
Abstract
Esophageal cancer is an aggressive tumor that has a high rate of incidence and mortality worldwide. It is the 10th most frequent type in Brazil, being squamous cell carcinoma (ESCC) the predominant subtype. There is currently an incessant search to identify the frequently altered genes associated with esophageal squamous cell carcinoma biology that could be druggable. This study aimed to analyze the somatic mutation profile of a large panel of cancer-related genes in Brazilian ESCC. In a series of 46 ESCC diagnoses at Barretos Cancer Hospital, DNA isolated from paired fresh-frozen and blood tissue, a panel of 150 cancer-related genes was analyzed by next-generation sequencing. The genes with the highest frequency of mutations were TP53 (39/46, 84.8%), followed by NOTCH1 (7/46, 15.2%), NFE2L2 (5/46, 10.8%), RB1 (3/46, 6.5%), PTEN (3/46, 6.5%), CDKN2A (3/46, 6.5%), PTCH1 (2/46, 4.3%) and PIK3CA (2/46, 4.3%). There was no significant association between molecular and patients’ clinicopathological features. Applying an evolutionary action score of p53 (EAp53), we observed that 14 (35.9%) TP53 mutations were classified as high-risk, yet no association with overall survival was observed. Concluding, this the largest mutation profile of Brazilian ESCC patients, which helps in the elucidation of the major cancer-related genes in this population.
Similar content being viewed by others

Introduction
Esophageal cancer is an aggressive tumor with high incidence and mortality rates1,2. Worldwide estimates show that esophageal cancer is the seventh most common type, with around 572,000 esophageal cancer cases in 20181,2. In Brazil, esophageal cancer is among the ten most common tumor types, ranking 6th among men and 15th among women3. According to the National Cancer Institute (INCA), for the year 2020–2021, it is estimated that about 11390 new cases, 8690 in men and 2700 in women, also presenting mortality rates close to the incidence rates3. Esophageal cancer's poor prognosis is due to a lack of specific symptoms in the early stages of the disease and late diagnosis, with less than 20% of cases showing an overall five-year survival4,5. However, when diagnosed early, the five-year survival rate increases to 80–90%5.
Histologically, 90% of cases are esophageal squamous cell carcinoma (ESCC) and 12% of adenocarcinoma (EADC)5. The highest incidence of ESCC occurs in northern Iran to central north China and in developing countries such as Brazil, whereas EADC occurs more frequently in developed countries3,5,6,7. ESCC affects the middle third of the esophagus and shares several characteristics with head and neck squamous cell carcinoma5,8. The main risk factors are tobacco and alcohol consumption (especially in combination), hot food and beverage intake5,9,10.
Recent extensive comprehensive molecular studies determine the genomic landscape of ESCC11,12,13,14,15,16,17. The TP53 was the most frequent mutated gene observed all over the studies, and genes involved in other essential cancer pathways, such as the cell cycle, PI3K, and NOTCH pathways, were also reported11,12,13,14,15,16,17. Of note, the integrative TCGA consortium identified three molecular subtypes: ESCC1, associated with alterations in the NRF2 pathway, which regulates adaptation to oxidative stressors, and gene expression profile resembles lung cancer and head and neck squamous carcinoma; ESCC2, with higher NOTCH1 mutation rate and deregulation of apoptosis: and ESCC3 with upregulation of the PI3K pathway and lower TP53 mutation rate17. Interestingly, the three ESCC subtypes tended geographic associations, being the ESCC1 more prevalent in the Asian population, the ESCC2 in Eastern European and South American (Brazil), and the ESCC3 observed only in North America17. The characterization of genes involved in ESCC tumorigenesis is crucial to understand its biology and help identify putative cancer biomarkers and targeted therapies13.
The molecular profile of ESCC is mainly unknown in Brazil, being mainly restricted to TP53 mutation status7,18. Therefore, this study aimed to identify the somatic mutational profile of Brazilian ESCC patients by analyzing the entire coding region of a panel of 150 cancer-related genes by next-generation sequencing.
Results
Description of the clinicopathological features
The summary of clinicopathological features of the esophageal squamous cell carcinoma studied is reported in Table 1. We observed that the population was primarily composed of men (39 cases, 84.8%) with an average age of 60 years (minimum 39 years and maximum 77 years); most of them consumed alcohol (37 cases, 82.2%) and tobacco (36 cases, 80.0%). The most used these substances in combination (32 cases, 71.1%). Most patients presented moderately differentiated tumors (26 cases, 59.1%), whose clinical stage was most advanced III and IV (36 cases, 87.8%). Patients' median survival concerning the tumor diagnosis data until the last information (death or follow-up) was 9.40 months.
Description of the mutation profile
We sequenced the whole coding region of 150 cancer-related genes in 46 cases of esophageal squamous cell carcinoma. The mean read depth of sequencing was 911 × per gene and 310.9 × per variant. We found a mean of 1.9 driver mutations per patient (range 0–7), and we identified at least one driver somatic variant in 42 tumor samples. Driver mutations in single genes were found in 41.3% of tumors (19/46), whereas 26.1% (12/46) showed driver mutations in two genes, 17.4% (8/46) in three genes, 6.5% (3/46) in four or more genes and 8.7% (4/46) showed driver mutations in none of the genes analyzed. In total, 25 genes were found to harbor driver somatic mutations (Fig. 1). A complete list of variants (missense, frameshift, nonsense, in-frame, and splice mutations) identified is presented in Supplementary Table S1.

Oncoplot of the distribution of mutations found in cases of esophageal squamous cell carcinoma. The upper graph shows the mutation frequency for each tumor sample. The left graph shows the frequency of samples with mutations. The central graph shows the types of mutations in each tumor sample. The lower part of the figure shows the clinical-pathological data (tumor stage in the diagnosis and histological differentiation) of each sample.
The gene with the highest frequency of driver mutations was TP53 (39 cases, 85%), followed by NOTCH1 (5 cases, 11%), NFE2L2 (5 cases, 11%), RB1 (3 cases, 7%), PTEN (3 cases, 7%), CDKN2A (3 cases, 7%), PTCH1 (1 case, 2%) and PIK3CA (2 cases, 4%) (Fig. 1). Below, we describe in more detail the most affected genes and pathways.
Genes involved in the cell cycle
Of the 25 genes with driver mutations identified, ten were involved in the cell cycle (Fig. 1; Supplementary Table S1). Overall, 41 (89.1%) cases showed mutation of this crucial cancer pathway.
A total of 44 somatic TP53 mutations were identified in 39 tumors (85%) (Fig. 1; Supplementary Table S1). The mutations were 28 missense, 9 nonsense, 4 splice sites, and 3 frameshift. The most frequent changes were p.His193Arg, p.His179Arg, p.Arg248Trp, p.Arg273Leu, p.Tyr107Asp, and p.Tyr220Cys, all of them previously reported. We also identified two changes (p.Thr211AsnfsTer5 and p.Thr256HisfsTer8) that have not been previously identified. (Fig. 2A).

Lollipop plot of the genes with the highest frequency of mutations.
Five cases (11%) showed somatic mutations in the NFE2L2 gene (Figs. 1, 2 and Supplementary Table S1). The mutations were all missense, being p.Arg34Gln and p.Val32Gly in 2 cases, and p.Glu79Lys and p.Leu30Phe, which was present in one case each (Fig. 2B). Three tumors (7%) exhibited somatic changes in the RB1 gene (Fig. 1; Supplementary Table S1). The mutations were two frameshifts and one splice site (Fig. 2D). The CDKN2A gene was mutated in 3 cases (7%) (Fig. 1; Supplementary Table S1). The types of changes identified in the CDKN2A gene were two nonsense and one frameshift (Fig. 2E). We also observed the PTCH1 gene's somatic mutation in two tumors (2%) (Fig. 1; Supplementary Table S1). The types of changes identified were one missense and one nonsense. Moreover, we observed mutations in TERT, STAT3, DDX3X, CDK12, and BAP1 genes in one case each (Supplementary Table S1).
NOTCH signaling pathway alterations
The Notch signaling pathway involving the genes NOTCH1 and FBXW7 was altered in 6 cases (13%) (Fig. 1; Supplementary Table S1). The most affected gene was NOTCH1 (11%), and the types of changes identified were 3 missense, one frameshift, and one splice site (Fig. 2C). Only one missense mutation was identified in FBXW7.
PIK3-AKT signaling pathway alterations
The PIK3-AKT signaling pathway involving the genes PTEN, PIK3CA, and TSC2 was altered in 6 (13%) cases (Fig. 1; Supplementary Table S1).
PTEN was the gene with the most mutations identified with alterations in 3 cases (7%). The types of changes identified were one missense, one nonsense, and one translocation (Fig. 2F). Mutations in the PIK3CA gene were identified in 2 cases (4%), at hotspot codons p.Glu542Lys and p.Glu545Lys in one case each. The TSC2 gene was also mutated in one case (2%), and the alteration was a p.Thr831Met.
MAPK signaling pathway alterations
The MAPK signaling pathway, with genes KRAS, NF1, and BRAF, was altered in 3 ESCC (7%) (Fig. 1; Supplementary Table S1). All three genes were changed in one case each (2%). All changes were missense type. The type of alterations in the KRAS gene was the p.Gly12Ser; in the NF1 gene, it was p.Pro2115Leu, and in the BRAF gene, it was p.Arg260Cys, outside the known hotspot V600 region.
Genes involved in the DNA repair
Of the 25 mutated genes, only 2 (MSH6 and PALB2) were involved in the DNA repair (Fig. 1; Supplementary Table S1).
RTKs signaling pathway alterations
All four genes of the RTKs signaling pathway had a mutation each (2%). The RET gene is the missense p.Arg1050Gln mutation; the EPHA7 gene harbor the splice site p.Val777Leu mutation and the ERBB2 gene have a missense mutation. In the KIT gene, the only mutation was for p.Val825Gly.
Classification of TP53 mutations
TP53 mutations were further classified using the evolutionary action score of p53 (EAp53), which considers only missense mutations such as “low-risk” and “high-risk”19. Of the 23 mutations, we observe 14/23 (60%) as high-risk and 9 /23 (40%) as low-risk (Table 2). A second classification was done following Poeta et al., which categorize mutations as “disruptive” and “non-disruptive”20,21, and we observed 21/39 (54%) as disruptive and 18/39 (46%) as non-disruptive (Table 2).
Association of mutation profile and clinicopathological features
Next, we performed an analysis of the association between clinicopathological features and mutation status. Due to the small number of cases analyzed, this analysis was performed only for genes harboring more than 10% of mutations, namely TP53, NFE2L2, and NOTCH1 (Table 3). No significant association was found (Table 3). We also analyzed the association of TP53 categories of (EAp53 risk score and disruptive and non-disruptive) with patients’ clinicopathological features, yet, no significant association was found (Table 4).
In addition, a Kaplan–Meier survival analysis was performed, and no association was observed with TP53, NFE2L2, and NOTCH1, neither with TP53 EAp53 score and disruptive and non-disruptive categories (Table 5 and Fig. 3).

Kaplan–Meier curve for assessing the estimated overall survival probability of follow-up time among esophageal squamous cell carcinoma patients concerning mutation profile. A – TP53 gene; B – NOTCH1 gene; C- NFE2L2 gene; D – Eap53 score; E- Disruptive and non-disruptive classification of TP53 mutations.
Discussion
In the present study, we performed, for the first time, the somatic mutational profile of a panel of 150 cancer-related genes in a series of 46 Brazilian esophageal squamous cell carcinomas. We identified 25 genes with alterations, the genes with the highest frequencies being TP53, NOTCH1, and NFE2L2.
The frequency of mutation observed was compared with the literature, and similar frequencies were observed with other populations (Table 6). As expected, the TP53 gene was the highest mutated (84.8%, 39 cases). In our study population, its frequency was similar to that reported in other studies such as TCGA14,15,16,17,22,23,24,25 (Table 6) and higher than previously reported in the Brazilian ESCC population (34–40%)18,24,26. This discrepancy with previous Brazilian studies is probably due to the distinct methodologies used since Sanger sequencing and hotspot TP53 regions were used on those studies, at variance with all TP53 coding regions and NGS used in the present study. We also applied for the first time a risk-system score based on Evolutionary Trace (ET) method proposed and validated in head and neck tumors (EAp53)19 to evaluate its impact in ESCC. We showed that 36% of TP53 mutation in squamous cell carcinoma would be considered high-risk; however, there was no significant association between the clinical and pathological condition of the patient and the prognosis. This lack of significance could be related to the limited number of cases analyzed, and further studies are needed to validate our findings. We also classified TP53 mutations in disruptive and non-disruptive, as reported by Poeta et al.20 and Molina et al.21, for head and neck and lung cancer, respectively. We observed that 54% were disruptive, yet no significant statistical association was observed with the clinical-pathological characteristics and patient prognosis.
Another important gene of the cell cycle is the CDKN2A gene, also known as p16, which the loss of its function can result from homozygous deletions and mutations. According to the TCGA and genomic studies, the significant alteration is homozygous deletion, leading to 76% loss of function of the CDKN2A gene in ESCC17. At variance with high copy number alterations, CDKN2A mutation frequency is much lower, ranging from 3–8% (Table 6).
The second most mutated gene was NOTCH1 (11%), which is part of the NOTCH signaling pathway, important to regulate the cell cycle senescence27. In esophageal squamous cell carcinoma, the frequency of mutations in this gene varies from 8 to 19% (Table 6), so our population is within the reported range. These findings are in concordance with the ESCC2 TCGA molecular subtype, observed in the 15 patients from South America (Brazil) analyzed17.
The NFE2L2 gene encodes the NRF2 protein, a transcription factor regulating the expression of antioxidant proteins that protect against damage caused by injuries and inflammation28. Genetic deletion of NRF2 the susceptibility to the development of cancer increases, causing the tumor cell to survive the oxidative stress caused by chemoradiation leading to resistance to treatment29,30,31. The frequency of mutations in the NRF2 gene has been reported in 3–17% of ESCC (Table 6). Driver mutations in NRF2 are believed to be late events in the process of developing esophageal squamous cell carcinoma22,32. In our population, we observed a frequency of 10.8% of alterations in this gene, and in accordance with TCGA, it is associated with the ESCC1 subtype, typical of squamous carcinomas associated with tobacco exposure17.
The PI3K-AKT signaling is a complex pathway that regulates cell growth, proliferation, motility, apoptosis, and cell growth and is often related to the development of esophageal squamous cell carcinoma through driver mutations in the PIK3CA gene15,16,33. The frequency of mutations reported in the literature varied from 4 in the present to 13% (Table 6). Recently our group analyzed another series of 38 ESCC and observed a frequency of 10.5% of PIK3CA mutations34. The majority of the mutations are present in the hotspot exons 9 (E542K and E545K) and 20 (H1047R)34,35,36. (Supplementary Table S1). Another gene of this PI3K-AKT pathway is the PTEN, which is frequently mutated in some tumor types, such as melanomas and glioblastomas, yet its mutation rate in ESCC is lower, varying from 1 to 6.5%, as observed in our Brazilian series (Table 6). Of note, the TCGA ESCC3 subgroup is characterized by upregulation of this pathway, and in our study, it was alternated in 13% (PTEN, PIK3CA, and TSC2).
The KRAS gene is an essential biomarker in cancer, mainly because it predicts the efficacy in therapies targeting the growth factor EGFR in tumors such as colorectal cancer37,38. According to the TCGA, the frequency of mutations in the KRAS gene is low, 7%, which is in line with the results obtained in our study population, which was 2%17.
ESCC is usually diagnosed late, and the minority of patients can benefit from treatments such as chemotherapy and radiation therapy39. Target therapies are important approaches for several tumors, including ESCC39. Significantly, in the present work, the identification of patients harboring PIK3CA mutations could potentially benefit from PI3K and mTOR inhibitors, such as buparlisib, alpelisib, and everolimus40,41. Moreover, the recent development of anti-KRAS agents, such as sotorasib and adagrasib42,43, can bring some hope for patients with KRAS mutations (Table 6).
The present study has several limitations, being the relatively small number of cases analyzed the major issue, and it could explain the lack of significant association of mutation status and patients’ clinical-pathological features. Additionally, a limited panel of 150 cancer-related genes, not whole-genome nor whole-exome sequencing, was performed, so a complete picture of the mutated landscape is lacking. Therefore, further studies with a larger population and broader mutation analysis are needed. Despite these issues, we performed paired germline/tumor analysis, and it is the first to the somatic landscape of an admixture population such as the Brazilian ESCC population. Our findings align with the frequencies reported in other populations, namely Occidental and Asian, and will contribute to understanding the mutational profile of esophageal squamous cell carcinoma in Brazil.
Material and methods
Tissue samples
Forty-six patients diagnosed with esophageal squamous cell carcinoma treated at the Barretos Cancer Hospital's upper-digestive department, Barretos, SP, Brazil, were evaluated. The main clinicopathological features were collected from patients' medical records.
The tumor and blood samples were obtained from biopsy or surgery and immediately processed and stored at − 80 °C in the Barretos Cancer Hospital Biobank. The present study was approved by the Barretos Cancer Hospital Institutional Review Board (Project No. 1.454.967/2016), and all patients included signed an Informed Consent Form. All methods were performed following the relevant guidelines and regulations.
DNA isolation
Tumor DNA was isolated from fresh-frozen tissue using QIAsymphony DNA Mini Kit following the Tissue_200 protocol for automated isolation in the QIAsymphony (QIAGEN, Hilden, Germany). DNA from leukocytes of peripheral blood was isolated using the QIAmp DNA Blood Mini Kit (QIAGEN, Hilden, Germany), following the manufacturer's instructions. DNA quantity and quality were assessed by Qubit (Thermo Scientific, Wilmington, DE, USA).
Mutation profile
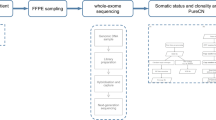
The mutation profile of a commercial panel of 150 cancer-related genes was conducted at Mendelics Genetics company (São Paulo, SP, Brazil, https://www.mendelics.com/oncologia/) as previously reported44. The panel analyzed all coding sequence of the following genes: ABL1, AKT1, AKT2, AKT3, ALK, APC, AR, ARAF, ATM, AURKA, AURKB, AXIN1, AXL, BAP1, BARD1, BCL2, BCL2L1, BCL2L2, BLM, BRAF, BRCA1, BRCA2, BRD2, BRD3, BRD4, BRDT, BRIP1, BTK, CBFB, CCND1, CCND2, CCND3, CCNE1, CD22, CD274, CD79A, CD79B, CDH1, CDK12, CDK4, CDKN1A, CDKN2A, CHEK1, CHEK2, CREBBP, CRKL, DDR2, DDX3X, EGFR, EPHA7, ERBB2, ERBB3, ERBB4, ESR1, EZH2, FAM175A, FAS, FBXW7, FGFR1, FGFR2, FGFR3, FGFR4, FHIT, FLT1, FLT3, FLT4, FRS2, GATA2, GNA11, GNAQ, HDAC1, HDAC4, HDAC7, HGF, HRAS, IDH1, IDH2, IGF1R, JAK1, JAK2, JAK3, KDR, KIT, KRAS, MAP2K1, MAP2K2, MAP2K4, MAP3K1, MAPK1, MCL1, MDM2, MET, MLH1, MPL, MRE11A, MS4A1, MSH2, MSH6, MTOR, MUTYH, MYC, MYD88, NBN, NF1, NF2, NFE2L2, NFKBIA, NOTCH1, NOTCH2, NOTCH3, NRAS, NT5C2, NTRK1, PALB2, PDGFRA, PDGFRB, PDK1, PIK3CA, PIK3CB, PIK3CG, PMS2, PTCH1, PTEN, RAC1, RAD51, RAF1, RANBP2, RARA, RB1, RET, RICTOR, ROS1, RRM1, RUNX1, SDHB, SMO, SOX2, SRC, STAT3, STAT5B, STK11, TERC, TERT, TGFBR2, TP53, TSC1, TSC2, VEGFA, WT1, and XPO1. For sequencing, paired tumor and blood DNA libraries were prepared using Nextera Rapid Capture Custom Enrichment kit (Illumina, San Diego, CA, USA). Qubit Fluorometer quantified libraries, and Agilent 2100 Bioanalyzer evaluated their quality. The cluster generation and sequencing were performed in Illumina HiSeq 4000 following the manufacturer's instructions. Paired-end reads from Illumina sequencing were obtained by script bcl2fastq (v. 2.17.1.14), and data pre-processing was performed following the recommended best practices, i.e., alignment against the human genome reference build GRCh37 using Burrows-Wheeler Aligner (BWA, version 0.7.13), duplicates were marked, and a further base quality score recalibration step was applied45.
The VarScan2 algorithm called the somatic variants46. The variants with artifacts due to indel reads at their position, or less than 10% or more than 90% of variant supporting reads on one strand, were removed. The variants were further filtered to remove those with fewer than ten reads covering the variant and less than 5% of the variant allele frequency.
A second algorithm was applied using the somatic SNV and indell caller MuTect2 from GATK. MuTect2 combines the somatic genotyping engine of the original MuTect47 with the assembly-based machinery of HaplotypeCaller provided by GATK48, detecting somatic mutations using a Bayesian classifier approach. The variants were detected by comparing the likelihood of the site to be to sequencing noise and filtered by the alterations of the normal paired control (blood) from the same patient, by a pool of normals of 291 local samples using the same NGS technology and according to the allelic frequencies provided by the Genome Aggregation Database (gnomAD) datasets49 to reduce miscalled germline calls. In this study, only variants found by both variant callers (VarScan2 and MuTect2) were considered. Finally, the annotation of variants was done using Variant Effect Prediction50.
To identify driver mutations in tumors, we applied the Cancer Genome Interpreter-CGI51. Briefly, CGI annotates potential driver mutations detected in tumors by identifying known tumorigenic variants and classifying variants of unknown significance via OncodriveMUT51. After the CGI classification, we maintained variants classified as tumorigenic and variants predicted as Tier1 or Tier2 for tumorigenesis. Variants that were not classified as cancer driver mutation or not predicted as Tier1 or Tier2 as driver by the OncodriveMut algorithm were excluded. Therefore, mutations identified as polymorphism (high allele frequency) or predicted as neutral or passenger for oncogenesis and found in DNA sequence outside coding regions were excluded.
Classification of TP53 mutations
To assess the impact of TP53 missense mutations, we used a risk-system score based on the Evolutionary Trace (ET) method proposed and validated in head and neck tumors by Neskey et al., called evolutionary action score of p53 (EAp53) (http://mammoth.bcm.tmc.edu/cgi-bin/panos/EAp53.cgi)19. In this system, mutations are scored from 0 to 100, with higher scores representing more deleterious mutations. A threshold score of 75 is used to classify variants as low-risk (EAp score < 75) or as high-risk (EAp score > 75). Mutations classified as high risk was associated with a poor prognosis, decreased survival, and increased development of distant metastases in head and neck tumors19,52.
Additionally, we evaluated a second classification proposed for head and neck tumors by Poeta et al.20 and validated in lung cancer by Molina et al.21. According to this system, TP53 mutations are divided into “disruptive” and “non-disruptive”. Disruptive mutations are stop-codon all over the coding region and missense mutations within the L2 and L3 sites, codons 163–195 and 236–251 with an amino acid polarity shift. Non-disruptive mutations are missense mutations within the L2 and L3 sites and do not change in polarity between the amino acids.
Statistical analysis
Characterization of the study population was analyzed through frequency tables for qualitative variables and measures of central tendency and dispersion (mean, standard deviation, minimum, and maximum) for the quantitative variables. Regarding the clinicopathological association analyzes with profile mutation (TP53, NFE2L2, and NOTCH1) and classification of TP53 mutations (EAp53 score and disruptive and non-disruptive), we used the Mann–Whitney test for age, and for other categorical variables, the Chi-square test or Fisher's exact test.
The level of significance adopted was 5% (p ≤ 0.05). Statistical analyses were performed using the SPSS software v.21.0 (SPSS, Chicago, IL).
Data availability
Data that support the findings are available upon reasonable request and with the permission of Dr. Rui Manuel Reis.
References
Ferlay, J. et al. Estimating the global cancer incidence and mortality in 2018: GLOBOCAN sources and methods. Int. J. Cancer 144, 1941–1953. https://doi.org/10.1002/ijc.31937 (2019).
Bray, F. et al. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 68, 394–424. https://doi.org/10.3322/caac.21492 (2018).
INCA. Estimativa 2020: Incidência de Câncer no Brasil, http://www.inca.gov.br/estimativa/2016/ (2020).
Wu, S. G. et al. Surgery combined with radiotherapy improved survival in metastatic esophageal cancer in a surveillance epidemiology and end results population-based study. Sci. Rep. 6, 28280. https://doi.org/10.1038/srep28280 (2016).
Abnet, C. C., Arnold, M. & Wei, W. Q. Epidemiology of esophageal squamous cell carcinoma. Gastroenterology 154, 360–373. https://doi.org/10.1053/j.gastro.2017.08.023 (2018).
Mahboubi, E. et al. Oesophageal cancer studies in the Caspian Littoral of Iran: the Caspian cancer registry. Br. J. Cancer 28, 197–214. https://doi.org/10.1038/bjc.1973.138 (1973).
Hull, R. et al. A multinational review: Oesophageal cancer in low to middle-income countries. Oncol. Lett. 20, 42. https://doi.org/10.3892/ol.2020.11902 (2020).
Tirumani, H. et al. Esophageal Carcinoma: Current concepts in the role of imaging in staging and management. Can. Assoc. Radiol. J. 66, 130–139. https://doi.org/10.1016/j.carj.2014.08.006 (2015).
Islami, F. et al. High-temperature beverages and foods and esophageal cancer risk: A systematic review. Int. J. Cancer 125, 491–524. https://doi.org/10.1002/ijc.24445 (2009).
Prabhu, A., Obi, K. O. & Rubenstein, J. H. The synergistic effects of alcohol and tobacco consumption on the risk of esophageal squamous cell carcinoma: a meta-analysis. Am. J. Gastroenterol. 109, 822–827. https://doi.org/10.1038/ajg.2014.71 (2014).
Cheng, C. et al. Whole-genome sequencing reveals diverse models of structural variations in esophageal squamous cell Carcinoma. Am. J. Hum. Genet. 98, 256–274. https://doi.org/10.1016/j.ajhg.2015.12.013 (2016).
Qin, H. D. et al. Genomic characterization of esophageal squamous cell Carcinoma reveals critical genes underlying tumorigenesis and poor prognosis. Am. J. Hum. Genet. 98, 709–727. https://doi.org/10.1016/j.ajhg.2016.02.021 (2016).
Sawada, G. et al. Genomic landscape of esophageal squamous cell Carcinoma in a Japanese population. Gastroenterology 150, 1171–1182. https://doi.org/10.1053/j.gastro.2016.01.035 (2016).
Song, Y. et al. Identification of genomic alterations in oesophageal squamous cell cancer. Nature 509, 91–95. https://doi.org/10.1038/nature13176 (2014).
Lin, D. C. et al. Genomic and molecular characterization of esophageal squamous cell carcinoma. Nat. Genet. 46, 467–473. https://doi.org/10.1038/ng.2935 (2014).
Gao, Y. B. et al. Genetic landscape of esophageal squamous cell carcinoma. Nat Genet 46, 1097–1102. https://doi.org/10.1038/ng.3076 (2014).
Cancer Genome Atlas Research, N. et al. Integrated genomic characterization of oesophageal carcinoma. Nature 541, 169–175, https://doi.org/10.1038/nature20805 (2017).
Lacerda, C. F. et al. Mutational profile of TP53 in esophageal squamous cell carcinoma associated with chagasic megaesophagus. Dis. Esophagus 30, 1–9. https://doi.org/10.1093/dote/dow040 (2017).
Neskey, D. M. et al. Evolutionary action score of TP53 identifies high-risk mutations associated with decreased survival and increased distant metastases in head and neck cancer. Cancer Res. 75, 1527–1536. https://doi.org/10.1158/0008-5472.CAN-14-2735 (2015).
Poeta, M. L. et al. TP53 mutations and survival in squamous-cell carcinoma of the head and neck. N. Engl. J. Med. 357, 2552–2561. https://doi.org/10.1056/NEJMoa073770 (2007).
Molina-Vila, M. A. et al. Nondisruptive p53 mutations are associated with shorter survival in patients with advanced non-small cell lung cancer. Clin. Cancer Res. 20, 4647–4659. https://doi.org/10.1158/1078-0432.CCR-13-2391 (2014).
Chen, X. X. et al. Genomic comparison of esophageal squamous cell carcinoma and its precursor lesions by multi-region whole-exome sequencing. Nat. Commun. 8, 524. https://doi.org/10.1038/s41467-017-00650-0 (2017).
Deng, J. et al. Comparative genomic analysis of esophageal squamous cell carcinoma between Asian and Caucasian patient populations. Nat. Commun. 8, 1533. https://doi.org/10.1038/s41467-017-01730-x (2017).
Putz, A. et al. TP53 mutation pattern of esophageal squamous cell carcinomas in a high risk area (Southern Brazil): Role of life style factors. Int. J. Cancer 98, 99–105. https://doi.org/10.1002/ijc.10128 (2002).
Hu, N. et al. Genomic landscape of somatic alterations in esophageal squamous cell carcinoma and gastric cancer. Cancer Res. 76, 1714–1723. https://doi.org/10.1158/0008-5472.CAN-15-0338 (2016).
Rossini, A. et al. TP53 mutation profile of esophageal squamous cell carcinomas of patients from Southeastern Brazil. Mutat. Res. 696, 10–15. https://doi.org/10.1016/j.mrgentox.2009.11.005 (2010).
Kagawa, S. et al. Cellular senescence checkpoint function determines differential Notch1-dependent oncogenic and tumor-suppressor activities. Oncogene 34, 2347–2359. https://doi.org/10.1038/onc.2014.169 (2015).
Kensler, T. W., Wakabayashi, N. & Biswal, S. Cell survival responses to environmental stresses via the Keap1-Nrf2-ARE pathway. Annu. Rev. Pharmacol. Toxicol. 47, 89–116. https://doi.org/10.1146/annurev.pharmtox.46.120604.141046 (2007).
Satoh, H., Moriguchi, T., Takai, J., Ebina, M. & Yamamoto, M. Nrf2 prevents initiation but accelerates progression through the Kras signaling pathway during lung carcinogenesis. Cancer Res. 73, 4158–4168. https://doi.org/10.1158/0008-5472.CAN-12-4499 (2013).
Wang, X. J. et al. Nrf2 enhances resistance of cancer cells to chemotherapeutic drugs, the dark side of Nrf2. Carcinogenesis 29, 1235–1243. https://doi.org/10.1093/carcin/bgn095 (2008).
Sporn, M. B. & Liby, K. T. NRF2 and cancer: The good, the bad and the importance of context. Nat. Rev. Cancer 12, 564–571. https://doi.org/10.1038/nrc3278 (2012).
Hao, J. J. et al. Spatial intratumoral heterogeneity and temporal clonal evolution in esophageal squamous cell carcinoma. Nat. Genet. 48, 1500–1507. https://doi.org/10.1038/ng.3683 (2016).
Fruman, D. A. et al. The PI3K pathway in human disease. Cell 170, 605–635. https://doi.org/10.1016/j.cell.2017.07.029 (2017).
Munari, F. F. et al. PIK3CA mutations are frequent in esophageal squamous cell carcinoma associated with chagasic megaesophagus and are associated with a worse patient outcome. Infect. Agent Cancer 13, 43. https://doi.org/10.1186/s13027-018-0216-3 (2018).
Alqahtani, A., Ayesh, H. S. K. & Halawani, H. PIK3CA gene mutations in solid malignancies: Association with clinicopathological parameters and prognosis. Cancers (Basel) https://doi.org/10.3390/cancers12010093 (2019).
Samuels, Y. et al. High frequency of mutations of the PIK3CA gene in human cancers. Science 304, 554. https://doi.org/10.1126/science.1096502 (2004).
Winer, E. et al. Clinical cancer advances 2008: Major research advances in cancer treatment, prevention, and screening—A report from the American Society of Clinical Oncology. J. Clin. Oncol. 27, 812–826. https://doi.org/10.1200/JCO.2008.21.2134 (2009).
Karapetis, C. S. et al. K-ras mutations and benefit from cetuximab in advanced colorectal cancer. N. Engl. J. Med. 359, 1757–1765. https://doi.org/10.1056/NEJMoa0804385 (2008).
Zhang, L. et al. Targeted therapy in esophageal cancer. Expert Rev. Gastroenterol. Hepatol. 10, 595–604. https://doi.org/10.1586/17474124.2016.1140036 (2016).
Zhang, L. et al. Genomic analyses reveal mutational signatures and frequently altered genes in esophageal squamous cell carcinoma. Am. J. Hum. Genet. 96, 597–611. https://doi.org/10.1016/j.ajhg.2015.02.017 (2015).
Du, P. et al. Comprehensive genomic analysis of Oesophageal Squamous Cell Carcinoma reveals clinical relevance. Sci. Rep. 7, 15324. https://doi.org/10.1038/s41598-017-14909-5 (2017).
Canon, J. et al. The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature 575, 217–223. https://doi.org/10.1038/s41586-019-1694-1 (2019).
Heist, L. et al. Activity and safety of Adagrasib (MRTX849) in advanced/metastatic non-small-cell lung cancer (NSCLC) harboring KRAS G12C mutation. Eur. J. Cancer https://doi.org/10.1016/S0959-8049(20)31076-5 (2020).
Dos Santos, W. et al. Mutation profiling of cancer drivers in Brazilian colorectal cancer. Sci. Rep. 9, 13687. https://doi.org/10.1038/s41598-019-49611-1 (2019).
Van der Auwera, G. A. et al. From FastQ data to high confidence variant calls: The genome analysis toolkit best practices pipeline. Curr. Protoc. Bioinform. https://doi.org/10.1002/0471250953.bi1110s43 (2013).
Koboldt, D. C. et al. VarScan 2: Somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 22, 568–576. https://doi.org/10.1101/gr.129684.111 (2012).
Cibulskis, K. et al. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat. Biotechnol. 31, 213–219. https://doi.org/10.1038/nbt.2514 (2013).
McKenna, A. et al. The genome analysis toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303. https://doi.org/10.1101/gr.107524.110 (2010).
Karczewski, K. J. et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 581, 434–443. https://doi.org/10.1038/s41586-020-2308-7 (2020).
McLaren, W. et al. The ensembl variant effect predictor. Genome Biol. 17, 122. https://doi.org/10.1186/s13059-016-0974-4 (2016).
Tamborero, D. et al. Cancer genome interpreter annotates the biological and clinical relevance of tumor alterations. Genome Med. 10, 25. https://doi.org/10.1186/s13073-018-0531-8 (2018).
Katsonis, P. & Lichtarge, O. A formal perturbation equation between genotype and phenotype determines the evolutionary action of protein-coding variations on fitness. Genome Res. 24, 2050–2058. https://doi.org/10.1101/gr.176214.114 (2014).
Acknowledgements
This study was financially supported by the Public Ministry of Labor Campinas (Research, Prevention, and Education of Occupational Cancer, Brazil). FFM was supported by Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP) [Grant No. 2015/20077-3 to FFM]. The authors would like to thank Barretos cancer Hospital Biobank for helping in frozen tissue and blood processing. We also would like to thank Dr. Pedro Ferreira from I3S, Porto, Portugal, for the study's initial discussions.
Author information
Authors and Affiliations
Contributions
F.F.M. performed experiments, analyzed the data, and wrote the draft of the manuscript; W.S. and A.F.E. performed experiments, analyzed the data. A.C.C. contributed to the interpretation of the results. P.A.P., D.B., D.R.W., C.S., D.P.G., and A.L.F. edited and revised the manuscript critically for important intellectual content. R.M.R. conceived the original idea, supervised the project, and wrote the manuscript. All authors critically reviewed and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Munari, F.F., dos Santos, W., Evangelista, A.F. et al. Profile of esophageal squamous cell carcinoma mutations in Brazilian patients. Sci Rep 11, 20596 (2021). https://doi.org/10.1038/s41598-021-00208-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-021-00208-7
This article is cited by
-
EZH2 regulates oncomiR-200c and EMT markers in esophageal squamous cell carcinomas
Scientific Reports (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
