
Abstract
In vitro gonad culture systems have proven useful to investigate intrinsic mechanisms of sexual reproduction in animals. Here we describe development of an in vitro culture method for coral ovaries. Mesenterial tissues containing both ovaries and mesenterial filaments were microscopically isolated from the scleractinian coral, Fimbriaphyllia ancora, and culture conditions were optimized. M199 diluted 10× (10% M199, pH 8.1) and supplemented with 25 mM HEPES and the antibiotics, ampicillin, penicillin and streptomycin, supported oocyte survival and maintained the structural integrity of ovaries during short-term culture (~ 6 days). Addition of a commercial antibiotic–antimycotic solution (Anti–Anti) and fetal bovine serum adversely affected ovary maintenance and caused tissue disintegration. Characterization of cultured ovaries showed that there is no difference in cell proliferation of ovarian somatic cells between culture Days 1 and 6. Moreover, the presence of oogonia and expression of a major yolk protein, vitellogenin, were confirmed in ovaries cultured for 6 days. This system will be useful for studying effects of a wide range of substances on coral oogenesis.
Similar content being viewed by others
Introduction
Sexual reproduction of scleractinian corals is a biological phenomenon that continues to fascinate researchers and the general public. Since the 1980s, many studies have investigated sexual reproduction in corals worldwide, resulting in improved understanding of environmental factors influencing gametogenesis and spawning/brooding1,2,3,4. Nonetheless, there remain many unanswered questions about intrinsic mechanisms controlling coral sexual reproduction.
Corals are marine organisms belonging to the phylum Cnidaria. Like other cnidarians (sea anemones, jellyfish, hydra, etc.), corals are diploblastic, having a relatively simple body structure. Complex organs corresponding to vertebrate brains, blood vessels, and intestines are lacking in corals5,6. Corals reproduce both asexually and sexually. For sexual reproduction, many corals develop gametes within polyps over a period of several months to a year and release them at specific times7. Coral germ cells generally develop within specific regions of mesenterial tissues in the polyp7. These regions are usually small swellings, conventionally called gonads. Coral gonads are composed of several cell types, including germ cells, gonadal somatic cells, and neurons7,8.
In many animals, germ cell proliferation, differentiation, and/or maturation are regulated by intrinsic factors such as neuropeptides, steroid hormones, and biogenic amines9,10,11,12,13. Information about corresponding compounds in corals is important to better understand both basic and applied biology. For example, identifying similarities and differences between corals and vertebrates could provide insight into evolution of sexual reproduction in metazoans14. Additionally, coral propagation projects are being undertaken worldwide to restore damaged coral reefs15,16,17,18,19. Asexual propagation is the method commonly used worldwide19. Identification of intrinsic factors regulating coral gametogenesis would make it possible to induce sexual reproduction artificially, which could lead to efficient production of genetically diverse coral seedlings.
Our recent transcriptome analysis of coral gonads showed that genes encoding growth factors, neuropeptides, and neurotransmitter receptors are expressed in the gonads20, leading us to hypothesize that coral gametogenesis is regulated by those intrinsic factors. To test this hypothesis, functional analysis of genes and proteins is essential. However, development of related techniques for corals has been slow. Although gene knockdown or knockout has recently been accomplished in Acropora species21,22, these techniques are only applicable during the embryonic stage.
In vitro tissue and organ culture systems are powerful tools for investigating intrinsic mechanisms of sexual reproduction. For example, in fish, the mechanism of hormonal regulation during gametogenesis has been investigated by adding steroids or growth factors to the culture systems and observing responses of germ cells at the molecular and cellular levels23,24,25. Establishment of a similar in vitro culture system for coral gonads should provide a useful platform to study effects of a wide range of substances on coral gametogenesis.
There have been no reports of in vitro culture techniques for coral gonads. Therefore, we developed a technique to culture ovaries in vitro for short periods while maintaining oocyte survival and their three-dimensional structure. A coral species, Fimbriaphyllia ancora, which is widely distributed in the Indo-Pacific Ocean, was used. This species is gonochoric (separate male and female organisms) and has large polyps (3–5 cm in diameter). Moreover, a technique for isolating mesenterial tissues encompassing ovaries is well established26. Furthermore, antibodies identifying germ cells or yolk proteins in the ovaries are available for this species26,27. We first optimized culture conditions by assessing effects of antibiotics/antifungal agents, types of culture medium, medium pH, and fetal bovine serum on oocytes, using histological analysis. We then characterized ovarian cells cultured under optimized conditions.
Materials and methods
Sampling of experimental animals
Three large female colonies of F. ancora were selected and labeled at Nanwan Bay in southern Taiwan (21°57′N, 120°46′E). In March and April 2016, before the spawning season in May, scuba divers collected coral segments (~ 5–6 cm in length) from each labeled colony with approval from the Kenting National Park administration. Collected fragments were subsequently transferred to an aquarium at National Taiwan Ocean University (NTOU). Dissection of the coral and gonad isolation were carried out in accordance with guidelines for Institutional Animal Care and Use from NTOU.
Isolation and observation of mesenterial tissues
Gonads and associated mesenterial filaments (mesenterial tissue) were microscopically isolated from individual polyps and pooled in a culture dish. In order to minimize mechanical damage, ovaries were not separated from mesenterial filaments. Isolated mesenterial tissues were washed three times with filtered sterile seawater (FSW, sterile, pH 8.1) supplemented with antibiotics (50 µg/mL ampicillin, 50 U/mL penicillin and 50 µg/mL streptomycin) to inhibit bacterial growth. All procedures were performed in a laminar flow hood. Observations employed a stereomicroscope (SZX16; Olympus) or a fluorescence microscope (IX71SF1, Olympus). Endogenous red fluorescent protein (RFP) of oocytes was used as an oocyte marker and observed with a U-MWIG 2 filter (excitation wavelength 520–550 nm, emission wavelength 580 nm) on a fluorescence microscope28.
To examine effects of antibiotics and antifungals, 50 µg/mL ampicillin, 50 U/mL penicillin, 50 µg/mL streptomycin, or Antibiotic–Antimycotic (Anti–Anti, containing 100 units/mL of penicillin, 100 µg/mL of streptomycin, and 0.25 µg/mL of Amphotericin B, Thermo Fisher scientific, Waltham, MA) were selected, according to previous studies29,30. They were added individually, or in combinations of three antibiotics, or all together, and isolated mesenterial tissues were maintained under each condition for 6 days. As a control, tissues were maintained in FSW containing no antibiotics (Table 1).
To investigate effects of culture medium, 4 commercially available culture media, Dulbecco’s modified Eagle’s medium (DMEM), Medium 199 (M199), RPMI 1640 medium (RPMI), Leibovitz’s L-15 medium (L-15) (all purchased from Thermo Fisher Scientific) were tested as used previously29,30. These media were diluted 10× with FSW29, and supplemented with 25 mM HEPES and antibiotics (50 µg/mL ampicillin, 50 U/mL penicillin, and 50 µg/mL streptomycin). To investigate effects of medium pH, 10% M199 (diluted with FSW, containing 25 mM HEPES and antibiotics) was adjusted to pH 7.8, 8.1, or 8.4. Effects of fetal bovine serum (FBS, Thermo Fisher Scientific) were also investigated and isolated tissues were cultured in 10% M199 (diluted with FSW, pH 8.1, containing 25 mM HEPES and antibiotics) supplemented with 1, 5, or 10% FBS. For each experiment, 3–4 isolated mesenterial tissues were cultured for 6 days in 3-cm laboratory dishes with 7.5 mL of culture media at 26 °C under standard atmospheric pressure with a 12-h light/dark cycle (light intensity: 17–19.5 μmol m−2 s−1). Culture media were replaced every 2 days. Cultures were observed and photographed under microscopes as described above at 0, 3, and 6 days of culture.
Histological analysis
Samples were fixed with 20% Zinc Formal‐Fixx (Thermo Shandon, Pittsburgh, PA) for 16 h at room temperature, and preserved in 70% ethanol before use. Fixed samples were embedded in paraplast (Thermo Fisher Scientific), sectioned at a thickness of 4 µm, and stained with haematoxylin and eosin Y (HE, Thermo Fisher Scientific). Since approximately 600 serial sections comprise an entire isolated ovary, 10–30 sections were randomly photographed for each ovary, and used for subsequent analyses. To evaluate ovarian status, we developed two indicators, ovarian integrity and oocyte abnormality. Ovarian integrity is the ratio of ovarian area occupied by oocytes. This value (%) was estimated by measuring the total ovarian area and the area occupied by oocytes (oocyte area) on the ovarian section. Integrity of freshly isolated ovaries was 50–60% and this value decreased as oocytes disappeared from the ovary by cell death. Oocyte abnormality is the ratio of oocytes exhibiting morphological abnormalities (disintegrating cell membranes, appearance of unidentified vesicles, severe cytoplasmic deviations) in all oocytes of an ovary. The value (%) was determined by direct observation of histological sections. Three ovaries from three different colonies (9 ovaries in total) were analyzed. The total ovarian area and the area occupied by oocytes (oocyte area) in ovarian sections were determined by manual area measurements using ImageJ software (National Institutes of Health, Bethesda, MD).
EdU assay
5-Ethynyl-2′-deoxyuridine (EdU, 50 µM) was added to the cultures at day 0 or day 5 of culture for 24 h. This labeled proliferating cells during days 0–1 or days 5–6 of culture. After EdU incubation, mesenterial tissues were washed three times with FSW supplemented with antibiotics (50 µg/mL ampicillin, 50 U/mL penicillin and 50 µg/mL streptomycin) and replaced with new culture media. Sample fixation and preparation of paraffin sections were performed as described in the section above (see “Histological analysis”). A Click-iT EdU Colorimetric IHC Detection Kit (C10644: Invitrogen, OR) was used to visualize proliferating cells. Eight to ten sections were randomly selected from each ovary, and percentages of ovarian somatic cells labeled with EdU were determined. Three ovaries from three different colonies (9 ovaries in total) were analyzed.
RNA extraction, cDNA synthesis, and quantitative reverse transcription PCR
Ovaries were isolated from mesenterial tissues, and total RNA was extracted with TRIzol reagent (Thermo Fisher Scientific) following the manufacturer's protocol. First‐strand cDNA was synthesized from 1 μg of DNase‐treated RNA using SuperScript III reverse transcriptase (Thermo Fisher Scientific). Transcript levels of a major yolk protein, vitellogenin (Vg, GenBank accession no. KC777188), were determined by quantitative real‐time RT‐PCR, and compared between uncultured and cultured ovaries. F. ancora β‐actin (GenBank accession no. JQ968408) was used as a reference gene. Primers (Table 2) were designed using Primer Express 3.0 software (Applied Biosystems, USA) and primer efficiency was determined with a fivefold dilution series of the template cDNA. Only primer pairs that worked at 90–100% efficiency were used for quantitative PCR analysis. PCR was performed using a Bio‐Rad CFX Connect Real‐Time PCR detection system (Bio‐Rad Laboratories, Hercules, CA) with iQTM SYBR Green Supermix (Bio‐Rad Laboratories). Two‐step RT‐PCR was performed using the following conditions: (a) 1 cycle of 95 °C for 5 min, and (b) 40 cycles of 95 °C for 15 s and 60 °C for 1 min. Calculations were performed using the 2−△△Ct method31. Reactions in which the template was omitted were used as negative controls for each primer set.
Immunohistochemical analysis
Fixation of samples and preparation of paraffin sections were carried out as described in the section above (see Histological analysis). Immunohistochemistry was performed according to our previous methodology32. For primary antibody reactions, sections were incubated with affinity-purified anti-F. ancora piwi antibody (a marker for early stage germ cells, 1:400027) or anti-F. ancora vitellogenin (Vg) antibody (1:400026). For secondary antibody reactions, sections were incubated with a biotinylated goat anti-guinea pig IgG antibody (1:4000, Vector Laboratories, Burlingame, CA). Immunoreactive signals were visualized with 3,3′-diaminobenzidine (DAB; Sigma-Aldrich).
Statistical analysis
Data are reported as means ± standard errors (SE). For comparisons of three or more groups, statistical significance was analyzed using Kruskal–Wallis tests (SPSS), followed by Dunn–Bonferroni pairwise comparisons. Statistical significance was analyzed with Mann–Whitney U-tests for comparisons between two groups. Statistical significance was set at p < 0.05.
Results
Morphological and histological characteristics of isolated mesenterial tissues in cultures and changes while maintained in seawater
Isolated mesenterial tissues, which include both ovaries and mesenterial filaments (Fig. 1A) were cultured. In culture, ovaries were generally observed as distinct swellings with pinkish color (Fig. 1A), and oocytes exhibited endogenous RFP28 (Fig. 1B). Mesenterial filaments exhibited slow movement, contracting and extending (Fig. 1B). Histologically, ovaries comprised oocytes with surrounding layers of ovarian somatic cells, and oocytes were filled with cytoplasm with evenly distributed small oil droplets (Fig. 1C).

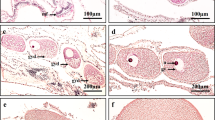
Morphological and histological characteristics of isolated mesenterial tissues and their changes during maintenance in FSW. (A) Brightfield view of freshly isolated mesenterial tissue containing an ovary and mesenterial filaments (Non-cultured). (B) Endogenous expression of oocyte RFP, observed with the U-MWIG 2 (RFP) filter of a fluorescence microscope. (C) HE section of the freshly isolated ovary (Non-cultured). (D) Brightfield view of mesenterial tissue maintained in FSW for 6 days. (E) U-MWIG 2 (RFP) filter view of an ovary maintained in FSW for 6 days. (F) HE section of an ovary maintained in FSW for 6 days. Oocyte disappearance was observed (arrows). (G–I) Morphological abnormalities in oocytes: disintegration of cell membranes (G), appearance of unidentified vesicles (H), and severe cytoplasmic deviations with unidentified vesicles (I). Scale bars = 500 μm (A,B,D,E,F); 200 μm (C,F,G,H,I).
When tissues were maintained in FSW for 6 days, contours of some oocytes, observed with RFP, became unclear (Fig. 1D,E). By contrast, no apparent changes were observed in morphological or behavioral characteristics of mesenterial filaments (Fig. 1D). Histological analysis of ovaries showed that some oocytes disappeared (Fig. 1F), or exhibited morphological abnormalities, such as disintegrating cell membranes (Fig. 1G), appearance of unidentified vesicles (Fig. 1H), or severe cytoplasmic aberrations with unidentified vesicles (Fig. 1I). Thus, oocytes degraded and/or underwent cell death during maintenance under FSW for 6 days. This raised the possibility that some nutrients essential for oocyte survival were missing in these cultures.
Optimization of culture conditions
We therefore sought to optimize in vitro culture conditions that support oocyte survival. First, we attempted to identify suitable antibiotics and/or antifungal agents. Corals harbor a huge diversity of microorganisms (symbiotic algae, bacteria, etc.)33. Suppressing microbial growth is thus essential for any in vitro coral culture. Although addition of antibiotics and/or antifungal agents has been commonly used to reduce bacterial overgrowth, some of these additives induce disintegration of coral tissues34,35. Since disintegration of ovarian structure not only adversely affects oocyte survival, but is also incompatible with our objectives, we attempted to identify antibiotics and/or antifungal agents that permit maintenance of the structural integrity of whole mesenterial tissues. Addition of a commercial antibiotic–antimycotic solution (Anti–Anti) to the cultures (FSW), induced disintegration of mesenterial tissue (Fig. 2,B). In contrast, when three antibiotics (ampicillin, penicillin, and streptomycin) were added singly or in combination, little tissue disintegration was observed (Fig. 2C).

Effects of a commercially available antibiotic–antimycotic solution or antibiotics on mesenterial tissue. (A) Brightfield view of mesenterial tissue maintained in FSW for 3 days (Control). (B) Mesenterial tissue maintained in FSW supplemented with antibiotic–antimycotic solution (Anti–Anti, from Thermo Fisher Scientific) for 3 days. (C) Mesenterial tissue maintained in FSW supplemented with 50 μg/mL ampicillin, 50 U/mL penicillin and 50 μg/mL streptomycin for 3 days. Note that addition of Anti–Anti induced deformation of mesenterial tissues (arrows). Scale bars = 1 mm.
Next, we investigated effects of 4 commercially available culture media (DMEM, M199, RPMI, and L-15) on F. ancora ovaries in the presence of the three antibiotics. To reduce microbial growth, all test media were diluted with FSW 1:10. HEPES (25 mM) was added to all media and the pH was maintained at 8.1. M199 effectively supports ovarian maintenance in culture. Microscopically, no differences were observed in ovarian appearance or oocytes, as indicated by RFP in uncultured (Fig. 3A,A′) and cultured ovaries in 10% M199 for 6 days (Fig. 3B,B′). In other groups, apparent disappearance of oocytes and disintegration of mesenterial tissues was observed in some samples (Fig. 3C–E,C′–E′). Histological analysis further demonstrated that ovaries cultured in 10% M199 had higher integrity, estimated by a higher ratio of area occupied by oocytes in ovaries, compared with those in other media (Kruskal–Wallis test, H = 29.148, df = 4, p < 0.05, Fig. 3F–J,F′–J′,K). Ratios of abnormal oocytes in ovaries cultured in 10% M199 were lower than those of other media (Kruskal–Wallis test, H = 22.577, df = 4, p < 0.05, Fig. 3F–J,F′–J′,L). No bacterial overgrowth was observed during the culture period. Based on this, 10% M199 was selected as the basic culture medium for F. ancora ovaries.

Effects of 4 commercially available culture media and medium pH. Brightfield views of freshly isolated mesenterial tissues (A) and mesenterial tissues cultured in M199 (B), DMEM (C), RPMI (D), or L-15 (E) for 6 days. (A′–E′) U-MWIG 2 (RFP) filter views of the same field as (A–E). HE sections of freshly isolated mesenterial tissue (F) and mesenterial tissues cultured in M199 (G), DMEM (H), RPMI (I), and L-15 (J) for 6 days. (F′–J′) Higher magnification views of the insets shown in (F′–J′). Effects of the 4 commercially available culture media (K,L) and medium pH (M,N) on ovarian integrity and oocyte status. Ovarian integrity was estimated by the ratio of the area occupied by oocytes in ovaries. Disappearance and abnormalities of oocytes in ovaries decrease this value. Oocyte status (oocyte abnormality) was examined histologically. Data are shown as means ± SE. Groups with different letters are significantly different (Kruskal–Wallis test, p < 0.05 and Dunn-Bonferroni post hoc test, p < 0.05). Scale bars = 500 μm (A–E,A′–E′); 200 μm (F–J,F′–J′).
For further improvement of culture conditions, we next investigated effects of medium pH and addition of FBS on F. ancora ovaries. No significant differences were observed among ovaries cultured at pH 7.8, 8.1, or 8.4 (Ovarian integrity, Kruskal–Wallis test, H = 5.174, df = 3, p > 0.05, Fig. 3M and oocyte abnormality, Kruskal–Wallis test, H = 5.282, df = 3, p > 0.05, Fig. 3N). Addition of FBS (1–10%) adversely affected ovary maintenance in culture and caused disintegration of mesenterial tissue structure within 3 days (Fig. S1).
Characterization of ovaries cultured under optimized conditions
Based on the above results, we then utilized 10% M199 (pH 8.1) supplemented with 25 mM HEPES and antibiotics (50 µg/mL ampicillin, 50 U/mL penicillin, 50 µg/mL streptomycin) as the culture medium for F. ancora ovaries. In this medium, structural integrity of ovaries and mesenterial filaments, as well as oocyte survival were well maintained at least for 6 days. Notably, mesenterial filaments continued to manifest slow movements (Fig. 4A and S2). Further characterization of cultured ovaries showed that oogonia were also maintained in cultured ovaries, as assessed by immunohistochemical detection (Fig. 4B,C). The EdU assay showed that there is no difference in cell proliferation activity of ovarian somatic cells between culture days 1 and 6 (Mann–Whitney, p > 0.05, Fig. 4D–F). mRNA expression of the yolk protein, vitellogenin (Vg), in cultured ovaries was also detected on culture days 3 and 6, although expression levels were lower than in uncultured ovaries (Kruskal–Wallis test, H = 8.856, df = 2, p < 0.05, Fig. 4G). Immunohistochemically, Vg protein was also detected in both somatic cells and oocytes of cultured ovaries, as in uncultured ovaries (Fig. 4H,I). These results indicate that coral ovaries can be cultured under our conditions for at least 6 days, maintaining ovarian structural integrity, proliferative activity of ovarian somatic cells, and some ovarian functions.

Status of ovaries cultured in optimized conditions. (A) Time-lapse images of mesenterial tissues (ovary and mesenterial filaments) cultured for 6 days in optimized conditions. The indicated mesenterial filaments (arrowheads) exhibited slow movements (see also Fig. S2). (B,C) Immunohistochemical detection of oogonia with anti-Eapiwi antibodies in an uncultured ovary (B) and an ovary cultured for 6 days (C). (D–F) Proliferating cells in cultured ovaries detected with an EdU assay. EdU was added to culture media (50 µM) at day 0 or day 5 of culture for 24 h. (D) The percentage of EdU-positive cells in ovarian somatic cells at day 1 and 6 of culture. Data are shown as means ± SE (n = 9 ovaries). EdU-positive cells (proliferating cells) detected in ovaries at culture Day 1 (E) or day 6 (F). Arrows indicate detected proliferating cells. (G–I) Expression of a major yolk protein, vitellogenin (Vg), in cultured ovaries. (G) Expression of Vg mRNA in uncultured ovaries and ovaries cultured in M199 for 3 days and 6 days. Data are shown as means ± SEs (n = 9) of relative values with those of uncultured ovaries. Groups with different letters are significantly different (p < 0.05). (H,I) Immunohistochemical detection of Vg in uncultured ovaries (H) and ovaries cultured for 6 days in M199 (I). Scale bars = 50 μm (B,C,E,F,H,I); 10 μm (insets in B,C,E,F).
Discussion
During the past four decades, progress has been made in culturing coral tissues/cells in vitro36,37. Although most coral tissues/cells have only been cultured as primary cultures without active growth, culture systems have been utilized to study coral-algal symbiosis30,38, biomineralization34,39,40,41,42,43, stress responses to heat44, and responses to marine pollutants45. Most recently, the first coral cell line was established from embryos of Acropora tenuis, and it is expected to be a very useful tool for comprehensive understanding of the coral organism and coral-algal symbiosis at the cellular and molecular levels46. Although the number of reports on coral tissue/cell culture is gradually increasing, to the best of our knowledge, there have been no published studies regarding in vitro culture of coral gonads/germline cells. The lack of research may reflect difficulties in sample preparation. Since most corals undergo sexual reproduction during a limited period every year47,48,49,50, this limits the time in which experiments can be carried out. Additionally, isolation techniques for live gonads have so far been reported only in F. ancora. In this study, despite the limited sampling period and the number of colonies we were permitted to sample, we successfully developed a culture system to enable oocyte survival and to maintain ovarian structure in culture for short periods. We also developed two indicators for assessing ovarian status, ovarian integrity and oocyte abnormality. Although the obtained data are still preliminary, and there is much room for improvement, the findings and techniques established in this study will facilitate studies of sexual reproduction in corals.
Culture media used for coral cell/tissue culture differ among species and target cell/tissue types. Conventionally, filtered sterile natural seawater (FSW), artificial seawater (ASW), or ASW supplemented with appropriate concentrations of commercial culture media (DMEM, L-15, M199, RPMI, etc.) have been used29,30,37. We found that 10% M199 improves maintenance of oocytes, as well as ovarian integrity in culture. M199 was first developed in 1950 for nutritional studies of chick embryo muscle fibroblasts51,52, and has been used in cell/tissue culture systems with a broad range of animals, including marine invertebrates such as sponges, corals, shrimp, abalones, and sea urchins29,53,54,55,56. M199 contains all 20 amino acids, and also more vitamins (ascorbic acid, vitamin A, vitamin D2) and additives (ATP, ADP, cholesterol, and guanine hydrochloride, etc.) than other media that were tested in this study. Vitamins are organic compounds that act as co-enzymes to augment catalytic activity57 and antioxidants58. Appropriate vitamin supplements improve cell viability, growth, and cell proliferation and differentiation of mammalian cells59,60. ATP comprises the primary means of energy storage and transfer in cells, driving a broad range of biological processes61. Exogenous ATP supplemented in the culture medium improves cell proliferation in rat astrocytes62 and has also been used to increase collagen and proteoglycan synthesis in chondrocyte tissue 3D culture63. At present, it is still unclear which components in M199 are responsible for improved performance of F. ancora ovarian cultures, because little is known about the nutrient and growth requirements of cnidarian tissue/cells culture37. It is probable that some amino acids, vitamins, or other additives in M199 medium solely or synergistically affect coral ovaries, and support survival and basal ovarian functions in culture.
FBS contains a variety of proteins, polypeptides, amino acids, glucose, growth factors, hormones, etc. that are essential for maintenance, attachment, growth and proliferation of cells64. FBS has been used in a broad range of cell/tissue cultures for both vertebrate and invertebrate cells64. In most studies of coral cell culture, FBS has been added to the mediums at a concentration of 5–20%29,30, and survival promoting effects were demonstrated in Acropora microphthalma endothelial cell culture30. However, we found that addition of FBS to culture media strongly induces disintegration of F. ancora ovaries. Since one of the objectives of this study was to maintain ovarian structure in vitro, we decided not to use FBS. Although it is currently unclear which factors in FBS cause this phenomenon, factors that inhibit intercellular adhesion of mesenterial tissues may be present.
The EdU assay demonstrated that proliferation of ovarian somatic cells was very low (~ 0.6%). This may be due to inherent characteristics of the ovaries at this stage. In a previous study, we reported a massive apoptotic phenomenon in gonadal somatic cells of F. ancora8. We found that while germ cells developed during gametogenesis, many gonadal somatic cells died by apoptosis, resulting in a significant decrease in the thickness and density of the gonadal somatic cell layer8. In the present study, ovaries were prepared a month or two before spawning. At this stage, active cell proliferation of gonadal somatic cells may not be occurring or may have been suppressed, as gonadal structure was being transformed to facilitate gamete release by reducing the number of somatic cells.
The mRNA level of yolk protein decreased in ovarian cultures from culture days 3–6. Although currently nothing is known about factors regulating yolk synthesis in corals, decreased expression levels are probably due to either the paucity of factors needed for maintenance of yolk synthesis or the existence of inhibitory factors in culture. Our previous study demonstrated that vitellogenin is produced by ovarian somatic cells26. Notably, in this study, no histological abnormality was observed in ovarian somatic cells, and cell proliferation appeared to be maintained during the culture period. These findings imply that our current culture conditions can at least support survival and proliferation of ovarian somatic cells but cannot fully support vitellogenin synthesis.
Although decreased expression of the vitellogenin gene was observed, it was indeed expressed in cultured ovaries. In future studies, vitellogenin expression may be used as an indicator to further improve culture conditions or to identify factors that promote yolk formation. For instance, by culturing ovaries under different temperatures, light intensities, wavelengths, pHs, etc., and by examining expression of vitellogenin, it may be possible to identify environmental factors that influence yolk formation. Similarly, it may also be possible to identify hormone-like substances that promote yolk formation, by culturing ovaries in the presence of sex steroids, neuropeptides, and monoamines, which, based on our previous transcriptomic and biochemical studies of F. ancora, are thought to exist20,65,66. Currently, the time that ovaries can be maintained in culture is limited to approximately 1 week, and in the case of prolonged cultivation, disappearance and/or morphological abnormalities of oocytes are observed after 9–10 days of culture. Nevertheless, because gene responses are usually detected within a few hours to a few days67,68,69, the established culture system could be used to identify those factors.
In summary, we have described the development of an in vitro tissue culture system for coral ovaries using the stony coral F. ancora. Our culture conditions support oocyte survival and structural integrity of ovaries for a short period (~ 6 days). This system will be a useful tool for investigating intrinsic mechanisms underlying coral oogenesis.
References
Harrison, P. L. & Wallace, C. C. Reproduction, dispersal and recruitment of scleractinian corals. In Ecosystems of the World 25, Coral Reefs (eds Dubinsky, Z. & Stambler, N.) 133–207 (Elsevier, 1990).
Richmond, R. H. & Hunter, C. L. Reproduction and recruitment of corals: Comparisons among the Caribbean, the tropical Pacific, and the Red Sea. Mar. Ecol. Prog. Ser. 60, 185–203 (1990).
Baird, A. H., Guest, J. R. & Willis, B. L. Systematic and biogeographical patterns in the reproductive biology of scleractinian corals. Annu. Rev. Ecol. Evol. Syst. 40, 551–571 (2009).
Harrison, P. L. Sexual reproduction in scleractinian corals. In Coral Reefs: An Ecosystem in Transition (eds Dubinsky, Z. & Stambler, N.) 59–85 (Springer, 2011).
Pechenik, J. A. Biology of the Invertebrates 6th edn. (McGraw-Hill, Higher Education, 2010).
Brusca, R. C. & Brusca, G. J. Invertebrates (Basingstoke, 2003).
Shikina, S. et al. Involvement of GLWamide neuropeptides in polyp contraction of the adult stony coral Euphyllia ancora. Sci. Rep. 10, 9427 (2020).
Shikina, S., Chen, C. C., Chiu, Y. L., Tsai, P. H. & Chang, C. F. Apoptosis in gonadal somatic cells of scleractinian corals: Implications of structural adjustments for gamete production and release. Proc. Biol. Sci. 287, 20200578 (2020).
Licht, P., Breitenbach, G. L. & Congdon, J. D. Seasonal cycle in testicular activity, gonadotropin, and thyroxine in the painted turtle, Chrysemys picta, under natural conditions. Gen. Comp. Endocrinol. 59, 130–139 (1985).
Stamper, D. L. & Licht, P. Effect of gonadotropin-releasing hormone on gonadotropin biosyntheis. Biol. Reprod. 43, 420–426 (1990).
Hirschenhauser, K. et al. Seasonal relationships between plasma and fecal testosterone in response to GnRH in domestic ganders. Gen. Comp. Endocrinol. 118, 262–272 (2000).
Pierantoni, R., Cobellis, G., Meccariello, R. & Fasano, S. Evolutionary aspects of cellular communication in the vertebrate hypothalamo-hypophysio-gonadal axis. Int. Rev. Cytol. 218, 69–141 (2002).
Schulz, R. W. et al. Spermatogenesis in fish. Gen. Comp. Endocrinol. 165, 390–411 (2010).
Shikina, S. & Chang, C. F. Sexual reproduction in stony corals and insight into the evolution of oogenesis in Cnidaria. In The Cnidaria, Past, Present and Future The World of Medusa and Her Sisters (eds Goffredo, S. & Dubinsky, Z.) 49–268 (Springer, 2016).
Shafir, S., Van Rijn, J. & Rinkevich, B. Steps in the construction of underwater coral nursery, an essential component in reef restoration acts. Mar. Biol. 149, 679–687 (2006).
Shaish, L., Levy, G., Gomez, E. & Rinkevich, B. Fixed and suspended coral nurseries in the Philippines: Establishing the first step in the “gardening concept” of reef restoration. J. Exp. Mar. Biol. Ecol. 36, 86–97 (2008).
Johnson, M. E. et al. Caribbean Acropora restoration guide: best practices for propagation and population enhancement. In The Nature Conservancy, Arlington, VA, 52–53 https://dspace.mote.org/handle/2075/2910 (2011)
Rinkevich, B. Rebuilding coral reefs: Does active reef restoration lead to sustainable reefs?. Curr. Opin. Environ. Sustain. 7, 28–36 (2014).
Omori, M. Coral restoration research and technical developments: what we have learned so far. Mar. Biol. Res. 15, 377–409 (2019).
Chiu, Y. L., Shikina, S., Yoshioka, Y., Shinzato, C. & De Chang, C. F. novo transcriptome assembly from the gonads of a scleractinian coral, Euphyllia ancora: Molecular mechanisms underlying scleractinian gametogenesis. BMC Genom. 21, 732 (2020).
Yasuoka, Y., Shinzato, C. & Satoh, N. The mesoderm-forming gene brachyury regulates ectoderm–endoderm demarcation in the coral Acropora digitifera. Curr. Biol. 26, 2885–2892 (2016).
Cleves, P. A., Strader, M. E., Bay, L. K., Pringle, J. R. & Matz, M. V. CRISPR/Cas9-mediated genome editing in a reef-building coral. Proc. Natl. Acad. Sci. U.S.A. 115, 5235–5240 (2018).
Miura, T., Yamauchi, K., Takahashi, H. & Nagahama, Y. Hormonal induction of all stages of spermatogenesis in vitro in the male Japanese eel (Anguilla japonica). Proc. Natl. Acad. Sci. U.S.A. 88, 5774–5778 (1991).
Bouma, G. J., Cloud, J. G. & Nagler, J. J. An in vitro system for the long-term tissue culture of juvenile rainbow trout (Oncorhynchus mykiss) testis. J. Exp. Zool. A Comp. Exp. Biol. 303, 698–703 (2005).
Leal, M. C. et al. Zebrafish primary testis tissue culture: An approach to study testis function ex vivo. Gen. Comp. Endocrinol. 162, 134–138 (2009).
Shikina, S. et al. Yolk formation in a stony coral Euphyllia ancora Cnidaria, Anthozoa: Insight into the evolution of Vitellogenesis in nonbilaterian animals. Endocrinology 154, 3447–3459 (2013).
Shikina, S. et al. Localization of early germ cells in a stony coral, Euphyllia ancora: Potential implications for a germline stem cell system in coral gametogenesis. Coral Reefs 34, 639–653 (2015).
Shikina, S. et al. Oocytes express an endogenous red fluorescent protein in a stony coral, Euphyllia ancora: A potential involvement in coral oogenesis. Sci. Rep. 6, 25868 (2016).
Frank, U., Rabinowitz, C. & Rinkevich, B. In vitro establishment of continuous cell cultures and cell lines from ten colonial cnidarians. Mar. Biol. 120, 491–499 (1994).
Kopecky, E. J. & Ostrander, G. K. Isolation and primary culture of viable multicellular endothelial isolates from hard corals. In Vitro Cell. Dev. Biol. Anim. 35, 616–624 (1999).
Livak, K. J. & Schmittgen, T. D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25, 402–408 (2001).
Shikina, S. et al. Germ cell development in the scleractinian coral Euphyllia ancora (Cnidaria, Anthozoa). PLoS ONE 7, 69 (2012).
Peixoto, R. S., Rosado, P. M., Leite, D. C., Rosado, A. S. & Bourne, D. G. Beneficial microorganisms for corals (BMC): Proposed mechanisms for coral health and resilience. Front. Microbiol. 8, 341 (2017).
Reyes-Bermudez, A. & Miller, D. J. In vitro culture of cells derived from larvae of the staghorn coral Acropora millepora. Coral Reefs 28, 859 (2009).
Nowotny, J. D., Connelly, M. T. & Traylor-Knowles, N. Novel methods to establish whole-body primary cell cultures for the cnidarians Nematostella vectensis and Pocillopora damicornis. Sci. Rep. 11, 4086 (2021).
Rinkevich, B. Marine invertebrate cell cultures: New millennium trends. Mar. Biotechnol. 7, 429–439 (2005).
Domart-Coulon, I. & Ostrander, G. K. Coral cell and tissue culture methods. In Diseases of Coral (eds Woodley, C. M. et al.) 489–505 (Wiley, 2015).
Gates, R. D., Baghdasarian, G. & Muscatine, L. Temperature stress causes host cell detachment in symbiotic cnidarians: Implication for coral bleaching. Biol. Bull. 182, 324–332 (1992).
Kingsley, R. J., Bernhardt, A. M., Wilbur, K. M. & Watabe, N. Scleroblast cultures from the gorgonian Leptogorgia virgulata (Lamarck) (Coelenterata: Gorgonacea). In Vitro Cell. Dev. Biol. 23, 297–302 (1987).
Domart-Coulon, I. J., Elbert, D. C., Scully, E. P., Calimlim, P. S. & Ostrander, G. K. Aragonite crystallization in primary cell cultures of multicellular isolates from a hard coral, Pocillopora damicornis. Proc. Natl. Acad. Sci. U.S.A. 98, 11885–11890 (2001).
Domart-Coulon, I. J. et al. A basidiomycete isolated from the skeleton of Pocillopora damicornis (Scleractinia) selectively stimulates short-term survival of coral skeletogenic cells. Mar. Biol. 144, 583–592 (2004).
Helman, Y. et al. Extracellular matrix production and calcium carbonate precipitation by coral cells in vitro. Proc. Natl. Acad. Sci. U.S.A. 105, 54–58 (2008).
Auzoux-Bordenave, S. & Domart-Coulon, I. Short review marine invertebrate cell cultures as tools for biomineralization studies. J. Sci. Hal. Aquat. 2, 42–47 (2010).
Nesa, B. & Hidaka, M. High zooxanthella density shortens the survival time of coral cell aggregates under thermal stress. J. Exp. Mar. Biol. Ecol. 368, 81–87 (2009).
Downs, C. A., Fauth, J. E., Downs, V. D. & Ostrander, G. K. In vitro cell-toxicity screening as an alternative animal model for coral toxicology: Effects of heat stress, sulfide, rotenone, cyanide, and cuprous oxide on cell viability and mitochondrial function. Ecotoxicology 19, 171–184 (2010).
Kawamura, K., Nishitsuji, K., Shoguchi, E., Fujiwara, S. & Satoh, N. Establishing sustainable cell lines of a coral, Acropora tenuis. Mar. Biotechnol. 23, 1–16 (2021).
Harrison, P. L. et al. Mass spawning in tropical reef corals. Science 223, 1186–1189 (1984).
Willis, B. L., Babcock, R. C., Harrison, P. L., Oliver, J. K. & Wallace, C. C. Patterns in the mass spawning of corals on the Great Barrier Reef from 1981 to 1984. In Proceedings of the Fifth International Coral Reef Congress (Tahiti, French Polynesia, 27 May–1 June) 343–348 (1985).
Babcock, R. C. et al. Synchronous spawnings of 105 scleractinian coral species on the Great Barrier Reef. Mar. Biol. 90, 379–394 (1986).
Guest, J. R., Baird, A. H., Goh, B. P. L. & Chou, L. M. Seasonal reproduction in equatorial reef corals. Invertebr. Reprod. Dev. 48, 207–218 (2005).
Morgan, J. F., Morton, H. J. & Parker, R. C. The nutrition of animal cells in tissue culture. I. Initial studies on a synthetic medium. Proc. Soc. Exp. Biol. Med. 73, 1–8 (1950).
Morgan, J. F., Campbell, E. & Morton, H. J. The nutrition of animal tissues cultivated in vitro. I. A survey of natural materials as supplements to synthetic medium 199. J. Natl. Cancer Inst. 16, 557–567 (1995).
Ghosh, D., Ray, A. R. & Dasmahapatra, A. K. Primary culture of prawn hepatocytes in serum free media. In Vitro Cell. Dev. Biol. Anim. 31, 811–813 (1995).
Suja, C. P., Sukumaran, N. & Dharmaraj, S. Effect of culture media and tissue extracts in the mantle explant culture of abalone, Haliotis varia Linnaeus. Aquaculture 271, 516–522 (2007).
Mercurio, S., Di Benedetto, C., Sugni, M. & Candia Carnevali, M. D. Primary cell cultures from sea urchin ovaries: a new experimental tool. In Vitro Cell. Dev. Biol. Anim. 50, 139–145 (2014).
Conkling, M. et al. Breakthrough in marine invertebrate cell culture: Sponge cells divide rapidly in improved nutrient medium. Sci. Rep. 9, 17321 (2019).
Freeland-Graves, J. H. & Bavik, C. Coenzymes. In Encyclopedia of Food Sciences and Nutrition 2nd edn (eds Caballero, B. et al.) 1475–1481 (Elsevier, 2003).
Sinbad, O. O., Folorunsho, A. A., Olabisi, O. L., Ayoola, O. A. & Temitope, E. J. Vitamins as antioxidants. J. Food Nutr. Res. 2, 214–235 (2019).
Büntemeyer, H. & Lehmann, J. The role of vitamins in cell culture media. In Animal Cell Technology: From Target to Market (eds Lindner-Olsson, E. et al.) 204–206 (Springer, 2001).
Choi, H. S. et al. Vitamin D insufficiency in Korea-a greater threat to younger generation: The Korea National Health and Nutrition Examination Survey (KNHANES) 2008. J. Clin. Endocrinol. Metab. 96, 643–651 (2011).
Fitz, J. G. Regulation of cellular ATP release. Trans. Am. Clin. Climatol. Assoc. 118, 199–208 (2007).
Ciccarelli, R. et al. Effects of exogenous ATP and related analogues on the proliferation rate of dissociated primary cultures of rat astrocytes. J. Neurosci. Res. 39, 556–566 (1994).
Bow, J. K. Using Adenosine Triphosphate (ATP) as a substitute for mechanical stimulation for tissue engineering applications. https://qspace.library.queensu.ca/handle/1974/6290 (2012).
Gstraunthaler, G. Alternatives to the use of fetal bovine serum: serum-free cell culture. Altex 20, 275–281 (2003).
Twan, W. H., Hwang, J. S. & Chang, C. F. Sex steroids in scleractinian coral, Euphyllia ancora: Implication in mass spawning. Biol. Reprod. 68, 2255–2260 (2003).
Twan, W. H. et al. Hormones and reproduction in scleractinian corals. Comp. Biochem. Physiol. A Mol. Integr. Physiol. 144, 247–253 (2006).
Mak, A. S. et al. Vitellogenesis in the red crab Charybdis feriatus: Hepatopancreas-specific expression and farnesoic acid stimulation of vitellogenin gene expression. Mol. Reprod. Dev. 70, 288–300 (2005).
Tsutsui, N. et al. The effects of crustacean hyperglycemic hormone-family peptides on vitellogenin gene expression in the kuruma prawn, Marsupenaeus japonicus. Gen. Comp. Endocrinol. 144, 232–239 (2005).
Tsutsui, N., Ohira, T., Kawazoe, I., Takahashi, A. & Wilder, M. N. Purification of sinus gland peptides having vitellogenesis-inhibiting activity from the whiteleg shrimp Litopenaeus vannamei. Mar. Biotechnol. (NY) 9, 360–309 (2007).
Acknowledgements
This research was supported by a grant from the Ministry of Science and Technology, Taiwan MOST 108-2628-B-019-003 to SS. We gratefully acknowledge colleagues and divers who helped us to collect samples. We especially thank Dr. Pung-Pung Hwang from Academia Sinica, Taiwan, who served as a scientific advisor.
Author information
Authors and Affiliations
Contributions
S.S. conceived and designed the study. Y.L.C. and S.S. performed sample collection and lab work. Y.L.C. and S.S. analyzed data. Y.L.C., C.F.C., and S.S. wrote the manuscript text and prepared figures. All authors gave final approval for publication and agree to be held accountable for the work performed.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Chiu, YL., Chang, CF. & Shikina, S. Development of an in vitro tissue culture system for hammer coral (Fimbriaphyllia ancora) ovaries. Sci Rep 11, 24338 (2021). https://doi.org/10.1038/s41598-021-03810-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-021-03810-x
This article is cited by
-
What are the toxicity thresholds of chemical pollutants for tropical reef-building corals? A systematic review
Environmental Evidence (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
