic Processes in Physics and Chemistry 3rd edn, 171 (Elsevier, New York, 2007).
Yanofsky, C. & Crawford, I. P. The tryptophan operon. In Escherichia coli and Salmonella typhimurium 1st edn (eds Ingraham, J. L. et al.) 1453–1472 (American Society of Microbiology, Washington, 1987).
Yang, S. W. & Nash, H. A. Comparison of protein binding to DNA in vivo and in vitro: defining an effective intracellular target. EMBO J. 14, 6292–6300 (1995).
Gunsalus, R. P., Miguel, A. G. & Gunsalus, G. L. Intracellular trp repressor levels in Escherichia coli. J. Bacteriol. 167, 272–278 (1986).
Afek, A. & Lukatsky, D. B. Nonspecific protein-DNA binding is widespread in the yeast genome. Biophys. J. 102, 1881–1888 (2012).
Afek, A. & Lukatsky, D. B. Genome-wide organization of eukaryotic preinitiation complex is influenced by nonconsensus protein-DNA binding. Biophys. J. 104, 1107–1115 (2013).
Afek, A. & Lukatsky, D. B. Positive and negative design for nonconsensus protein-DNA binding affinity in the vicinity of functional binding sites. Biophys. J. 105, 1653–1660 (2013).
Afek, A., Schipper, J. L., Horton, J., Gordaˆn, R. & Lukatsky, D. B. Protein-DNA binding in the absence of specific base-pair recognition. Proc. Natl. Acad. Sci. USA 111, 17140–17145 (2014).
Schurr, J. M. The one-dimensional diffusion coefficient of proteins absorbed on DNA hydrodynamic considerations. Biophys. Chem. 9, 413–414 (1979).
Jeltsch, A. & Pingoud, A. Kinetic characterization of linear diffusion of the restriction endonuclease EcoRV on DNA. Biochemistry 37, 2160–2169 (1998).
Slutsky, M. & Mimy, L. Kinetics of protein-DNA interaction: Facilitated Target Location in Sequence-dependent potential. Biophys. J. 87, 4021–4035 (2004).
Mondal, A. & Bhattacherjee, A. Searching target sites on DNA by proteins: Role of DNA dynamics under confinement. Nucleic acids Res. 43, 9176–9186 (2015).
Zandarashvilia, L. et al. Balancing between affinity and speed in target DNA search by zinc-finger proteins via modulation of dynamic conformational ensemble. Proc. Natl. Acad. Sci. USA 112, E5142–E5149 (2015).
Acknowledgements
We thank the late Dr. J. Tomizawa of the National Institute of Genetics for his critical help in constructing the logic to interpret the observations and for helpful comments on the manuscript. The proposal of chemical ratchet has been made in collaboration with Dr. M. Toda (Nara Women’s University), Dr. S. Nara (Okayama University), T. Komatsuzaki (Hokkaido University), and Dr. K. Kamagata (Tohoku University). We thank Dr. J. Carey (Princeton) for providing the TrpR protein and encouraging discussion and Ms. Harriet Sallach for close reading of the manuscript. Supported by grants from the Japan Society for the Promotion of Science for Young Scientists (T. K.) and by grants from The Ministry of Education, Culture, Sports, Science and Technology (N. S.).
Author information
Authors and Affiliations
Contributions
T.K. has done all the bench works, including methodology. N.S. designed the experiment and all the desk works.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kinebuchi, T., Shimamoto, N. One-dimensional diffusion of TrpR along DNA enhances its affinity for the operator by chemical ratchet mechanism. Sci Rep 11, 4255 (2021). https://doi.org/10.1038/s41598-021-83156-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-021-83156-6
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
