
Abstract
Deficiency of P18 can significantly improve the self-renewal potential of hematopoietic stem cells (HSC) and the success of long-term engraftment. However, the effects of P18 overexpression, which is involved in the inhibitory effects of RUNX1b at the early stage of hematopoiesis, have not been examined in detail. In this study, we established inducible P18/hESC lines and monitored the effects of P18 overexpression on hematopoietic differentiation. Induction of P18 from day 0 (D0) dramatically decreased production of CD34highCD43− cells and derivative populations, but not that of CD34lowCD43− cells, changed the cell cycle status and apoptosis of KDR+ cells and downregulated the key hematopoietic genes at D4, which might cause the severe blockage of hematopoietic differentiation at the early stage. By contrast, induction of P18 from D10 dramatically increased production of classic hematopoietic populations and changed the cell cycle status and apoptosis of CD45+ cells at D14. These effects can be counteracted by inhibition of TGF-β or NF-κB signaling respectively. This is the first evidence that P18 promotes hematopoiesis, a rare property among cyclin-dependent kinase inhibitors (CKIs).
Similar content being viewed by others


Introduction
Hematopoiesis can be divided into primitive and definitive hematopoiesis, which originate from the yolk sac (YS) and the aorta/gonad/mesonephros (AGM) region, respectively1,2,3,4. Cell differentiation, including development of the hematopoietic lineage, is closely related to the cell cycle and is regulated by multiple cell cycle factors5,6. Among those factors, cyclin-dependent kinase inhibitors (CKIs) can manipulate the cell cycle and induce cell cycle arrest7. CKIs can be divided into two families: the Cip/Kip family, including p21Cip1/Waf1/Sdi1 (P21), p27Kip1 (P27), and p57Kip2 (P57), and the INK4 family, including p16INK4a (P16), p15INK4b (P15), p18INK4c (P18), and p19INK4d.2 (P19). P15, P18, P19, and P27 are expressed in all cell lines8. Members of the INK4 family play important roles in hematopoietic differentiation, which is downstream of the TGF-β/SMAD signaling pathway and is also controlled by RUNX1, especially in adulthood9,10,11.
Among the members of the INK4 family, p18INK4c (cyclin-dependent kinase inhibitor 2C, CDKN2C) is focally expressed during embryonic development12,13 and has important functions during hematopoiesis14,15. By interacting with the Cyclin D–CDK4/CDK6 complex, p18INK4c blocks the activation of the CDK kinases, thus playing a key role in controlling the G1 phase of the cell cycle. This interaction is associated with G1 arrest16.
Deletion or inhibition of the P18 gene can boost the self-renewal potential of hematopoietic stem cells and strikingly improve long-term engraftment by increasing self-renewal and differentiation of primitive hematopoietic cells in murine transplant models16,17,18. In comparison with the wild-type HSCs of unmanipulated young mice, P18-deficient HSCs can maintain their competitiveness and retain the potential for multi-lineage differentiation after multiple rounds of continuous bone marrow transplantation for as long as 3 years16. In addition, deletion of P18 significantly slows hematopoietic exhaustion caused by deletion of P2119. Relative to other CKIs, such as P21, P27, and P16, deletion of P18 has a stronger pro-hematopoietic effect, increasing the self-renewal time of hematopoietic stem cells and obtaining the advantages of transplantation20,21. Proliferation of hematopoietic progenitor cells (HPCs) is reduced in P18−/− mouse bone marrow, indicating that deletion of P18 has a positive effect on HPCs in vivo22. To date, however, the effects of P18 overexpression on hematopoiesis have not been systematically explored.
In previous research, we found that overexpression of RUNX1b during early hematopoiesis prevents the generation of CD34+ cells1. Subsequent studies revealed that RUNX1b overexpression also changes the status of the cell cycle and increases the expression of some cell cycle regulators, including P18 (unpublished data). Hence, we sought to elucidate the function of P18 on different stages of hematopoiesis using a mature inducible expression system based on piggyBac transposon and the AGM-S3 co-culture system that we established previously1,23,24. Our findings provide the first evidence that P18 overexpression can promote hematopoiesis, providing insight into the molecular mechanisms underlying CKI activity during hematopoietic differentiation.
Material and methods
Co-culture of hESCs with AGM-S3 cells
This study was approved by the institutional ethics committee of Institute of Blood Transfusion, Chinese Academy of Medical Sciences and Peking Union Medical College (CAMS & PUMC). AGM-S3 cells (provided by Prof. Tatsutoshi Nakahata) were plated in 12-well plates at 1 × 105 cells per well and cultured in an incubator containing 5% CO2 at 37 °C. After the cells had grown to 80–100% confluence, they were irradiated with 13 Gy of X-rays. Undifferentiated hESCs (provided by Prof. Tao Cheng) were cut into small squares containing 0.5–1 × 103 cells each by 200 μl tips, which were inoculated into 12-well plates (25 pieces per well). hPSC maintenance medium (Dulbecco’s modified Eagle’s medium (DMEM) with high glucose, F-12 nutrient mixture, 20% knockout serum replacement (KSR; Gibco), 1% L-glutamine, 1% non-essential amino acid solution (NEAA; Gibco), and 5 ng/ml basic FGF(b-FGF; Wako)) was used for the co-culture system for the first 3 days, and then replaced with hematopoiesis-inducing medium (Iscove’s modified Dulbecco’s medium (IMDM) containing 10% fetal bovine serum (FBS; Hyclone), 1% NEAA (Gibco), 60 ng/ml ascorbic acid (Sigma), and 20 ng/ml vascular endothelial growth factor (VEGF; Wako)); this day was defined as D0. The co-cultures were grown for up to 14 days with 5% CO2 at 37 °C, with a medium replacement once per day. The detailed procedure was defined previously1,23,24.
Confirming the inhibitory effects of P18 overexpression and the antagonistic effects of inhibition of TGF-β signaling at the early stage of hematopoiesis
The establishment of P18 (CDKN2C) inducible hESC lines (referred as P18/hESCs) was described in “Supplemental materials and methods”. D0-induced P18/hESCs co-cultured with AGM-S3 cells were treated without induction or with DOX from D0, D2, D4, D6, D8, D10, D12, or with DOX and 0.33 μM RepSox (Selleck Inc, dissolved in DMSO) from D0, as previously described1. An equal volume of DMSO was added to control samples. Treated co-cultures were subjected to cell-cycle analysis at D4 and flow cytometry using 7-AAD and anti-CD34/CD43 antibodies (D8) or 7-AAD and anti-CD34/CD43/CD45, CD71/GPA, or CD34/CD43/GPA/CD41a antibodies (D14). Untreated co-cultures were used as negative controls. The detail information of flow cytometry was described in “Supplemental materials and methods”.
Hematopoietic colony-forming assays
D14 P18/hESC co-cultured cells induced with DOX from D0, D6, or D10, or not induced, were dissociated into single cells with 0.25% trypsin–EDTA solution, centrifuged at 400g for 5 min, and resuspended in 400 μl of IMDM medium. After cell counting. 5 × 104 cells in suspension were mixed well on methylcellulose (H4320, STEM CELL) containing 1% Antibiotic–Antimycotic (Gibco) and cytokines, as previously described23,24,25, and then 1.1 ml of the mixture was divided into each 35-mm Petri dishes at a final concentration of 5 × 104 cells per dish. The cells were incubated for 12–14 days at 37 °C in a 5% CO2/95% humidity incubator. Colony forming unit–erythrocyte (CFU-E) was determined after 7 days, and other types of colonies were counted after 12–14 days.
Further hematopoietic culture analysis
Non-induced or induced P18/hESC co-cultures at D2 or D6 were dissociated by treatment with 0.05% trypsin EDTA solution and stained with 7-AAD and anti-KDR antibodies (D2) or anti-CD34/CD43 (D6). KDR+ cells at D2 or CD34highCD43- and CD34lowCD43- cells at D6 were sorted using a BD FACSJazz Cell Sorter, and their purity was confirmed by flow cytometry analysis. 1 × 104 sorted KDR+ cells were re-plated on irradiated AGM-S3 cells in each well of a 24-well plate, and 1 × 104 sorted CD34highCD43− and CD34lowCD43− cells were re-plated in FLHD (full-lineage hematopoietic differentiation, IMDM containing 10% FBS, 100 ng/ml SCF, 100 ng/ml IL-6, 10 ng/ml IL-3, 10 ng/ml FL, 10 ng/ml TPO, and 4 IU/ml EPO) medium in each well of a 48-well plate, further cultured with or without DOX induction for 11 or 14 days respectively. Half of the media was replaced every day, and flow cytometry analysis was performed. The detail information of flow cytometry and sorting was described in “Supplemental materials and methods”.
Confirming the activation of NF-κB signaling and the antagonistic effects of NF-κB signaling inhibition on P18 overexpression at the late stage of hematopoiesis
D10-induced P18/hESCs co-cultured with AGM-S3 cells were detected by qRT-PCR at D14 using primers for NFKB1 and NFKB2. QNZ, an inhibitor of the NF-κB signaling pathway (Selleck Inc) was dissolved in DMSO; siRNAs against NFKB1 (Sangon Biotech Inc, Shanghai, China) were described previously23,24. D10-induced P18/hESC co-cultures were treated from D10 with 20 nM QNZ (or an equal volume of DMSO) or 20 nM siRNA against NFKB1 (siRNA NFKB-1 vs NFKB-3 = 1:1 mixture or an equal concentration of control siRNA), and changed fresh media every day. At D14, the cells were evaluated by qRT-PCR, flow cytometry, and cell cycle analysis; untreated co-cultures were used as negative controls. qPCR primer pairs are listed in Table S1. siRNA sequences are listed in Table S3. The detail information of cell cycle analysis and flow cytometry was described in “Supplemental materials and methods”.
Statistical analysis
All experimental data are described as means ± SD, and statistical significance was evaluated using Student’s t-test. p < 0.05 was considered statistically significant. FlowJo 10 (https://www.flowjo.com/solutions/flowjo/downloads/) and GraphPad Prism5 (https://www.graphpad.com/scientific-software/prism/) were used for data analysis.
Method statement
All methods were carried out in accordance with relevant guidelines and regulations.
Results
Overexpression of RUNX1b upregulates P18 in co-culture on D4
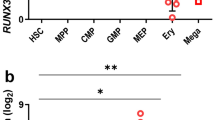
In co-cultures of RUNX1b/hESCs (Fig. 1a) with AGM-S3 cells, overexpression of RUNX1b at the early stage can block the mesoderm–hemogenesis transition, and treatment with 0.33 µM RepSox partially alleviates this blockage1. qRT-PCR (Fig. 1b) and western blotting (Fig. 1c) revealed that when RUNX1b/hESCs were induced at day 0 (D0), expression of P18 was upregulated at D4 (see Experimental Procedures for the definition of D0). All of these effects can be counteracted by addition of 0.33 µM RepSox from D0 (Fig. 1b,c). Together, these results indicated that P18 might be relevant to the inhibitory effects of RUNX1b on hematopoiesis.

Involvement of P18 is involved in inhibitory effects on the mesoderm–hemogenesis transition by RUNX1b, and establishment of inducible P18 transgenic hESC lines. D0-induced RUNX1b/hESCs (a) were co-cultured with AGM-S3 cells at D4. (b) qRT-PCR analysis and (c) Western blotting (WB) analysis revealed that when DOX was added from D0, P18 was upregulated at the mRNA and protein levels at D4, and that these effects can be counteracted by addition of 0.33 μM RepSox from D0. (d) Schematic diagram of the piggyBac vector used to induce P18 overexpression. TRE, tet-on regulation element; CMV Mini, minimum promoter of cytomegalovirus; T2A, Thosea asigna virus 2A peptide. (e) After P18/hESCs was treated with DOX for 48 h, co-expression of GFP was observed by fluorescence microscopy. (f) qRT-PCR analysis and (g) WB analysis confirmed that inducible expression of P18 was highly stringent and effective at the mRNA and protein levels. GAPDH was used as an internal control. (h) WB analysis with anti-SOX2, -OCT4, and -NANOG antibodies confirmed the normal pluripotency of P18/hESCs. The loading control was GAPDH. All results are expressed as means ± SD of three repeated experiments, and p < 0.05 was considered significant (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001).
P18/hESCs exhibit inducible P18 overexpression and normal pluripotency
After P18/hESCs were induced with DOX for 48 h, fluorescence imaging, qRT-PCR, and western blotting confirmed that P18 overexpression had been successfully achieved and stringently controlled (Fig. 1d–g). Western blotting revealed that OCT4, SOX2, and NANOG were normally expressed in P18/hESCs with or without DOX treatment, confirming that these cells had retained their normal pluripotency (Fig. 1h).
CD34+CD43− can be obviously divided into CD34lowCD43− and CD34highCD43− subpopulations with different traits during hematopoietic differentiation
We found that CD34+CD43− cells can be clearly divided into two subpopulations: CD34lowCD43− and CD34highCD43− cells. To investigate the difference in hematopoietic differentiation between them, these two subpopulations were sorted at D6, and subsequently resuspended in FLHD (full-lineage hematopoietic differentiation) medium without DOX induction (Fig. 2a). The results of flow cytometry analysis showed that compared with CD34lowCD43-, in FLHD medium CD34highCD43- subpopulations can produce more CD34 + CD43 + cells, which expression level of CD34 protein was much higher.

Overexpression of P18 from D0 inhibits hematopoiesis in co-culture with AGM-S3 cells. (a) P18/hESCs were cultured with AGM-S3 cells, 1 × 104 CD34HighCD43− and CD34LowCD43− cells were sorted from non-induced co-cultures at D6, and then re–re-plated in FLHD medium in each well of a 48-well plate without DOX induction for 14 days. (b) P18/hESCs were treated with or without DOX from D0, D2, D4, or D6, and then subjected to flow cytometry at D8 using 7-AAD and anti-CD34/CD43 antibodies. The GFP+ fractions of non-induced co-cultures and of co-cultures treated with DOX were compared. Overexpression of P18 at the early stage, especially from D0, led to significantly reduced production of CD34highCD43−, CD34−CD43+, and CD34+CD43+ cells at D8. (c) When P18 was induced from D0 to D8 or only from D0 to D2, production of the CD34highCD43−, CD34−CD43+, and CD34+CD43+ populations were also reduced to a similar degree at D8. All results are expressed as means ± SD of the three repeated experiments, and p < 0.05 was considered significant (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001).
Overexpression of P18 at the early stage blocks hematopoietic differentiation and the transition from CD34lowCD43− cells to CD34highCD43− cells
P18/hESCs treated with DOX starting from D0, D2, D4, or D6, or without DOX (as a negative control) were subjected to flow cytometry analysis at D8 to compare non-induced co-cultures and the GFP+ fractions of co-cultures treated with DOX. Overexpression of P18 at the early stage, especially from D0, significantly decreased production of CD34highCD43−, CD34−CD43+, and CD34+CD43+ populations at D8; these effects gradually weakened when P18 overexpression was initiated later, disappearing after D6 (Fig. 2b, Fig. S1a). When P18 was induced from D0 to D8 or only from D0 to D2, production of these populations was reduced to similar degrees at D8 (Fig. 2c, Fig. S1b). By contrast, the CD34lowCD43− population was not significantly affected. These results indicated that overexpression of P18 at the early stage severely blocks development of CD34highCD43− and its derived populations, except for the CD34lowCD43− population.
To investigate the inhibitory effects of P18 overexpression on generation of CD34+ cells, we sorted KDR+ cells at D2 from non-induced or induced P18/hESCs co-cultured with AGM-S3, and subsequently cultured them on irradiated AGM-S3 cells with or without DOX (Fig. 3). Flow cytometry revealed that production of CD34highCD43− cells was significantly decreased by overexpression of P18 from D0 to D2, whereas production of CD34lowCD43− cells was not significantly affected.

Overexpression of P18 at the early stage blocks formation of CD34highCD43− cells but not CD34lowCD43− cells. KDR+ cells were sorted from non-induced (a) or induced (b) P18/hESCs co-cultured with AGM-S3 at D2. About 1 × 104 sorted cells were re-plated on irradiated AGM-S3 (24 wells) treated with or without DOX, and FACS analysis with 7-AAD and anti-CD34/CD43 antibody was performed after 11 days. (c) Production of CD34highCD43− cells was dramatically decreased by overexpression of P18 from D0 to D2, whereas production of CD34lowCD43− cells was not severely affected. All results were expressed as means ± SD of three repeated experiments, and p < 0.05 was considered significant (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001).
Inhibition of TGF-β signaling counteracts the inhibitory effects of P18 overexpression at the early stage and alters cell-cycle status and apoptosis
P18/hESCs co-cultured with AGM-S3 cells were treated with DOX from D0 and subjected to flow cytometry at D4, D8, or D14. The results revealed that production of the CD34highCD43−, CD34−CD43+, and CD34+CD43+ populations were reduced at D8 and D14 (Fig. 4a,b, Fig. S2a,b), as confirmed by colony formation assays (Fig. 7a). Cell-cycle analysis of D4 co-cultures revealed that the proportion of KDR+ cells in G2/M phase decreased significantly, whereas the proportions of G0/G1 and S phase cells increased significantly (Fig. 4c). Apoptosis analysis of D4 co-cultures revealed that the proportion of KDR+ cells in apoptosis increased significantly (Fig. 4d, Fig. S5). qRT-PCR analysis at D4 demonstrated that expression of KDR was stable while important hematopoiesis related genes were downregulated with DOX induction (Fig. 4e). Treatment with both DOX and 0.33 μM RepSox counteracted all of these effects (Fig. 4). Thus, the inhibitory effects of P18 overexpression on the early stage of hematopoiesis might involve the TGF-β signaling pathway and alteration of cell-cycle status and transcription profile of key hematopoietic genes.

The effects of P18 overexpression from the early stage on hematopoietic differentiation can be counteracted by inhibition of TGF-β signaling. When P18/hESCs co-cultured with AGM-S3 cells were treated with DOX from D0, flow cytometry with 7-AAD and combination of anti-CD34/CD43 antibodies at (a) D8 or (b) D14 revealed that production of CD34highCD43−, CD34−CD43+, and CD34+CD43+ populations was reduced. (c) Cell cycle analysis at D4 indicated that the proportion of KDR+ cells in G2/M phase decreased significantly, whereas the proportions of cells in G0/G1 and S phases increased significantly. (d) P18/hESC cocultured with AGM-S3 cells were treated with or without DOX, or with both DOX and 0.33 μM RepSox started from D0. At D4 these cocultures were performed apoptosis analysis by corresponding kit using 7-AAD and anti-KDR antibody. The results indicated that P18 overexpression from D0 increased the apoptosis of co-cultures at D4, which can be counteracted by the inhibition of TGF-β signaling. (e) Co-cultured P18/hESCs were treated without or with DOX or with both DOX and 0.33 μM RepSox from D0, and analyzed by qRT-PCR at D4. The expression of KDR, which is related to mesoderm induction, was stable (i), while important hematopoiesis related genes were downregulated (ii). All of these effects can be counteracted by addition of 0.33 μM RepSox from D0. All results were expressed as means ± SD of three repeated experiments, and p < 0.05 was considered significant (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001).
Overexpression of P18 at the late stage promotes hematopoietic differentiation
P18/hESCs co-cultured with AGM-S3 cells treated with DOX from D6 or later, especially from D10, significantly promoted hematopoietic differentiation at the late stage (Fig. 5, Fig. S3). Flow cytometry at D14 revealed that the populations of CD34+CD43+, CD34−CD43+, CD34−CD45+, CD34+CD45+, GPA+CD71+, and erythroid-megakaryocytic progenitor (EMkP)-like cells were dramatically increased by induction of P18 from D10, indicating that P18 overexpression strongly promotes hematopoietic differentiation at the late stage.

Overexpression of P18 from D10 promotes hematopoietic differentiation. P18/hESCs were co-cultured with AGM-S3, and P18 was induced from D0, D2, D4, D6, D8, D10, or D12. Flow cytometry at D14 with 7-AAD and the antibodies against (a) CD34/CD43, (b) CD34/CD45, (c) CD71/GPA, (d) CD34/CD43/GPA/CD41a (For EMkP) revealed that most of the aforementioned cell populations were dramatically expanded by induction of P18 from D10, especially the CD34−CD43+, CD34−CD45+, GPA+CD71+, and EMkP populations, indicating that P18 overexpression strongly promoted hematopoietic differentiation at the late stage. All results were expressed as means ± SD of three repeated experiments, and p < 0.05 was considered significant (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001).
P18 overexpression from D10 upregulates NF-κB signaling, promotes hematopoietic differentiation, and changes cell-cycle status and apoptosis, and the most effects can be counteracted by inhibition of NF-κB signaling
When P18/hESCs co-cultured with AGM-S3 cells were treated with DOX from D10, qRT-PCR at D14 revealed that expression of both NFKB1 and NFKB2 was significantly higher than in non-induced cells (Fig. 6a). Overexpression of P18 from D10 stimulated hematopoietic populations at D14, including CD34+CD43+, CD34−CD43+, CD34−CD45+, CD34−CD45+, GPA+CD71+ cells. It also increased the proportion of cells in G0/G1 phase, but decreased the proportions of CD45+ cells in S and G2/M phases. Treatment with 20 nM QNZ or 20 nM siRNA against NFKB1 eliminated this increase (Fig. 6b,c, Fig. S4). Overexpression of P18 from D10 increased the proportion of CD45+ cells in apoptosis. Treatment with 20 nM QNZ or 20 nM siRNA against NFKB1 further increased the proportion of CD45+ cells in apoptosis (Fig. 6d, Fig. S6). Together, these observations revealed that P18 overexpression from D10 promotes hematopoietic differentiation, which is closely related to NF-κB signaling, potentially by altering cell-cycle status.

P18 overexpression from D10 promotes hematopoietic differentiation in a manner that involves NF-κB signaling and might result from alteration of cell cycle statues. (a) qRT-PCR detection at D14 revealed that when P18/hESCs co-cultured with AGM-S3 cells were treated with DOX from D10, the expression of both NFKB1 and NFKB2 was significantly higher than in non-induced cells, indicating that at the later stage, NF-κB signaling was up-regulated by P18 overexpression. (b) P18/hESCs co-cultured with AGM-S3 cells were treated from D10 with 20 nM QNZ (an inhibitor of NF-κB signaling) or 20 nM siRNA against NFKB1, and flow cytometry at D14 revealed that both treatments attenuated the positive effects of D10-induced P18 overexpression on the populations of CD34−CD43+, CD34+CD43+, CD34−CD45+, CD34+CD45+, and CD71+GPA+. (c) Analyses of cell cycle status revealed that P18 induction from D10 increased the proportion of CD45+ cells in G0/G1 phase but decreased the proportions of cells in S and G2/M phases; these effects were counteracted by treatment with QNZ or siRNA against NFKB1. (d) P18/hESCs co-cultured with AGM-S3 cells were treated with 20 nM QNZ (or an equal volume of DMSO as control) or 20 nM siRNA against NFKB1 (or an equal concentration of control siRNA) started from D10. At D14 these cocultures were performed apoptosis analysis by corresponding kit using 7-AAD and anti-CD45 antibody. The results indicated that P18 overexpression from D10 increased the apoptosis of co-cultures at D14, which can be further promoted with the inhibition of NF-κB signaling. All results were expressed as means ± SD of three repeated experiments, and p < 0.05 was considered significant (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001).
Overexpression of P18 blocks colony formation at the early stage and promotes it at the late stage
To further confirm that P18 overexpression had a negative effect on hematopoiesis, we performed hematopoietic colony assays on co-cultures at D14. P18 overexpression induced from D0 significantly blocked formation of CFU-GM, CFU-E, CFU-Mix, and BFU-E colonies; however, this inhibitory effect weakened or even disappeared when P18 was overexpressed from D6, and formation of all colony types was promoted by addition of DOX from D10 (Fig. 7a). The morphologies of typical hematopoietic colonies were examined by phase-contrast microscopy (Fig. 7b i–iv). BFU-E cells were confirmed by May–Grunwald–Giemsa staining (MGG) (Fig. 7b v).

Hematopoietic colony-forming assays. Co-cultured P18/hESCs were either not induced or treated with DOX from D0, D6, or D10. At D14, the cells were subjected to an assay to determine their hematopoietic potentials. (a) Numbers of colonies derived from 5 × 104 co-cultured cells. p < 0.05 was considered significant. (b) Typical morphologies of CFU-GM (i), CFU-Mix (ii), CFU-E (iii), and BFU-E (iv) colonies. Scale bars, 100 µm. MGG staining of cells in BFU-E colonies (v). Scale bar, 10 µm. CFU-E colony forming unit–erythrocyte, BFU-E burst-forming unit–erythroid, CFU-GM colony forming unit–granulocyte/macrophage, CFU-Mix colony forming unit–mixture. All results are expressed as means ± SD of three repeated experiments, and p < 0.05 was considered significant. (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001).
Discussion
P18 (CDKN2C) is a key CKI during the cell cycle. Knockout, knockdown or chemical inhibition of P18 in mouse models promotes the generation of HSCs17,20,21. To date, however, the effects of up-regulation of P18 on hematopoiesis have not been deeply explored. Previously, we reported that RUNX1b overexpression at the early stage blocks human hematopoiesis1. Here, we observed that in RUNX1b/hESC co-cultures, expression of P18 at D4 was upregulated when RUNX1b was induced from D0, and that this effect could be counteracted by addition of the TGF-β signaling inhibitor RepSox from D0 (Fig. 1b,c), which indicated that P18 might be relevant to the inhibitory effects of RUNX1b on hematopoiesis and the rescue effects of inhibiting TGF-β signaling inhibition, but both of these phenomena require further exploration. In this study, we have established an inducible P18/hESC line to investigate in detail the function of P18 overexpression (Fig. 1d–h).
The primary results showed us some key clues. At the early stage P18 overexpression can block the early hematopoietic differentiation, which is similar to the effects of RUNX1b/c that can be partially rescued by inhibition of TGF-β signaling1. At the late stage it can broadly promote the hematopoietic differentiation, which is similar to the effects of HOXA9 and HOXC423,24 that can be counteracted by inhibition of NF-κB signal pathway, and enhance the expression level of key genes of this pathway. Therefore, we speculated that P18 function ought to be closely related to TGF-β and NF-κB signaling at the early and late stage of hematopoiesis respectively according to our previous researches and performed further exploration based it.
In the cocultures with hESC and AGM-S3 at D8, CD34+CD43− cells can be produced and obviously divided into two sub-populations: CD34highCD43− and CD34lowCD43− cells. Compared with CD34lowCD43− cells, CD34highCD43− cells were able to produce more CD34+CD43+ cells expression level of CD34 protein, which indicated they might have different traits during hematopoietic differentiation (Fig. 2a). The CD34highCD43− cells might be the target population of P18, and that overexpression of P18 at the early stage (especially from D0) can severely decrease the production of these cells and their derivative populations, such as CD34+CD43+ and CD34−CD43+ cells at D8, which effect weakened when it was initiated after D2 and disappeared when it was initiated after D6. The CD34+CD43+ and CD34−CD43+ populations exhibited similar patterns (Fig. 2b). By contrast, the production of CD34lowCD43− cells was not significantly influenced. In addition, we found that the production of all of the aforementioned populations were influenced in a similar degree between the ones induced from D0 to D2 or from D0 to D8 (Fig. 2c). Overexpression of P18 from D0 to D2 was sufficient to prevent KDR+ cells in D2 cocultures from producing CD34high cells, but had no obvious influence on the production of CD34low cells (Fig. 3). When induction of P18 started from D4, the blockage disappeared. Above observation strongly indicated that the blockage of transition from the CD34low to CD34high sub-population happened mostly at the earliest stage of mesoderm induction (D0–D2) and not later than D4. Together, P18 overexpression in mesodermal populations (mainly KDR+ cells) at the early stage inhibited formation of the CD34high sub-population.
In a previous study, we showed that inhibition of TGF-β signaling partially rescued inhibition of hematopoietic differentiation by RUNX1b1, and the function of P18 at the early stage involves TGF-β signaling, a point that warranted investigation. The inhibition of TGF-β signaling could counteract most effects of P18 overexpression from D0 (Fig. 4a,b). The qRT-PCR analysis of hematopoiesis or mesoderm-related genes indicated that P18 overexpression probably blocked the mesoderm–hemogenesis transition while not significantly influenced the induction of mesoderm (Fig. 4d). For all these effects could be counteracted by inhibition of TGF-β signaling it is possible that together with TGF-β signaling, P18 overexpression from D0 significantly altered cell-cycle status, blocking the transition from CD34low to CD34high cells. We inclined to think that with D0–DOX treatment the D4 coculture cells were arrested in G1 stage and their proliferation was significantly retarded, which lead to severe blockage of the transition from CD34lowCD43− cells to CD34highCD43− cells and later significant less production of HSPCs than the control one. It ought to be the main reason that their D14 cocultures produced less colonies compared to the ones with D6-DOX treatment or untreated control ones. The P18 overexpression started after D6 had not such blockage effects, and the emerge of HSPC started at around D10 in AGM coculture system, at which stage P18 overexpression had a promote effects on hematopoiesis. Therefore, the cell cycle status of HSPCs (if in quiescent) might help to these prohibitory effects, but probably not be the main reason.
It is surprising that according to the results of flow cytometry the induction of P18 from D6 or later significantly promoted the production of the aforementioned populations at D8 (Fig. 2a), and that induction of P18 from D10 significantly promoted the production of classic hematopoietic populations at D14 (Fig. 5). Colony formation assays also confirmed such tendency of hematopoietic potentials changed by P18 overexpression at the late stages in functional level, consistent with the results of flow cytometry (Fig. 7). This observation indicated that from the middle to the late stage of hematopoiesis, P18 overexpression broadly promotes hematopoiesis, including myelogenesis and erythrogenesis. NFKB1 and NFKB2, were upregulated when P18 was induced overexpressed from D10 (Fig. 6a). When NF-κB signaling was inhibited by its inhibitor (QNZ) or by siRNA against NFKB1, except for further increased apoptosis, all other effects of P18 overexpression at the late stage were counteracted (Fig. 6b,c), which was very similar to the phenomenon caused by HOXC4 and HOXA9 overexpression at the late stage23,24, and indicated that these genes probably share common mechanisms to promote hematopoiesis with the aid of NF-κB signaling.
Xenotransplantation studies should be very helpful to elucidate the more detail function of p18 on hematopoiesis and show the changes of their potentials of hematopoietic differentiation and lineage bias caused by P18 overexpression and the inhibition of TGF-β or NF-κB signaling at different stages though we have no enough technique conditions to perform it. Nevertheless, the proofs from flow cytometry and colony formation assays ought to be enough to discover these phenomena, among which the promotion effects of P18 overexpression on hematopoiesis has been rare reported in a functional study of CKIs.
Because few previous studies had investigated the effects of P18 overexpression, we carefully monitored its effects on different stages of hematopoietic differentiation and sought to identify the relevant signal pathways. The negative effects at the early stage and the positive effects at the late stage both involved in changes in cell cycle status. Overexpression of P18 significantly decreased the proportion of D4 KDR+ cells or D14 CD45+ cells in G2/M and increased the proportions of these cells in G0/G1; these effects can be counteracted by the inhibition of TGF-β or NF-κB signaling. To the cells in S stage, the changes of cell cycle status caused by P18 overexpression was different at the different stages, which were also counteracted by the inhibition of the corresponding signaling (Figs. 4c, 6c). This indicated that both signaling pathways are involved in the effects of P18 overexpression on cell cycle status, with distinct consequences in different stages and contexts.
From the view of apoptosis, the P18 overexpression at the early or late stage can both promote the apoptosis. Inhibition of TGF-β signaling can counteract it at early stage while NF-κB signaling can promote it at the late stage (Figs. 4d, 6d). It is very interesting that the members of INK4 family was only reported to negatively regulate apoptosis26. Some INK4 member, such as P16, was inactive by hypermethylation and homozygous deletions in leukemia-lymphoma cell lines, and ectopic expression of P16 in such cell line will lead to growth inhibition, arrest in G1 without apoptosis and rare differentiation27. It is very similar to the effects of P18 in hematopoiesis at the early stage except that P18 overexpression can increase the apoptosis, which was also closely related to TGF-β signaling. But in renal tubular epithelial cells (LLC-PK1) p18 overexpression reduced the percentage of apoptotic cells significantly28, which indicated that it effects on apoptosis seem distinctive in different context and cell type.
p18 overexpression from D0 can block transition from CD34lowCD43− cells to CD34highCD43− cells so as to prevent the emerging of hematopoietic endothelium and their derived hematopoietic stem/progenitor cells. p18 overexpression from D10 can promote the development of hematopoietic stem/progenitor cells and other progenitors of classic hematopoietic lineage. It is reasonable to speculate that both of them ought to be caused by similar effects that G1 arrest, proliferation blockage and apoptosis were increased, but they influence the production of hematopoietic stem/progenitor cells with contrary style, which detail mechanism need further exploration. The primary mechanisms and functions of p18 overexpression in hematopoiesis were summarized in Fig. 8.

The diagram of control mechanisms for the functions of P18 on hematopoiesis. The RUNX1b overexpression can upregulate TGF-β signaling and P18, and the inhibition of TGF-β signaling can restore the expression level of P18. The P18 overexpression at D0 can severely decrease the product of CD34highCD43− cells and their derived populations but not CD34lowCD43− cells that can only be blocked by RUNX1b overexpression at D0. The P18 overexpression at D10 can significantly increase the product of classic hematopoietic populations, which can be counteracted by the inhibition of NF-κB signaling. P18 has distinctive function on hematopoietic differentiation at different stage, which are both related to the change of cell cycle status.
At the early stage of AGM-S3 co-culture, overexpression of P18 and P21 had opposing effects on the proportions of cells in S and G2/M, but similar inhibitory effects on hematopoietic differentiation. All of these effects could be counteracted by inhibition of TGF-β signaling, as in the case of RUNX1b1,29. Overexpression of any of these genes increased the proportion of cells in G0/G1, indicating that TGF-β signaling plays a key role in G1 arrest. Stimulation of TGF-β signaling mediates cell-cycle arrest and up-regulates expression of P1530,31,32.In our co-culture system, RUNX1b overexpression stimulated TGF-β signaling, which upregulates not only P15 (unpublished data) but also P18 and P21. P18, P15 and other members of the INK4 family contain repeated ankyrin motifs and can regulate G1 phase by inhibiting CDK4/6 and interfering with cyclin–CDK assembly, thereby inducing G1 arrest15,33,34. P21, which belongs to the Cip/Kip family, does not contain repeated ankyrin motifs and therefore cannot specifically bind to CDK4 and CDK6; consequently, its mechanism is distinct from that of P1819; however, the two proteins appear to have similar effects on hematopoiesis at the early stage. It is reasonable to speculate that with the help of TGF-β signaling, RUNX1 might coordinate and organize various CKIs (such as P15, P18, and P21) to control early hematopoietic differentiation via distinct mechanisms.
In contrast to P21, P18 overexpression at the late stage efficiently promotes hematopoietic differentiation, potentially due to differences in protein structure15,34. Similar effects can be observed following overexpression of HOXA9 and HOXC4 at the same stage23,24. Overexpression of these factors changed the cell cycle status with different styles while the inhibition of NF-κB signaling can counteract all these changes except for apoptosis, which reveal it involved in the function of P18 on hematopoietic differentiation through the controlling of cell cycle status but not apoptosis. This study provides the first evidence that P18 overexpression promotes hematopoiesis, a property that is very rare among CKIs. The underlying mechanism, which requires further exploration, may have important clinical applications.
Data availability
All date generated or analyzed during this study are included in the published article and its supplementary information files.
References
Chen, B. et al. Inducible overexpression of RUNX1b/c in human embryonic stem cells blocks early hematopoiesis from mesoderm. J. Mol. Cell Biol. 9(4), 262–273 (2017).
Lux, C. T., Yoshimoto, M., Mcgrath, K., Conway, S. J. & Yoder, M. C. All primitive and definitive hematopoietic progenitor cells emerging before E10 in the mouse embryo are products of the yolk sac. Blood 111(7), 3435–3438 (2008).
Matsuoka, S., Tsuji, K., Hisakawa, H., Xu, M. & Nakahata, T. Generation of definitive hematopoietic stem cells from murine early yolk sac and paraaortic splanchnopleures by aorta-gonad-mesonephros region-derived stromal cells. Blood 98(1), 6–12 (2001).
Medvinsky, A. & Dzierzak, E. Definitive hematopoiesis is autonomously initiated by the AGM region. Cell 86(6), 897–906 (1996).
Furukawa, Y. Cell cycle control during hematopoietic cell differentiation. Hum. Cell. 10(3), 159–164 (1997).
Furukawa, Y. Cell cycle control genes and hematopoietic cell differentiation. Leuk. Lymphoma. 43(2), 225–231 (2002).
Schreiber, M. et al. Comparison of the effectiveness of adenovirus vectors expressing cyclin kinase inhibitors p16INK4A, p18INK4C, p19INK4D, p21(WAF1/CIP1) and p27KIP1 in inducing cell cycle arrest, apoptosis and inhibition of tumorigenicity. Oncogene 18(9), 1663–1676 (1999).
Kissa, K. & Herbomel, P. Blood stem cells emerge from aortic endothelium by a novel type of cell transition. Nature 464(7285), 112–115 (2010).
Ford, A. M. et al. The TEL-AML1 leukemia fusion gene dysregulates the TGF-beta pathway in early B lineage progenitor cells. J. Clin. Invest. 119(4), 826–836 (2009).
Sood, R., Talwar-Trikha, A., Chakrabarti, S. R. & Nucifora, G. MDS1/EVI1 enhances TGF-beta1 signaling and strengthens its growth-inhibitory effect but the leukemia-associated fusion protein AML1/MDS1/EVI1, product of the t(3;21), abrogates growth-inhibition in response to TGF-beta1. Leukemia 13(3), 348–357 (1999).
Zhang, L., Magli, A., Catanese, J., Xu, Z. & Perlingeiro, R. C. R. Modulation of TGF-beta signaling by endoglin in murine hemangioblast development and primitive hematopoiesis. Blood 118(1), 88–97 (2011).
Phelps, D. E. et al. Coupled transcriptional and translational control of cyclin-dependent kinase inhibitor p18INK4c expression during myogenesis. Mol. Cell Biol. 18(4), 2334–2343 (1998).
Zindy, F., Soares, H., Herzog, K. H., Morgan, J. & Roussel, M. F. Expression of INK4 inhibitors of cyclin D-dependent kinases during mouse brain development. Cell Growth Differ. 8(11), 1139–1150 (1997).
Guan, K. L. et al. Growth suppression by p18, a p16INK4/MTS1- and p14INK4B/MTS2-related CDK6 inhibitor, correlates with wild-type pRb function. Genes Dev. 8(24), 2939–2952 (1994).
Hirai, H., Roussel, M. F., Kato, J. Y., Ashmun, R. A. & Sherr, C. J. Novel INK4 proteins, p19 and p18, are specific inhibitors of the cyclin D-dependent kinases CDK4 and CDK6. Mol. Cell Biol. 15(5), 2672–2681 (1995).
Yuan, Y., Shen, H., Franklin, D. S., Scadden, D. T. & Cheng, T. In vivo self-renewing divisions of haematopoietic stem cells are increased in the absence of the early G1-phase inhibitor, p18INK4C. Nat. Cell Biol. 6(5), 436–442 (2004).
Gao, Y. et al. Small-molecule inhibitors targeting INK4 protein p18(INK4C) enhance ex vivo expansion of haematopoietic stem cells. Nat. Commun. 6, 6328 (2015).
Yuan, Y. et al. Hematopoietic stem cells are not the direct target of spontaneous leukemic transformation in p18(INK4C)-null reconstituted mice. Cancer Res. 66(1), 343–351 (2006).
Yu, H., Yuan, Y., Shen, H. & Cheng, T. Hematopoietic stem cell exhaustion impacted by p18 INK4C and p21 Cip1/Waf1 in opposite manners. Blood 107(3), 1200–1206 (2006).
Song, X. et al. Long-term engraftment of p18(INK4C)-deficient hematopoietic stem cells is enhanced in the sublethally-irradiated recipients. Sci. China C Life Sci. 49(4), 390–394 (2006).
Wang, Y. Y. et al. RNA interference reveals a requirement for both p18INK4c and p27Kip1 in B lymphopoiesis. J. Mol. Cell Biol. 2(4), 209–216 (2010).
Broxmeyer, H. E., Franklin, D. S., Cooper, S., Hangoc, G. & Mantel, C. Cyclin dependent kinase inhibitors differentially modulate synergistic cytokine responsiveness of hematopoietic progenitor cells. Stem Cells Dev. 21(10), 1597–1603 (2012).
Zeng, J. et al. HOXC4 up-regulates NF-κB signaling and promotes the cell proliferation to drive development of human hematopoiesis, especially CD43+ cells. Blood Sci. 2(4), 117–128 (2020).
Zeng, J. et al. Overexpression of HOXA9 upregulates NF-κB signaling to promote human hematopoiesis and alter the hematopoietic differentiation potentials. Cell Regeneration. 10, 9 (2020).
Chang, J. et al. Establishment of an in vitro system based on AGM-S3 co-culture for screening traditional herbal medicines that stimulate hematopoiesis. J. Ethnopharmacol. 240, 111938 (2019).
Ivanchuk, S. M. et al. The INK4A/ARF locus: Role in cell cycle control and apoptosis and implications for glioma growth. J. Neurooncol. 51, 219–229 (2001).
Drexler, H. Review of alterations of the cyclin-dependent kinase inhibitor INK4 family genes p15, p16, p18 and p19 in human leukemia–lymphoma cells. Leukemia 12, 845–859 (1998).
Zhang, Y. et al. Overexpression of p18INK4C in LLC-PK1 cells increases resistance to cisplatin-induced apoptosis. Pediatr. Nephrol. 26, 1291–1301 (2011).
Zeng, J. et al. Overexpression of p21 has inhibitory effect on human hematopoiesis by blocking generation of CD43 cells via cell-cycle regulation. Int. J. Stem Cells. 13(2), 202–211 (2020).
Hannon, G. J. & Beach, D. p15INK4B is a potential effector of TGF-beta-induced cell cycle arrest. Nature 371(6494), 257–261 (1994).
Reynisdottir, I. & Massague, J. The subcellular locations of p15(Ink4b) and p27(Kip1) coordinate their inhibitory interactions with cdk4 and cdk2. Genes Dev. 11(4), 492–503 (1997).
Reynisdottir, I., Polyak, K., Iavarone, A. & Massague, J. Kip/Cip and Ink4 Cdk inhibitors cooperate to induce cell cycle arrest in response to TGF-beta. Genes Dev. 9(15), 1831–1845 (1995).
Latres, E. et al. Limited overlapping roles of P15(INK4b) and P18(INK4c) cell cycle inhibitors in proliferation and tumorigenesis. EMBO J. 19(13), 3496–3506 (2000).
Sherr, C. J. & Roberts, J. M. Inhibitors of mammalian G1 cyclin-dependent kinases. Genes Dev. 9(10), 1149–1163 (1995).
Acknowledgements
We thank Professor Tao Cheng from the State Key Laboratory of Experimental Hematology, CAMS & PUMC for generously providing the H1 hESC line, and Professor Tatsutoshi Nakahata from Center for iPS Cell Research and Application (CiRA), Kyoto University for generously providing AGM-S3 cell line. This work was supported by awards from the CAMS Initiatives for Innovative Medicine (2016-I2M-1-018 to F. Ma, 2017-I2M-3-021 to J.X. Liu, 2020-I2M-CoV19-006 to Z. Liu), Sichuan Provincial Science and Technology Department Key R&D projects (020YFSY0023 to B. Chen), and the Chengdu Science and Technology Project-Technology Innovation R&D (2018-YF05-01341-SN to B. Chen).
Author information
Authors and Affiliations
Contributions
Conceived and designed the experiments: B.C. Performed the experiments: B.C., D.Y., L.Z., Y.L., J.Z., W.S., J.T., J.C., Y.D., X.P., Y.C., Y.Z., M.L., Q.Z. Analyzed and interpreted the data: B.C., D.Y., L.Z., Y.L., F.M. Contributed reagents, materials, analytical tools, or data: B.C., D.Y., L.Z., Y.L., J.Z., W.S., J.L., F.M.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Yi, D., Zhu, L., Liu, Y. et al. The distinct effects of P18 overexpression on different stages of hematopoiesis involve TGF-β and NF-κB signaling. Sci Rep 11, 24014 (2021). https://doi.org/10.1038/s41598-021-03263-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-021-03263-2
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
