
Abstract
The role of pyrite (FeS2) in the process of water treatment using metallic iron (Fe0) was investigated. FeS2 was used as a pH-shifting agent while methylene blue (MB) and methyl orange (MO) were used as an indicator of reactivity and model contaminant, respectively. The effect of the final pH value on the extent of MB discoloration was characterized using 5 g L−1 of a Fe0 specimen. pH variation was achieved by adding 0 to 30 g L−1 of FeS2. Quiescent batch experiments with Fe0/FeS2/sand systems (sand loading: 25 g L−1) and 20 mL of MB were performed for 41 days. Final pH values varied from 3.3 to 7.0. Results demonstrated that MB discoloration is only quantitative when the final pH value was larger than 4.5 and that adsorption and co-precipitation are the fundamental mechanisms of decontamination in Fe0/H2O systems. Such mechanisms are consistent with the effects of the pH value on the decontamination process.
Similar content being viewed by others

Introduction
The removal of anthropogenic and natural pollutants from aqueous systems is a major environmental concern1,2,3. Several technologies have been developed for water treatment over the past 170 years1,4,5,6,7,8. Technologies based on adsorption processes have been proven to be the most affordable and suitable for water treatment in low-income communities1,5. During the past three decades, metallic iron (Fe0) has been intensively used for in-situ environmental remediation9,10,11 and ex-situ water treatment12,13,14,15. However, controversy still exists on whether Fe0 acts as a reducing agent for several pollutants (e.g. selected chemicals)11 or a generator of contaminant scavengers (iron corrosion products–FeCPs) for all classes of pollutants (e.g. chemicals and pathogens)16,17,18. It is certain that, while undergoing oxidative dissolution, Fe0 induces contaminant removal in aqueous systems19,20,21,22,23, and this process can last for decades24,25,26,27. This ability to remove contaminants for prolonged periods has prompted the use of granular Fe0 for decentralized water treatment12,14,28,29,30.
Metallic iron (Fe0) and iron sulfide (FeS)-based materials (including pyrite–FeS2) are two important components of Fe0-based water treatment technology11,22,31,32,33. Both materials are reported to be stand-alone reducing agents that effectively degrade several aqueous contaminants34,35. In this context, Henderson and Demond34,36 have explicitly compared the suitability of both materials, and observed the superiority of FeS (including FeS2) over Fe0 with respect to the sustainability in terms of loss of permeability. During the past two decades, Fe0 and FeS2 have been often mixed in an effort to increase the efficiency of single-Fe0 systems22,33,37,38,39,40,41. However, FeS2 is mostly added to avoid the formation of a passive oxide scale (oxide film) which can hinder further reactions between the Fe0 and pollutants41,42. This application contradicts the successful use of FeS2 to improve the removal of non-reducible contaminants (e.g. As) in Fe0/H2O systems22. Thus, there is a need to understand the real mechanism by which FeS2 improves the efficiency of Fe0/H2O systems, irrespective of any redox transformation. The oxidative dissolution of both Fe0 (Eq. 1) and FeS2 (Eq. 2) typically releases Fe2+, which is also a stand-alone reducing agent for several contaminants35. Fe2+ from Eq. (1) and/or Eq. (2) can be further oxidized to Fe3+ (Eq. 3).
It is evident that both reactions depicted by Eqs. (1 and 3) consume acidity (H+), while reaction in Eq. 2 produces H+. According to the Le Chatelier’s principle, the reactions consuming H+ (Eq. 1 and 3) are accelerated by pyrite oxidation (Eq. 2). Here, the forward Fe0 dissolution is given priority as it is the main reactant, while less attention is paid to the possible inhibitory effect of FeS2 oxidation on Fe0 dissolution (production of Fe2+). In fact, using FeS2 to enhance iron corrosion is consistent with scientific principles (“Background to the experimental methodology”). However, this understanding provides no insights on the mechanisms of decontamination (adsorption, co-precipitation) and/or induced redox transformation of contaminants (degradation, precipitation). The stoichiometry of the reaction in Eq. (1) shows that 1 mol of Fe0 generates one mole of Fe2+ and one mole of H2, while 1 mol of FeS2 generates only one mole of Fe2+ (Eq. 2). Given that Fe2+ and H2 are stand-alone reducing agents, it is clear that there are more reducing agents (electron donors) in Fe0/H2O than in FeS2/H2O systems. The actual reductive characteristics of each system (i.e., Fe0/H2O, FeS2/H2O, and Fe0/FeS2/H2O) primarily depend on the relative dissolution kinetics of Fe0 and FeS2. According to their relative electrode potentials (-0.44 V for FeII/Fe0 versus 0.25 V for SIII/S-I), Fe0 should be transforming SIII species back to S-I ones. This would correspond to the blocking of oxidative dissolution of Fe0. Although this reaction is thermodynamically feasible, it is masked by the more kinetically favourable FeS2 oxidation by dissolved O2. In the long-term (i.e., over the 41 d investigated herein), this process could contribute to the FeII cycle within a Fe0/FeS2 /H2O system.
The presentation above demonstrates the extreme complexity of the Fe0/FeS2/H2O and accounts for the controversies in the literature on the role of FeS2 in enhancing the efficiency of Fe0/H2O systems22,33,41. Thus, a detailed investigation of the efficiency of the Fe0/H2O system as influenced by the presence of FeS2 is warranted. A critical evaluation of the Fe0 literature suggests that one major limitation has been to test the efficiency of the Fe0/H2O system on individual contaminants or groups of contaminants (e.g. As, dyes, halogenated carbons)22,28,33,41,43. The net result is that increased adsorption, co-precipitation, degradation or precipitation have been reported as the supposed removal mechanisms. An innovative approach was introduced by Miyajima and colleagues using methylene blue (MB) as an indicator of reactivity for the Fe0/H2O system (MB method)44,45. The MB method exploits the differential adsorptive behaviour of MB onto sand and iron oxides or iron-coated sand44,45,46,47. Accordingly, in parallel experiments using constant amounts of sand and different Fe0 specimens, the most reactive system has been the one exhibiting the least MB discoloration, or the one producing the largest amount of iron oxides47,48,49. Note that the exact nature of the oxide is not very important, but its coating activity is the key aspect for the MB method. For example, Banerji and Chaudhari12 did not add any sand in their Fe0 bed to avoid oxide loss by sand coating. The objective of the current study was to characterize the impact of FeS2 addition on the reactivity of the Fe0/H2O system using the MB method. Thus, six different FeS2 mass loadings were used to achieve different final pH values (Eq. 2) (4.5 ≤ pHfinal ≤ 5.2).
Material and methods
The present research is based on the chemistry of the Fe0/FeS2/sand/H2O system. Therefore, the operating mode of the system will be first discussed. In this study, various amounts of a reactive FeS2 mineral were added to a Fe0/sand mixture to investigate their effects on pH shifts and dye removal.
Background to the experimental methodology
At neutral pH values, immersed reactive Fe0 corrodes and generates solid iron corrosion products (FeCPs), which progressively coat the surface of sand. The process of iron corrosion causes a pH shift to higher values (Eq. 1). The extent of sand coating depends among other factors on: (i) the Fe0 intrinsic reactivity, (ii) the volume of the solution, (iii) the initial pH value of the solution, (iv) the Fe0/sand ratio, and (v) the duration of the experiment. Under given experimental conditions, the removal efficiency of the system for individual contaminants depends on the final pH value, the extent of sand coating, and the availability of “free” FeCPs. The final pH value determines the speciation of the contaminant and the surface charges of sand and FeCPs50.
When a FeS2 mineral is added to a Fe0/sand system (at a given Fe0:sand ratio) a pH shift to lower values occurs. The extent of pH shift depends on the FeS2 intrinsic reactivity and the amounts added. Lower pH values avoid or delay sand coating and modify the speciation of dissolved contaminants. It then follows that, when FeS2 is added to a Fe0/sand mixture, there are two counteracting processes controlling the pH value of the system37,38,39. Previous results observed with the FeS2 mineral used in the current study41 suggest that pyrite dissolution occurs with much rapid kinetics than Fe0 corrosion. Consequently, the system will not achieve a steady state before the initial pH of the FeS2-free system is achieved. The larger the pH shift the larger the amount of FeCPs generated, which will in turn precipitate at pH > 4.5 and induce contaminant removal by adsorption and co-precipitation.
The methodology used for characterizing the impact of FeS2 on the efficiency of Fe0/H2O systems comprises monitoring the discoloration of a methylene blue solution (MB method) by Fe0/sand systems amended with various FeS2 amounts. Clearly, the availability of FeCPs and their reactivity is modified by lowering the initial pH value to various extents while observing MB discoloration in systems having a final pH value between 4.0 and 5.0. The discoloration of methyl orange (MO) in parallel experiments is used to support the findings based on the MB method. This approach is radically different from the conventional approach testing dyes as model contaminants33,51. For example, Chen et al.33 recently investigated the removal of three different azo dyes (Orange II, Reactive Red X-3B and Amido Black 10B) in the Fe0/FeS2/H2O system. All the three dyes are negatively charged, and were explicitly reported to be removed via reductive transformations. Following the conventional approach, Chen et al.33 monitored the concentrations of dyes, iron and protons (pH value), and performed solid phase characterizations using scanning electron microscopy (SEM), energy dispersive X-ray spectroscopy (EDS) and X-ray photoelectron spectroscopy (XPS). On the contrary, the MB method does not imply such solid phase characterizations because all FeCPs are positively charged50 and the extent to which they cover sand is reflected in the extent of MB discoloration.
Solutions
Dyes
Methylene blue (MB) was used as a tracer of reactivity47, while methyl orange (MO) was a model organic contaminant52. Both dyes are widely used to characterize the suitability of various systems for water treatment46,52,53,54,55. The used dyes were of analytical grade. MB was supplied by Sinopharm Chemical Reagent Co. Ltd, Shanghai (China) and MO by Tianjin Chemical Reagent Research Institution Co. Ltd, Tianjin (China). The dyes were selected due to: (i) similarity in their molecular size, and (ii) differences in their affinity to positively charged iron oxides (Table 1)55. The initial dye concentration used was 10 mg L−1, equivalent to 31.5 μM for MB and 30.7 μM for MO. The working solutions were prepared by diluting concentrated stock solutions (3150 μM for MB and 3070 μM for MO) using deionized water. The pH values of the initial solutions were 6.5 (MB) and 7.0 (MO).
Iron
A standard iron solution (1000 mg L−1) from General Research Institute for Nonferrous Metals was used to calibrate the UV/VIS spectrophotometer used for analysis. In preparation for spectrophotometric analysis, ascorbic acid was used to reduce FeIII in solution to FeII. 1,10 orthophenanthroline was used as reagent for FeII complexation48,49,55. Other chemicals used in this study included L( +)-ascorbic acid and L-ascorbic acid sodium salt. Ascorbic acid also degrades dyes (in particular MO) and eliminates interference during iron determination.
Solid materials
Metallic iron (Fe0)
The Fe0 material was purchased from Shanghai Institute of Fine Technology (China). The material is available as scrap iron with a particle size between 0.05 and 5 mm. Its elemental composition as specified by the supplier was: Fe: > 99.99%; C: < 0.1%; N: < 0.1%; O: < 0.1%. Its kPhen value is 13 mg h−156. The kPhen value is the kinetic constant of Fe0 dissolution in a 2 mM 1,10 orthophenanthroline solution, and characterizes the material’s intrinsic reactivity57. The material was used without any further pre-treatment. Fe0 was proven as a powerful discoloration agent for MB specifically because the discoloration agents are progressively generated in-situ45,55. Therefore, the discoloration capacity of the used Fe0 cannot be exhausted within the experimental duration used in the current study (41 d).
Sand
The sand conformed to the China ISO standard, and was used as received without any further pre-treatment or characterization. The particle size was between 1.25 and 2.00 mm. Sand was used because it is cheap and readily available and is widely used as admixing agent to prevent rapid permeability loss in Fe0/H2O systems58.
Pyrite (FeS2)
The FeS2 mineral was from Tongling City, Anhui province, China. The particle size was between 38 and 48 μm. Its weight composition was 46.0% Fe and 52.2% S, which is equivalent to a purity of 98.2%41. FeS2 was used because of its demonstrated suitability as a pH shifting agent in Fe0/H2O systems37,41,59.
Dye discoloration experiments
Quiescent batch experiments were conducted in glass test tubes for an experimental duration of 41 d. Dye discoloration was initiated by adding 20.0 mL of the dye solution to a test tube containing 0.1 g of Fe0, 0.0 to 0.6 g of FeS2, 0.0 or 0.5 g of sand, and Fe0/FeS2/sand mixtures containing varying FeS2 loadings. Table 2 summarizes the aggregate content of the 8 Fe0/FeS2/sand systems and one operational reference (blank experiment), giving a total of 9 experimental treatments. Note that the pure Fe0 system (0.1 g of Fe0) is regarded as a Fe0/FeS2/sand system without FeS2 nor sand. The addition of sand was meant to avoid the compaction of the materials by gelatinous FeCPs via cementation55.
The efficiency of individual Fe0 systems for dye discoloration was characterized at laboratory temperature (about 25 ± 2 °C). The final pH value, the iron concentrations and the residual dye concentrations were recorded. All experiments were carried out in triplicates under laboratory (oxic) conditions. The test tubes were protected from direct sunlight.
Analytical methods
Aqueous dye and iron concentrations were determined by a 752 UV/VIS spectrophotometer (automatic) (Shanghai Jing Hua Technology Instrument Co. LTD). The working wavelengths for MB, MO and iron were 664.5, 464.0 and 510.0 nm, respectively. Cuvettes with a 1.0 cm light path were used. The iron determination followed the 1,10 orthophenanthroline method60. The spectrophotometer was calibrated for dye concentrations ≤ 15.0 mg L−1 and iron concentration ≤ 10.0 mg L−1. The pH value was measured by combined glass electrodes (INESA Scientific Instrument Co. China).
Presentation of experimental results
In order to characterize the magnitude of the tested systems for dye discoloration, the discoloration efficiency (E) was calculated (Eq. (4)). After the determination of the residual dye concentration (Ct), the corresponding percent dye discoloration efficiency (E value) was calculated as:
where C0 is the initial aqueous dye concentration (about 10.0 mg L−1), while Ct gives the final dye concentration at sampling time (t). The operational initial concentration (C0) for each case was acquired from a triplicate control experiment without additive materials (blank). This procedure was mainly meant to account for experimental errors due to dye adsorption onto the walls of the test tubes.
Results and discussion
Dye discoloration in single-aggregate and ternary-aggregate systems
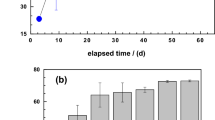
Figure 1a compares the extent of dye discoloration in the four investigated systems: (i) single-Fe0, (ii) single-FeS2, (iii) single-sand, and (iv) Fe0/FeS2/sand. Figure 1b shows the final pH variation with varying FeS2 doses in the ternary Fe0/FeS2/sand systems. Figure 1a clearly shows that there was no MO discoloration in the pure sand system (E = 0%). The E values for both dyes in all other systems were larger than 30%. The uniqueness of the single-sand system (100% sand) relative to the other three systems is that it contains no in-situ generated FeCPs (Table 3). Therefore, only sand with its negatively charged surface46,50 is available for dye discoloration via pure electrostatic interactions. Strong surface interactions with positively charged species is thus responsible for the observed MB discoloration, but no MO discoloration occurs in the single-sand system44,61. As quiescent batch experiments were performed (no advection), diffusive mass transfer in the bulk solution and/or in the pores of generated FeCPs are the rate-limiting steps for the discoloration process.

Changes of the dye discoloration efficiency (E values) in single-aggregate and ternary system (a) and changes of final pH value as a function of the FeS2 dose (b). Experimental conditions: V = 20 mL, miron = 0.0 or 0.1 g, msand = 0.0 or 0.5 g, mpyrite 0 to 0.6 g, and t = 41 d. The lines are not fitting functions, they simply connect points to facilitate visualization.
The absence of MO discoloration in the single-sand system is the most important observation from these experiments. MO discoloration is observed in all other systems and is consistently more intensive than MB discoloration in single-Fe0 and single-FeS2 systems (“Dye discoloration in Fe0/sand/H2O systems”). For the Fe0/FeS2/sand system there is no pronounced difference in the discoloration of both dyes. It is recalled that the Fe0/FeS2/sand system contains 5 g L−1 Fe0, 25 g L−1 of sand and 20 g L−1 of FeS2. Thus, according to Table 2, the extent of dye discoloration depends on: (i) the availability of adsorption sites on inert sand (adsorption on sand), (ii) the extent to which sand is covered by in-situ generated FeCPs (selective adsorption on sand and/or FeCPs), and (iii) the extent to which excess FeCPs can be freely precipitated in the bulk solution (co-precipitation with FeCPs) (see Table 3). In this case, free precipitation herein means FeCPs that are not coating the sand surface (“Background to the experimental methodology”)37,38,44,61.
The merit of the experimental design is to demonstrate dye discoloration in a Fe0/sand/H2O systems as the FeS2 mass loadings vary from 0 to 30 g L−1. Figure 1b clearly shows that varying the FeS2 mass loading under the experimental conditions has resulted in various final pH values, ranging from 4.5 to 7.0 (Table 4). The results summarized in Table 4 also show lower pH values in all FeS2-containing systems with the single-FeS2 having a pH value of 3.3 ± 0.2 for both dyes. Notably, comparison of MO versus MB showed no profound difference in the final pH value (4.7 vs 4.8), the iron concentration (63 vs 65) and the E values (71 vs 73) in the ternary system with a FeS2 mass loading of 20 g L−1. An exception was the Fe0/H2O system without FeS2 (i.e., [FeS2] = 0 g L−1) for which a significant difference in the pH value was observed (6.5 for MB vs. 7.0 for MO). This corresponds to the pH value of the respective initial dye solutions (“Solutions”). The ternary-aggregate system used in this case comprised of 20 g L−1 FeS2. For a better illustration of the role of the FeS2 mineral in the process of dye discoloration in Fe0/H2O systems, three lower (5, 12 and 18 g L−1) and two higher (24 and 30 g L−1) FeS2 doses were also used.
Iron release in Fe0/sand/H2O systems
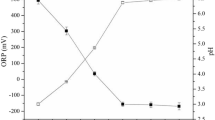
Figure 2 compares the iron concentration in the Fe0/sand/H2O systems as the FeS2 loadings vary from 0 to 30 g L−1. Figure 1b and Table 4 have shown a monotonous, but non-linear decrease of the pH value with increasing FeS2 mass loadings. Contrary, Fig. 2a shows a monotonous and linear increase of the iron concentration with increasing FeS2 mass loadings. For a constant FeS2 loading, the final pH values (Fig. 1b) and the [Fe] values (Fig. 2a) were almost the same for both dyes. This observation suggests that the differential behavior of MB and MO in interacting with the involved aggregates, particularly Fe0 and sand, is not reflected in changes of the pH value. Thus, the final pH value arises from the pseudo-steady state equilibrium between two antagonistic processes: (i) Fe0 corrosion which increases the pH value (Eq. 1), and (ii) FeS2 dissolution which decreases the pH value (“Background to the experimental methodology”). Figure 2b clearly shows that lower pH values correspond to higher [Fe] values. Specifically, the [Fe] values dropped from 100 mg L−1 for pH 4.7 to 0.7 mg L−1 for pH 7.0. A sharp decrease of the iron concentration between pH 4.7 and 5.5 is observed and corresponds to the solubility behavior of Fe under oxic conditions62,63. Again, there is no significant difference evident between both dyes.

Changes of the iron concentration as function of the pyrite dose (a) and the final pH value (b). Experimental conditions: V = 20 mL, miron = 0.1 g, msand = 0.5 g, mpyrite 0 to 0.6 g, and t = 41 d. The lines are not fitting functions, they simply connect points to facilitate visualization.
The fact that iron dissolution from any reactive material increases with decreasing pH value is intuitive (Fig. 2a). However, the extent to which iron is dissolved under any given operational conditions should be characterized (Fig. 2b), and their impact on the investigated process (dye discoloration in this case) discussed. Past efforts to characterize the Fe0/FeS2 system have not properly considered these issues as the final pH value was not always recorded and/or not used in discussing the results22,41. Moreover, in earlier efforts it was commonplace to vary both the initial pH value and the FeS2 loading (Table 5), thereby making it difficult to determine the effect of each parameter. By using only various FeS2 loadings to shift the pH value of a Fe0/sand/H2O system, the present study is an extension of earlier efforts from the early 2000s37,38,39. Moreover, the current study applied a recently developed tool using MB as an indicator of Fe0 reactivity44,45,47.
Dye discoloration in Fe0/sand/H2O systems
Figure 3a shows the extent of dye discoloration (E values) by the ternary system as the FeS2 loadings increase from 0 to 30 g L−1. Figure 3b depicts the variation of E values as a function of the final pH value. It is evident that there is a general linear decrease in E value with increasing FeS2 loading or decreasing pH values (Fig. 2a). However, three important issues have to be considered: (i) the highest E value for each system corresponds to [FeS2] = 0 g L−1 (Issue 1); (ii) for [FeS2] = 5 g L−1 and [FeS2] > 20 g L−1, there is a significant difference in the E values for both dyes (Issue 2), and (iii) for [FeS2] = 10, 15 and 20 g L−1, there is no profound difference in the E values for both dyes (Issue 3). It is evident that such results would be considered controversial if coming from different independent studies. That is why considering the experimental conditions is crucial in discussing experimental result from independent studies18,23. For the so-called bottle-point technique used herein64, relevant operational variables include: Fe0 pre-treatment, Fe0 particle size (mm, mm, nm), Fe0 mass loading, volume of used vessels, volume of solution, buffer application, mixing type (e.g. stirring, shaking), mixing intensities (e.g. 200 rpm), and experimental duration43. A more detailed discussion of the three issues is given below.

Changes of the dye discoloration efficiency (E values) as function of the pyrite dose (a) and the final pH value (b). Experimental conditions: V = 20 mL, miron = 0.1 g, msand = 0.5 g, mpyrite 0 to 0.6 g, and t = 41 d. The lines are not fitting functions, they simply connect points to facilitate visualization.
Issue 1 implies that FeS2 addition inhibits the efficiency of Fe0/H2O systems for dye discoloration. Similar results were reported by Noubactep et al.37,38,39 while investigating UVI removal in Fe0/H2O systems. Note that the single-FeS2S2 discolored both dyes (Fig. 1a). This observation raises questions about the assertion that FeS2 increases contaminant removal in Fe0/H2O systems22,33,41.
Issue 2 can be regarded as a striking feature as there is either a larger extent of MO discoloration relative to that of MB ([FeS2] = 5 g L−1) or the opposite ([FeS2] > 20 g L−1). Note that the ion-selectivity principle of the Fe0/H2O system implies that in the presence of FeCPs in aqueous systems, the anionic MO is better discolored than the cationic MB. This is obviously the case at [FeS2] = 5 g L−1 where enough FeCPs is generated to cover the surface of sand, thereby inducing a larger E value for MO than for MB. The observation that for Fe0/H2O systems without FeS2 (i.e., at [FeS2] = 0 g L−1), MO discoloration was only slightly higher than that of MB is also noteworthy. This indicates that for the experimental duration used in the current study (41 d), enough FeCPs were generated to co-precipitate both dyes. For further clarification of this issue, a binary Fe0/sand system should have been investigated, but this was beyond the scope of the current study. The higher MB discoloration relative to MO observed at [FeS2] > 20 g L−1 is explained by the formation of complexes between Fe and MO which delay co-precipitation55,61,65.
Finally, Issue 3 can also be regarded as a striking observation because despite all differences (solubility, affinity), there is no difference in the E values for the two dyes. It can be assumed that, under the experimental conditions, MB and MO which have almost the same molecular size (Table 1) are both discolored by co-precipitation66. This assumption is corroborated by results in Fig. 3b showing clearly that there is no quantitative dye discoloration (E > 60%) at pH < 4.5. This corresponds to the observations of Noubactep et al.37,38,39 for the Fe0/UVI/H2O system. The fact that MB, MO and UVI exhibited very similar behaviors in the Fe0/FeS2/H2O system is an indication that contaminant removal might be a pure positive side effect of aqueous iron corrosion. The most tangible proof for this assertion is the kinetics of Fe2+ oxidation by dissolved oxygen (O2). According to Langmuir67, the kinetics of this reaction increases by a factor 65 between pH 4.0 and 5.0. Thus, quantitative dye discoloration is observed only in systems where Fe2+ oxidation to Fe3+ was quantitative for the 41-d experimental period. The in-situ generated FeIII precipitates are good contaminant scavengers.
Mechanisms of contaminant removal in Fe0/H2O systems
This study has investigated the effect of FeS2 addition on the efficiency of Fe0/sand systems for MB and MO discoloration. No enhanced dye discoloration could be attributed to FeS2 addition at mass loading of 0 to 30 g L−1 for 41 d. Two questions arise: First, why is there no increased dye discoloration in a context where the expected pH shift and increased iron dissolution are evident? (Question 1). It is noteworthy that each individual aggregate (Fe0, FeS2, sand) tested herein can achieve MB discoloration as depicted in Fig. 1a. Second, why did the ternary system perform far lower that single- Fe0 systems? (Question 2). By applying a known experimental approach consisting of varying individual operational parameters to better understand complex systems17,68,69,70, and accounting for the relative slow kinetics of Fe0 and FeS2 dissolution37,38,39, this study has adopted a novel approach to answer Questions 1 and 2. Specifically, the current study assessed the role of FeS2 in enhancing contaminant removal in Fe0/H2O system. MB is used herein as an operational reactivity tracer (“Introduction”) and the achieved results corroborated earlier reports on U(VI) removal37,38,39, and account for discrepancies and inconsistencies reported in literature33,41,49,70.
The evidence that FeS2 oxidation produces acidity (Eq. 2) is corroborated in the current study (Fig. 2a). By consuming acidity, Fe0 (Eq. 1) and Fe2+ (Eq. 3) oxidation are accelerated by Eq. (2) (Le Chatelier’s principle). Fe3+ from Eq. (2) catalyses FeS2 oxidation and produced less soluble Fe(OH)3. Thus, mixing Fe0 and FeS2 can be regarded as continuously generating less soluble Fe(OH)3, until one of the reactants is depleted or until a pseudo-steady state is established. This work posits that Fe(OH)3 discolors the dye solutions mainly by co-precipitation66,70. Thus, dye discoloration is only quantitative when Fe(OH)3 precipitation is intensive (pH > 4.5). Having used quiescent systems, various final pH values could be achieved, thereby confirming the pH shifting function of FeS237,41,59. However, the extent of dye discoloration depends on the amount of free in-situ generated Fe(OH)355,70 which is determined by the kinetics of Fe2+ oxidation by dissolved O267. As expected, for a longer experimental duration (t > 41 d), the efficiency of the ternary mixture will surpass that of the single- Fe0 systems37,38,39,70. This answers Question 1, and demonstrates that enhanced dye discoloration needs more time to occur under quiescent conditions37,38,39. Accordingly, the documented delay of quantitative dye discoloration is not a negation of the view that FeS2 addition enhances the efficiency of Fe0/H2O system33,40,41,70. This study aims to better understand why Fe0/H2O systems are more efficient upon the addition of pyrite (FeS2) relative to those without pyrite.
In a ternary Fe0/FeS2/sand system, sand is non-reactive (inert) and is in-situ coated by iron oxides from the dissolution of the two other aggregates (“Background to the experimental methodology” and Table 2). This in-situ coating of sand delays the availability of free Fe(OH)3 for dye co-precipitation. Initially, MB and Fe2+/Fe3+ compete for adsorptive removal at the negatively charged sand surface70,71. Once the sand surface is completely coated, it will be no longer attractive for MB. This competition for active adsorption sites explains the observations in Fig. 3a. Note that neither Fe0 nor FeS2 are the discoloring agents, but rather the products of their oxidative dissolution which are variably available in the investigated systems (Table 3). To completely answer Question 2, the ternary mixture performed less than the single-aggregate systems because: (i) sand is in-situ coated, thereby retarding the availability of free Fe(OH)3, and (ii) the synergy of Fe0 and FeS2 has not yet produced enough free Fe(OH)3. The latter is the case whenever the pH value of the system has not exceeded 4.5 (Fig. 3b).
The presentation until this point has not addressed the redox properties of MB and MO. The thermodynamics predict MO reduction by Fe055,61. The results reported herein demonstrate that even the ion-selective nature of the individual dyes was not the key factor accounting for dye discoloration when the pH was lower than 4.5. Thus, regardless of any redox properties, the current work has demonstrated that Fe0-based systems are only efficient when the final pH value is larger than 4.5. Unlike the current study, several previous works have mostly failed to record the final pH values of their systems and use them in their discussion (Table 5).
Significance of the results
Fe0-based systems have been important components of the water treatment industry for the past 170 years8,27,70. Research reported before 1990 is not really considered by current active scientists whose starting point is the advent of in-situ permeable reactive barriers (PRBs), and the premise that Fe0 is an environmental reducing agent18,23,72,73. Conventional PRBs use micro-scale or granular Fe0 specimens (g Fe0). During the past two decades, some tools have been developed to improve the efficiency of g Fe0. In this regard, the following three tools have been introduced: (i) using nano-scale Fe0, (ii) alloying g Fe0 with metals such as Pd or Ni (also at nano-scale), and (iii) admixing another aggregate with g Fe011,24,25. The Fe0/FeS2/sand system investigated herein is part of the third category. It has been reported that in sulfide-containing environments, using g Fe0 results in the formation of iron sulfides which are conductive and sustain electron transfer from Fe0 to the contaminant22,74,75,76. On the other hand, such iron sulfides are stand-alone reducing agents for the reductive transformations of many contaminants70,77,78. Because Fe0 and FeS2 have in common the release of FeII species, it can be assumed that the material containing more Fe will be first passivated by FeIII species. However, when both materials are mixed, FeS2 accelerates Fe0 corrosion and none of both materials is really available for quantitative reductive transformation of other foreign species, including contaminants. Consequently, any observed enhancement of contaminant removal in a Fe0/H2O system by virtue of the presence of FeS2 is an indirect process. This assertion was elegantly demonstrated in the present study by slowing down the process of iron precipitation via addition of various FeS2 doses to the same Fe0/sand system for 41 d. It then follows that, FeS2 is mostly a pH shifting agent for the Fe0/H2O system37,59,70.
Table 5 reveals that all other investigations on the Fe0/FeS2 system were performed under shaken/stirred conditions. However, under such conditions, the target FeS2 intrinsic properties (including semi-conduction) are undermined. For example, how can FeS2 act as a ‘mediator’ for electron transfer’ from Fe0 to contaminants (Fig. 4) when the whole system is mechanically stirred at 400 rpm? Such a high stirring speed was explicitly selected to ensure that both Fe0 and FeS2 could be uniformly dispersed in the reaction solution22. This example clearly shows that using FeS2 to enhance the efficiency of Fe0/H2O systems is a simple tool to design more sustainable Fe0-based systems. However, current rationalization efforts are not really based on scientific principles22,33. Thus, only when the scientific principles are well-understood can better systems be designed8,27,70. A typical design problem is how to cope with the increased Fe0 dissolution specifically in column operations intrinsically prone to clogging79,80. Thus, in solving the enigma of the Fe0/FeS2/H2O system, this work leads to several avenues for sustaining the efficiency of conventional Fe0/H2O remediation systems. This result is especially important as Fe0-based (filtration) systems are an excellent candidate to help the international community to solve the long-lasting issue of universal safe drinking water8,14,15,27,29,30,81,82.

Schematic diagram of interactions between metallic iron (Fe0), pyrite (FeS2) and contaminants (RCl) in the remediation process: (a) Fe0/H2O and (b) Fe0/FeS2/H2O. In the Fe0/H2O system, anode and cathode are different sites on the same grain. In the Fe0/FeS2/H2O system, granular Fe0 is additionally the anode and FeS2 the cathode. The representation is based on the knowledge that Fe0 is not a reducing agent. Therefore, electrochemical contaminant reduction of RCl is possible in (a) and not in (b).
A further argument against the electrochemical nature of FeS2 in mediating electron transfer from Fe0 to contaminants (Fig. 4) is given by recent investigations in efforts to suppress FeS2 oxidation under environmental conditions83,84,85,86. For example, Seng et al.84 reported that Fe0 is able to stop FeS2 oxidation, and thus remediate acid mine drainage. In essence, Seng et al.84 investigated a contaminant-free Fe0/FeS2/H2O system (Table 6) and concluded that from the intrinsic properties the addition of Fe0 selectively suppress pyrite oxidation. Table 6 shows that the experimental conditions of Seng et al.84 are very close to those of remediation Fe0/FeS2/H2O systems. The only two distinct differences are: (i) the higher FeS2 mass loading (FeS2: Fe0 = 10), and (ii) the longer experimental duration (41 days versus < 10 h). By using an even longer experimental duration (41 days) and quiescent conditions (0 rpm), the present work has demonstrated the essential virtue of working under near-field conditions. In other words, it is fair to state that the Fe0/FeS2 literature is full of possibly reproducible results, but with low practical value. As already shown in Table 5, the variability of the operational conditions is a major issue and the significance of results of solid phase characterization is questionable. In fact, as seen in Table 6, a myriad of characterization tools were used to “confirm” the reducing properties of Fe0 for dyes33. In such studies species like methylene blue70 used herein as a ‘tracer’ of reactivity or arsenic22, and proven to be non-reducible in Fe0/H2O systems are quantitatively removed. The first merit of the MB method is to uncover these controversial views without solid phase analysis.
The conclusion of Seng et al.84 supports the view presented herein that the relative kinetics of Fe0 and FeS2 oxidation determinate the preponderance of processes in Fe0/FeS2/H2O systems70. However, the reported selectivity of the process is questionable as sand and other natural minerals are also covered with FeCPs under similar conditions29,87,88,89. As an example, Song et al.87 reported on increased CrVI reduction in Fe0/sand/H2O systems compared to Fe0/H2O ones. The extent of coating of each aggregate (e.g. gravel, peat, pyrite, sand) depends on both the intrinsic reactivity of used Fe0 and the relative proportion of available materials. In the light of the kinetic arguments given herein, a re-evaluation of published works is possible, for example, the data of Sheba et al.86 discussing the extent of degradation of chlorinated organic compounds (RCl) by Fe0/FeS2/H2O systems and reporting on differential mechanisms at different Fe0:FeS2 ratios. The discussion given herein clearly suggests that if there are differential removal mechanisms, it is due to the differential extent of pH shift. Future research should be designed based on the chemistry of the systems89.
Concluding remarks
The concept that adsorption and co-precipitation are the fundamental mechanisms of contaminant removal in Fe0/H2O systems is consistent with many experimental observations. In particular, quantitative dye discoloration was only observed for pH values corresponding to iron precipitation (hydroxide formation) (pH > 4.5, Fig. 3b), while selective dye discoloration promoted adsorptive removal. Further, while the role of the redox-mediated reactions in the discoloration of both dyes can only be speculatively discussed based on one’s results, it is established that the role of FeS2 is as follows: (i) shifting the pH to more acidic values, and (ii) enhancing contaminant removal by adsorption and co-precipitation during the subsequent pH increase by virtue of iron corrosion. Finally, negatively charged methyl orange (MO) showed no significant increase in discoloration relative to positively charged methylene blue (MB). Both MB and MO have a similar molecular size. This observation is consistent with the role of Fe0 as a generator of contaminant scavengers, and not as a reducing agent. This observation could explain why various As (AsIII and AsV)90 or Se (SeIV and SeVI)91 species are quantitatively removed in Fe0/H2O systems, but not by aged iron oxides. Further research is needed to investigate the phenomena highlighted in the current study using a wide range of contaminants commonly occurring in drinking water and wastewaters.
References
Ali, I. Water treatment by adsorption columns: Evaluation at ground level. Sep. Purif. Rev. 43, 175–405 (2014).
Howe, K.J.,Hand, D.W., Crittenden, J.C., Trussell, R.R. & Tchobanoglous, G. Principles of Water Treatment. (ed. John, W. & Sons, I.) 674 (2012).
D’Mello, J. P. F. A Handbook of Environmental Toxicology: Human Disorders and Ecotoxicology 608 (CABI Publishing, Wallingford, 2019).
Tucker, W. G. The purification of water by chemical treatment. Science 20, 34–38 (1892).
Shannon, M. A. et al. Science and technology for water purification in the coming decades. Nature 452, 301–310 (2008).
Antia, D. D. J. Desalination of water using ZVI, Fe0. Water 7, 3671–3831 (2015).
Gonzalez-Perez, A., Persson, K. M. & Lipnizki, F. Functional channel membranes for drinking water production. Water 10, 859 (2018).
Antia, D.D.J. Water treatment and desalination using the eco-materials n- Fe0 (ZVI), n-Fe3O4, n-FexOyHz[mH2O], and n-Fex[Cation]nOyHz[Anion]m [rH2O]. In Handbook of Nanomaterials and Nanocomposites for Energy and Environmental Applications, (ed. O.V. Kharissova et al.) (2020).
Gillham, R. W. & O’Hannesin, S. F. Enhanced degradation of halogenated aliphatics by zero-valent iron. Ground Water 32, 958–967 (1994).
Henderson, A. D. & Demond, A. H. Long-term performance of zero-valent iron permeable reactive barriers: A critical review. Environ. Eng. Sci. 30, 401–423 (2007).
Guan, X. et al. The limitations of applying zero-valent iron technology in contaminants sequestration and the corresponding countermeasures: The development in zero-valent iron technology in the last two decades (1994–2014). Water Res. 75, 230–308 (2015).
Hussam, A. & Munir, A. K. M. A simple and effective arsenic filter based on composite iron matrix: Development and deployment studies for groundwater of Bangladesh. J. Environ. Sci. Health A 42, 1869–1878 (2007).
Kowalski, K. P. & Søgaard, E. G. Implementation of zero-valent iron (ZVI) into drinking water supply—Role of the ZVI and biological processes. Chemosphere 117, 108–114 (2014).
Banerji, T. & Chaudhari, S. In A Cost-Effective Technology for Arsenic Removal: Case Study of Zerovalent Iron-Based IIT Bombay Arsenic Filter in West Bengal (eds Nath, K. & Sharma, V.) (Springer, Berlin, 2017).
Tepong-Tsindé, R. et al. Characterizing a newly designed steel-wool-based household filter for safe drinking water provision: Hydraulic conductivity and efficiency for pathogen removal. Processes 7, 966 (2019).
Noubactep, C. Metallic iron for environmental remediation: a review of reviews. Water Res. 85, 114–123 (2015).
Gheju, M. & Balcu, I. Sustaining the efficiency of the Fe(0)/H2O system for Cr(VI) removal by MnO2 amendment. Chemosphere 214, 389–398 (2019).
Hu, R., Gwenzi, W., Sipowo-Tala, V. R. & Noubactep, C. Water treatment using metallic iron: A tutorial review. Processes 7, 622 (2019).
Devonshire, E. The purification of water by means of metallic iron. J. Frankl. Inst. 129, 449–461 (1890).
Lauderdale, R. A. & Emmons, A. H. A method for decontaminating small volumes of radioactive water. J. Am. Water Works Assoc. 43, 327–331 (1951).
Zhou, H., He, Y., Lan, Y., Mao, J. & Chen, S. Influence of complex reagents on removal of chromium(VI) by zero-valent iron. Chemosphere 72, 870–874 (2008).
Du, M. et al. Effect of pyrite on enhancement of zero-valent iron corrosion for arsenic removal in water: A mechanistic study. Chemosphere 233, 744–753 (2019).
Hu, R. & Noubactep, C. Redirecting research on Fe0 for environmental remediation: The search for synergy. Int. J. Environ. Res. Public Health 16, 4465 (2019).
Phillips, D. H. et al. Ten year performance evaluation of a field-scale zero-valent iron permeable reactive barrier installed to remediate trichloroethene contaminated groundwater. Environ. Sci. Technol. 44, 3861–4386 (2010).
Wilkin, R. T. et al. Fifteen-year assessment of a permeable reactive barrier for treatment of chromate and trichloroethylene in groundwater. Sci. Tot. Environ. 468–469, 186–194 (2014).
Guan, Q. et al. Assessment of the use of a zerovalent iron permeable reactive barrier for nitrate removal from groundwater in the alluvial plain of the Dagu River, China. Environ. Earth Sci. 78, 244 (2019).
Noubactep, C. Metallic Iron for Environmental Remediation: Prospects and Limitations. In A Handbook of Environmental Toxicology: Human Disorders and Ecotoxicology (ed. D’Mello, J. P. F.) 531–544 (CAB International, Wallingford, 2020).
Noubactep, C., Schöner, A. & Woafo, P. Metallic iron filters for universal access to safe drinking water. Clean: Soil, Air, Water 37, 930–937 (2009).
Bradley, I., Straub, A., Maraccini, P., Markazi, S. & Nguyen, T. H. Iron oxide amended biosand filters for virus removal. Water Res. 45, 4501–4510 (2011).
Neumann, A. et al. Arsenic removal with composite iron matrix filters in Bangladesh: A field and laboratory study. Environ. Sci. Technol. 47, 4544–4554 (2013).
Yang, Y. et al. Utilization of iron sulfides for wastewater treatment: A critical review. Rev. Environ. Sci. Biotechnol. 20, 289–308 (2017).
Liu, H., Chen, Z., Guan, Y. & Xu, S. Role and application of iron in water treatment for nitrogen removal: A review. Chemosphere 204, 51–62 (2018).
Chen, K. et al. Pyrite enhanced the reactivity of zero valent iron for reductive removal of dyes. J. Chem. Technol. Biotechnol. 95, 1412–1420 (2020).
Henderson, A. D. & Demond, A. H. Impact of solids formation and gas production on the permeability of ZVI PRBs. J. Environ. Eng. 137, 689–696 (2011).
Jin, X., Chen, H., Yang, Q., Hu, Y. & Yang, Z. Dechlorination of carbon tetrachloride by sulfide-modified nanoscale zerovalent iron. Environ. Eng. Sci. 35, 560–567 (2018).
Henderson, A. D. & Demond, A. H. Permeability of iron sulfide (FeS)-based materials for groundwater remediation. Water Res. 47, 1267–1276 (2013).
Noubactep, C., Meinrath, G., Dietrich, P. & Merkel, B. Mitigating uranium in groundwater: Prospects and limitations. Environ. Sci. Technol. 37, 4304–4308 (2003).
Noubactep, C., Meinrath, G. & Merkel, J. B. Investigating the mechanism of uranium removal by zerovalent iron materials. Environ. Chem. 2, 235–302 (2005).
Noubactep, C., Schöner, A. & Meinrath, G. Mechanism of uranium (VI) fixation by elemental iron. J. Hazard Mater. 132, 202–212 (2006).
Lü, Y. et al. Synergetic effect of pyrite on Cr(VI) removal by zero valent iron in column experiments: An investigation of mechanisms. Chem. Eng. J. 349, 522–529 (2018).
Lü, Y. et al. The roles of pyrite for enhancing reductive removal of nitrobenzene by zero-valent iron. Appl. Catal. B: Environ. 302, 9–18 (2019).
Xie, Y. & Cwiertny, D. M. Use of dithionite to extend the reactive lifetime of nanoscale zero-valent iron treatment systems. Environ. Sci. Technol. 44, 8649–8655 (2010).
Naseri, E. et al. Making Fe0-based filters a universal solution for safe drinking water provision. Sustainability 9, 1230 (2017).
Miyajima, K. Optimizing the design of metallic iron filters for water treatment. Freiberg Online Geosci. 32, 1–60 (2012).
Miyajima, K. & Noubactep, C. Characterizing the impact of sand addition on the efficiency of granular iron for water treatment. Chem. Eng. J. 262, 891–896 (2015).
Mitchell, G., Poole, P. & Segrove, H. D. Adsorption of methylene blue by high-silica sands. Nature 176, 825–826 (1955).
Btatkeu-K, B. D., Tchatchueng, J. B., Noubactep, C. & Caré, S. Designing metallic iron based water filters: Light from methylene blue discoloration. J. Environ. Manag. 206, 567–573 (2016).
Btatkeu-K, B. D., Olvera-Vargas, H., Tchatchueng, J. B., Noubactep, C. & Caré, S. Determining the optimum Fe0 ratio for sustainable granular Fe0/sand water filters. Chem. Eng. J. 307, 265–274 (2014).
Btatkeu-K, B. D., Olvera-Vargas, H., Tchatchueng, J. B., Noubactep, C. & Caré, S. Characterizing the impact of MnO2 on the efficiency of Fe0-based filtration systems. Chem. Eng. J. 250, 420–422 (2014).
Kosmulski, M. Isoelectric points and points of zero charge of metal (hydr)oxides: 50 years after Parks’ review. Adv. Colloid and Interface Sci. 238, 1–61 (2016).
Frost, R. L., Xi, Y. & He, H. Synthesis, characterization of palygorskite supported zero-valent iron and its application for methylene blue adsorption. J. Colloid Interface Sci. 341, 153–161 (2010).
Rodríguez, A., García, J., Ovejero, G. & Mestanza, M. Adsorption of anionic and cationic dyes on activated carbon from aqueous solutions: Equilibrium and kinetics. J. Hazard. Mater. 172, 1311–1320 (2009).
Attia, A. A., Girgis, B. S. & Fathy, N. A. Removal of methylene blue by carbons derived from peach stones by H3PO4 activation: Batch and column studies. Dyes Pigm. 76, 282–289 (2008).
Huang, J. H., Huang, K. L., Liu, S. Q., Wang, A. T. & Yan, C. Adsorption of Rhodamine B and methyl orange on a hypercrosslinked polymeric adsorbent in aqueous solution. Colloids Surf. A. Physicochem. Eng. Aspects 330, 55–61 (2008).
Gatcha-Bandjun, N., Noubactep, C. & Loura, B. Mbenguela, Mitigation of contamination in effluents by metallic iron: The role of iron corrosion products. Environ. Technol. Innov. 8, 71–83 (2017).
Hu, R. et al. Characterizing the suitability of granular Fe0 for the water treatment industry. Processes 7, 652 (2019).
Lufingo, M., Ndé-Tchoupé, A. I., Hu, R., Njau, K. N. & Noubactep, C. A novel and facile method to characterize the suitability of metallic iron for water treatment. Water 11, 2465 (2019).
Varlikli, C. et al. Adsorption of dyes on Sahara Desert sand. J. Hazard. Mater. 170, 27–34 (2009).
Lipczynska-Kochany, E., Harms, S., Milburn, R., Sprah, G. & Nadarajah, N. Degradation of carbon tetrachloride in the presence of iron and sulphur containing compounds. Chemosphere 29, 1477–1489 (1994).
Saywell, L. G. & Cunningham, B. B. Determination of iron: Colorimetric o-phenanthroline method. Ind. Eng. Chem. Anal. Ed. 9, 67–69 (1937).
Phukan, M. Characterizing the Fe0/sand system by the extent of dye discoloration. Freiberg Online Geosci. 40, 1–70 (2015).
Liu, X. & Millero, F. J. The solubility of iron hydroxide in sodium chloride solutions. Geochim. Cosmochim. Acta 63, 3487–3497 (1999).
Lewis, A. E. Review of metal sulphide precipitation. Hydrometallurgy 104, 222–234 (2010).
Vidic, R. D., Suidan, M. T., Traegner, U. K. & Nakhla, G. F. Adsorption isotherms: illusive capacity and role of oxygen. War. Res. 24, 1187–1195 (1990).
Phukan, M., Noubactep, C. & Licha, T. Characterizing the ion-selective nature of Fe0-based filters using azo dyes. Chem. Eng. J. 259, 481–491 (2015).
Crawford, R. J., Harding, I. H. & Mainwaring, D. E. Adsorption and coprecipitation of single heavy metal ions onto the hydrated oxides of iron and chromium. Langmuir 9, 3050–3056 (1993).
Langmuir, D. In Aqueous Environmental Geochemistry (ed. Robert, S.) 600 (American Geophysical Union, Washington, 1997).
Lavine, B. K., Auslander, G. & Ritter, J. Polarographic studies of zero valent iron as a reductant for remediation of nitroaromatics in the environment. Microchem. J. 70, 69–83 (2001).
Ghauch, A., Abou Assi, H., Baydoun, H., Tuqan, A. M. & Bejjani, A. Fe0-based trimetallic systems for the removal of aqueous diclofenac: Mechanism and kinetics. Chem. Eng. J. 172, 1033–1044 (2011).
Xiao, M., Cui, X., Hu, R., Gwenzi, W. & Noubactep, C. Validating the efficiency of the FeS2 method for elucidating the mechanisms of contaminant removal using Fe0/H2O systems. Processes 8, 1162 (2020).
Btatkeu-K, B. D., Miyajima, K., Noubactep, C. & Caré, S. Testing the suitability of metallic iron for environmental remediation: Discoloration of methylene blue in column studies. Chem. Eng. J. 215–216, 959–968 (2013).
Chen, Z. X., Jin, X. Y., Chen, Z., Megharaj, M. & Naidu, R. Removal of methyl orange from aqueous solution using bentonite-supported nanoscale zero-valent iron. J. Colloid Interf. Sci. 363, 601–607 (2011).
Mwakabona, H. T., Ndé-Tchoupé, A. I., Njau, K. N., Noubactep, C. & Wydra, K. D. Metallic iron for safe drinking water provision: Considering a lost knowledge. Water Res. 117, 127–142 (2017).
Erdem, M. & Ozerdi, A. Kinetics and thermodynamics of Cd(II) adsorption onto pyrite and synthetic iron sulphide. Sep. Purif. Technol. 51, 240–246 (2006).
Jeong, H. Y. & Hayes, K. F. Reductive dechlorination of tetrachloroethylene and trichloroethylene by mackinawite (FeS) in the presence of metals: Reaction rates. Environ. Sci. Technol. 41, 6390–6396 (2007).
He, Y. T., Wilson, J. T. & Wilkin, R. T. Transformation of reactive iron minerals in a permeable reactive barrier (biowall) used to treat TCE in groundwater. Environ. Sci. Technol. 42, 6690–6696 (2008).
Lee, W. J. & Batchelor, B. Abiotic reductive dechlorination of chlorinated ethylenes by iron-bearing soil minerals: Pyriye and magnetite. Environ. Sci. Technol. 36, 5147–5154 (2002).
Lan, Y. & Butler, E. C. Iron-sulfide-associated products formed during reductive dechlorination of carbon tetrachloride. Environ. Sci. Technol. 50, 5489–5497 (2016).
Domga, R., Togue-Kamga, F., Noubactep, C. & Tchatchueng, J. B. Discussing porosity loss of Fe0 packed water filters at ground level. Chem. Eng. J. 263, 127–134 (2015).
Moraci, N., Lelo, D., Bilardi, S. & Calabrò, P. S. Modelling long-term hydraulic conductivity behaviour of zero valent iron column tests for permeable reactive barrier design. Can. Geotech. J. 53, 946–961 (2016).
Nanseu-Njiki, C. P., Gwenzi, W., Pengou, M., Rahman, M. A. & Noubactep, C. Fe0/H2O filtration systems for decentralized safe drinking water: Where to from here?. Water 11, 429 (2019).
Yang, H. et al. Designing the next generation of Fe0-based filters for decentralized safe drinking water treatment. Processes 8, 745 (2020).
Seng, S. Galvanic microencapsulation: a new technique to suppress pyrite oxidation. PhD Dissertation, Hokkaido University, Japan (2019).
Seng, S., Tabelin, C. B., Kojima, M., Hiroyoshi, N. & Ito, M. Galvanic microencapsulation (GME) using zero-valent aluminum and zero-valent iron to suppress pyrite oxidation. Mater. Trans. 60, 277–286 (2019).
Tabelin, C. B. et al. Development of advanced pyrite passivation strategies towards sustainable management of acid mine drainage. IOP Conf. Ser. Earth Environ. Sci. https://doi.org/10.1088/1755-1315/351/1/012010 (2019).
Shiba, M., Uddin, Md. A., Kato, Y. & Ono, T. Degradation of chlorinated organic compounds by mixed particles of iron/iron sulfide or iron/iron disulfide. Mater. Trans. 55, 708–712 (2014).
Song, D.-I., Kim, Y. H. & Shin, W. S. A simple mathematical analysis on the effect of sand in Cr(VI) reduction using zero valent iron. Korean J. Chem. Eng. 22, 67–69 (2005).
Jia, Y., Aagaard, P. & Breedveld, G. D. Sorption of triazoles to soil and iron minerals. Chemosphere 67, 250–258 (2007).
Cao, V. et al. Tracing the scientific history of Fe0-based environmental remediation prior to the advent of permeable reactive barriers. Processes 8, 977 (2020).
Wang, D. et al. Iron mesh-based metal organic framework filter for efficient arsenic removal. Environ. Sci. Technol. 52, 4275–4284 (2018).
Stefaniak, J., Dutta, A., Verbinnen, B., Shakya, M. & Rene, E. R. Selenium removal from mining and process wastewater: A systematic review of available technologies. J. Water Supply: Res. Technol. Aqua 67, 903–918 (2018).
Acknowledgements
The used pyrite mineral (FeS2) was kindly donated by Prof. Yimin Li from the College of Chemistry and Chemical Engineering, Shaoxing University, PR China. Bernard Owuraku Kanin (School of Earth Science and Engineering, Hohai University) is thanked for technical support. This work was supported by the Ministry of Science and Technology of China through the Program “Driving process and mechanism of three dimensional spatial distribution of high risk organic pollutants in multi field coupled sites” (Project Code: 2019YFC1804303) and “Research on Mechanism of Groundwater Exploitation and Seawater Intrusion in Coastal Areas” (Project Code: 20165037412), and the program “Postgraduate Research & Practice Innovation Program of Jiangsu Province” (Project Code: SJKY19_0519, 2019B60214). We acknowledge support by the German Research Foundation and the Open Access Publication Funds of the Göttingen University.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Contributions
X.C., M.X. and C.N. conceived the presented idea and developed the theory. X.C. and M.X. carried out the experiment. R.H. and C.N. supervised this work. W.G. supervised the redaction of the first draft by X.C. and M.X. All authors discussed the results and contributed to the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Hu, R., Cui, X., Xiao, M. et al. Characterizing the impact of pyrite addition on the efficiency of Fe0/H2O systems. Sci Rep 11, 2326 (2021). https://doi.org/10.1038/s41598-021-81649-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-021-81649-y
This article is cited by
-
Realizing the potential of metallic iron for the mitigation of toxics: flee or adapt?
Applied Water Science (2022)
-
Characterizing the impact of MnO2 addition on the efficiency of Fe0/H2O systems
Scientific Reports (2021)
-
The key role of contact time in elucidating the mechanisms of enhanced decontamination by Fe0/MnO2/sand systems
Scientific Reports (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
