
Abstract
T-cell acute lymphoblastic leukemia (T-ALL) is caused by the accumulation of multiple genetic alterations. To determine the frequency of common genetic mutations and possible prognostic markers in childhood T-ALL, we performed targeted sequencing of 67 genes across 64 cases treated according to Taiwan Pediatric Oncology Group protocols between January 2002 and December 2015. Together, 302 variants were identified in 60 genes including 233 single nucleotide variants and 69 indels. Sixty-four samples had a median number of six genetic lesions each (range 1–17). Thirteen genes had mutation frequencies > 10%, and 5 were > 20%, with the highest being NOTCH1 (70.31%). Protocadherins FAT1 (32.81%) and FAT3 (17.19%), and the ubiquitin ligase component FBXW7 (28.13%) had higher mutation frequencies than previously reported. Other mutation frequencies (PHF6, DNM2, DNMT3A, CNOT3, and WT1) were within previously reported ranges. Three epigenetic-related genes (KMT2D, DNMT3A, and EZH2) were mutated in our cohort. JAK-STAT signaling pathway genes had mutation frequencies of 3–13% and were observed in 23 cases (35.94%). Changes to genes in the ErbB signaling pathway were detected in 20 cases (31.25%). Patients with NOTCH1/FBXW7 mutations and RAS/PTEN germline exhibited better 5-year overall survival rates.
Similar content being viewed by others

Introduction
Around 15% of cases of childhood acute lymphoblastic leukemia (ALL) is T-cell ALL (T-ALL)1,2. Clinically, T-ALL is characterized by a high white cell count, a mediastinal mass, and an inferior outcome compared to the B-cell ALL. With improvement in chemotherapy, supportive care, minimal residual disease (MRD) detection, and the possibility of stem cell transplants, outcomes have gradually improved in the last 10 to 20 years3. T-ALL can be classified into subgroups according to the gene expression of various transcription factors, including TAL1, TLX, HOXA9/10, LMO2, and NKX2-14,5,6. Deletions of the CDKN2A locus are present in about 70% of T-ALLs7. Candidate gene sequencing identified several genetic mutations or alterations in T-ALL, including NOTCH1, JAK1, IL7R, ETV6, RUNX1, BCL11B, LEF1, PHF6, and WT18,9,10,11,12,13,14,15,16,17,18. Zhang et al. demonstrated the first comprehensive whole genome sequencing of 12 patients with early T-cell precursor ALL, and assessed their findings in another 94 T-ALL patient samples19. Liu et al. used integrated genome analysis to investigate T-ALL samples including whole genome sequencing, whole exome sequencing, and RNA-sequencing. The above studies discovered more complex and heterogeneous genetic somatic mutations in T-ALL, which involve numerous transcriptional, signaling, and epigenetic factor pathways in the pathogenesis of this disease19,20,21,22.
Half of the T-ALLs show an aberrant expression of transcriptional factors, whose specific breaking points result in the aberrant expression of genes that might require the use of whole genome sequencing (WGS) to identify20. The other cases of T-ALL might harbor chromosomal rearrangements, some of which might become therapeutic targets20. NOTCH1 mutations were first identified in more than 50% of T-ALL cases8, and many subsequent studies have discussed NOTCH1 and its related pathway as a prognostic marker in T-ALL23,24,25,26,27,28,29. Current sequencing efforts have identified several pathways of genetic alteration in T-ALL, including transcription factors, signaling pathways, epigenetics, mistranslation, and RNA stability, in addition to NOTCH1 signaling19,20,30,31. However, these alterations have rarely been used as prognostic stratifications in the treatment of patients or they do not confer consistent prognostic significance in different protocols10,14,16,17,18,19,32,33,34,35. Chemotherapy can be tailored according to minimal residual disease (MRD) data in childhood T-ALL, although MRD in T-ALL may not be good enough to predict final outcomes in the same way that B-ALL can be targeted based on ETV6-RUNX1 or hyperdiploidy2,36. Investigators hope to identify genetic markers to predict T-ALL outcomes more precisely37,38. Another benefit of sequencing for T-ALL is the identification of possible genetic alterations suitable for targeted therapy, such as a mutant JAK-STAT pathway39.
In this study, we used targeted next generation amplicon sequencing (NGS-based amplicon sequencing) to sequence multiple genes, which were reported to be involved in T-ALL, and investigated their prognostic impacts for patients with pediatric T-ALL under treatment with Taiwan Pediatric Oncology Group (TPOG) protocols in Taiwan.
Materials and methods
Patients and protocols
The diagnosis of T-ALL was based on bone marrow aspiration or peripheral blood and immune-phenotyping with monoclonal antibodies directed to T-lineage-associated antigens. Early T-cell precursor ALL (ETP-ALL) status was diagnosed based upon the criteria proposed by Coustan-Smith et al.40. Between January 2002 and December 2015, 64 pediatric patients with T-ALL were enrolled. All were treated according to the TPOG ALL protocols. Risk-directed TPOG protocols use multiple chemotherapeutic agents of different intensities. Patients with T-ALL were assigned to the very high-risk protocol. After 2013, MRD levels were added to the risk assignment for therapy. Events were defined as any relapse, secondary malignancy, or death. The Institutional Review Board of National Taiwan University Hospital approved the study and all of the participants or their guardians provided written, informed consent, in accordance with the Declaration of Helsinki. Details of the protocols are published elsewhere41,42. Genomic DNA was extracted from leukemic bone marrow or peripheral blood, as previously described42.
Determination of gene expression by real time quantitative PCR (RT-PCR)
For samples with available total RNA, cDNA was synthesized using Maxima First Strand cDNA Synthesis Kit (Thermo Fisher Scientific, Waltham, MA, USA) and qPCR was performed using SensiFast Probe No-ROX Kit (Meridian Bioscience, Cincinnati, OH, USA) using the StepOnePlus system (Applied Biosystems, Foster City, CA)4,5,6. We determined the expression of TAL1, TAL2, LYL1, TLX1, TLX3, LMO1, LMO2, NKX2-1, and HOXA oncogenes, using the YWHAZ gene as an internal control.
Amplicon design and sequencing data processing
NuGEN’s Ovation Target Enrichment System (NuGEN Technologies, Inc., San Carlos, California) was used to design probes and hybridize exon regions of 67 targeted genes. In all, 6465 designed probes of 50 base pairs (bps) were used to enrich targeted regions of 556,600 bps. Sample DNA was fragmented, ligated by adaptors and then hybridized with designed probes. After polymerase chain reaction (PCR) amplification for sequencing library enrichment, products were sequenced by Illumina HiSeq 4000 (Illumina, Inc., San Diego, California), with 150 paired-end reads.
Sequencing reads were examined for quality, and for adaptor or primer sequence contamination, by FastQC (v0.11.5). After removing unqualified bases (average quality base < 20 per 4-base sliding window) by Trimmomatic (v0.36), reads were aligned to human reference genome GRC37/hg19 by BWA (v0.7.17). In order to reduce the effect of systematic sequencing errors from sequencers, and the false positive rate in identifying variants, quality score recalibration and marked duplication were both applied to reads by GATK (v4.0). Finally, variants were identified by VarScan (v2.3.9). Variants meeting the following criteria were selected for further analysis: (1) with ≥ 10 reads; (2) ≥ 2 reads with alternative alleles; (3) alternative allele fraction (AF) ≥ 1%; (4) average base quality ≥ 15; (5) without > 90% reads supported by one strand (6) ≥ 7 samples with variants (7) carrier frequency < 1% in East Asian populations of the 1000 Genome Project.
Statistical analysis
In order to clarify whether genetic profiles of NOTCH1, FBXW7, RAS, and PTEN were prognostic markers, as previously reported, variants present in the COSMIC database (v70) were selected and survival analysis was carried out, to compare the difference of time to induction failure or survival between genetic risk groups. The Kaplan–Meier method was used to generate survival curves. The log-rank test was applied to compare the difference between survival curves. Adjusted hazard ratio was estimated from multivariate Cox proportional hazards regression, with clinical variates of onset age, gender, and white blood cell (WBC) count. All tests were two-tailed, and p-values < 0.05 were considered to be significant.
Ethics declaration
This study was conducted in accordance with the Declaration of Helsinki guidelines. Written informed consent was obtained from all study participants or their guardians.
Results
Clinical characteristics of the patients
Among the 64 children with T-ALL, 45 were male and 19 were female, with a male to female ratio of 2.4:1. The median age of the patients at diagnosis was 12.1 years (range 1.4–17.4 years). The median WBC count was 69.8 × 109 /L (range 0.6–1096 × 109/L). Twenty-two (34.4%) patients had central nervous system (CNS) leukemia at diagnosis. The clinical features of this cohort are given in Table 1.
Landscape of genetic alterations in T-cell acute lymphoblastic leukemia
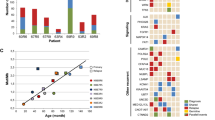
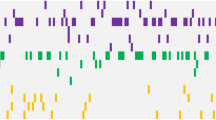
In all, 302 variants were identified in 60 genes, including 233 single nucleotide variants (SNVs) and 69 insertions and deletions (indels). These variants were all predicted to result in amino change and majority variants were single nucleotide variants (SNVs) (6.29% frameshift deletion, 8.61% frameshift insertion, 4.97% non-frameshift deletion, 2.32% non-frameshift insertion, 6.95% stop gain, and 70.86% nonsynonymous SNVs). The 64 samples showed a median number of 6 genetic lesions (range 1–17). Among 60 impacted genes, 13 had mutation frequencies > 10%, and 5 had > 20% carrier frequencies including NOTCH1 (70.31%), FAT1 (32.81%), FBXW7 (28.13%), KMT2D (28.13%) and NF1 (21.88%). As expected, the NOTCH1 gene had the highest mutation rate, with the majority of the mutations detected in exon 26 (35.09%) and exon 34 (42.11%). The details of the NOTCH1 mutations are shown in Fig. 1. Interestingly, protocadherins FAT1 (32.81%) and FAT3 (17.19%), and the ubiquitin ligase component FBXW7 (28.13%) had high mutation frequencies in our cohort. Mutation frequencies of PHF6 (14.06%), DNM2 (10.94%), DNMT3A (4.69%), CNOT3 (10.94%), and WT1 (3.13%) were in the range of those previously reported. Mutations in epigenetic-related genes KMT2D (28.13%), DNMT3A (4.69%), and EZH2 (4.69%) were detected in our patients. For T-ALL-related pathogenesis pathways, impacted genes involved in the JAK-STAT signaling pathways, such as EP300 (4.69%), STAT5B (9.38%), JAK1 (12.50%), JAK3 (7.81%), IL7R (4.69%), and PTPN11 (3.13%) had mutation frequencies in the range of 3%–13% and were observed in 23 cases (35.94%). Impacted genes involved in the ErbB signaling pathway were detected in 20 patients (31.25%), including NRAS (9.38%), KRAS (3.13%), BRAF (6.25%), STAT5B (9.38%), CBL (1.56%), and MTOR (7.81%). The genetic alterations in each of the gene expression subtypes are listed in Fig. 2. We observed a higher frequency of mutations in RAS signaling in ETP ALL cases (p = 0.04) (Fig. 2). The details of the sequencing results, including the gene list and mutational frequency, are listed in Supplementary Table 1.

Genetic profiling of NOTCH1 genes. Fifty-seven genetic mutations were detected in NOTCH1 and 70.31% of patients carried NOTCH1 mutations. EGF_CA calcium-binding EGF-like domain; Other: Ca2+ binding site; EGF EGF-like domain, NL domain found in Notch and Lin-12. Notch LNR domain. NOD and NODP NOTCH protein, Ank_2 ankyrin repeats, ANK repeat ANK repeat, ANK ankyrin repeats, Med25_SD1 mediator complex subunit 25 synapsin 1, DUF3454 domain of unknown function (DUF3454).

Genetic alteration profiling in 64 cases of T-cell acute lymphoblastic leukemia. Each column represents the genetic alterations of a single patient. AF alternative allele fraction, TRG T cell receptor-γ locus.
Paired samples show relapse-acquired mutations
When we compared the mutation status of 10 diagnosis-relapse pairs, the results showed that 29 genes exhibited mutation allele fraction (MAF) changes between diagnosis and relapse. The mutational status of diagnosis and relapse were quite different (Fig. 3). Noticeably, among these 29 genes, 70% of the patients maintained or acquired NOTCH1 or FBXW7 mutations during relapse. Both of these genes are involved in the NOTCH signaling pathway. In addition, 60% of the patients had obvious mutations in cell adhesion related genes after relapse. Finally, it is worth noting that 40% of the patients acquired TP53 mutations in this process.

Mutation allele fractions in 29 genes differ between diagnosis and relapse in 10 patients. MAF mutation allele fraction.
The prognostic relevance of genetic alterations in T-ALL, when treated according to the TPOG-ALL protocols
Because NOTCH1 and FBXW7 are commonly discussed prognostic markers in childhood T-ALL, we tested these two genotypes against the prognosis. Although patients with both gene mutations showed slightly better 5-year event-free survival (EFS) and overall survival (OS), this was not significant.
The genetic profiles of NOTCH1, FBWX7, RAS and PTEN have been reported to be prognostic genotypes in T-ALL in both adults and children. Patients with NOTCH1/FBXW7 mutations and RAS/PTEN germline have been considered as oncogenic low risk, whereas those with NOTCH1/FBXW7 germline and RAS/PTEN germline or NOTCH1, FBXW7, RAS, and PTEN mutations are classified as high risk37,38. In this cohort, patients with NOTCH1/FBXW7 mutations and RAS/PTEN germline had better 5-year EFS (65.3%; 95% CI 50.7%–84.2%) and OS (74.6%; 95% CI 60.8%–91.5%) than patients with other genetic combinations (EFS: 41.2%; 95% CI 26.0%–65.2%; OS: 44.7%; 95% CI 29.4%–67.8%), although only the 5-year OS has statistical significance (Fig. 4). In multivariate analysis of initial white cell counts, age of onset, and gender, patients with NOTCH1/FBXW7 mutations and RAS/PTEN germline had significantly better 5-year OS than patients with other genetic combinations (adjusted HR 0.36; 95% CI 0.15–0.89, p = 0.0268).

Event-free survival (A) and overall survival (B) of patients with T-cell acute lymphoblastic leukemia, stratified by genetic lesions in NOTCH1, FBWX7, RAS, and PTEN. Patients with NOTCH1/FBXW7 mutations and RAS/PTEN germline were considered to be a low risk group (n = 32) and the others were classified as a high-risk group (n = 32).
Discussion
Targeted sequencing is able to profile multiple genetic mutations, with different allele frequencies, at the same time. In this study, NOTCH1 mutations were the most common genetic alterations, accounting for more than 70% of mutations, followed by the JAK-STAT pathway (35.9%), which may be suitable for targeted therapy. Patients with NOTCH1/FBXW7 mutations and RAS/PTEN germline had a better 5-year EFS and OS than patients with other genetic combinations, although only the 5-year OS had statistical significance.
Some genetic alterations have been reported to be prognostic markers in childhood T-ALL24,25,43,44,45. The most common prognostic genetic alterations are to NOTCH1 signaling pathways. Patients with NOTCH1 mutations have previously been viewed as markers of good prognosis. However, prognostic impacts are not consistent in all related studies23,24,26,27,28,29. Two studies showed that the genetic alterations of NOTCH1, FBXW7, PTEN, and RAS were identified with prognostic value in adult T-ALL37,46. These oncogenic mutations, combined with MRD, might improve outcome prediction in cases of pediatric T-ALL when treated according to the protocol known as FRALLE2000T38. However, it was impossible to validate these oncogenic alterations in another 145 pediatric T-ALL patients treated in the UKALL 2003 trial25. There may be several reasons for the discrepancy between the two T-ALL cohorts. The UKALL cohort was relatively small, different molecular techniques were used and the results may have been interpreted differently. The incidence of PTEN abnormality was higher in the UKALL cohort (22%) than the FRALLE2000T cohort (14%) was. In our study, unfortunately, we lacked the MRD data for most patients, which would be required to validate its prognostic value. Another study from Taiwan, using Sanger sequencing, showed that PHF6 was an independent prognostic marker, after multivariate analysis47. However, the authors did not investigate the prognostic value of mutations to NOTCH1/FBXW7 and the RAS/PTEN germline in their cohort.
Another important strength of targeted sequencing is the ability to identify these targetable genetic alterations22. There are several possible targeted therapies for T-ALL, which have been investigated recently. For example, NOTCH1 is the most common genetic alteration and there are available drugs that interfere with the activation of NOTCH1 by the r-secretase complex in childhood T-ALL22,48,49,50. Antibodies targeting this pathway also showed some promising preclinical results51,52. Most importantly, the JAK-STAT pathway might be the most promising target due to the availability of drugs focused on this pathway and the fact that one third of patients have alterations in this pathway10,22,30,32,47. Our findings suggest that the genotyping of NOTCH1, FBXW7, RAS, and PTEN should be considered for risk-directed therapies, alongside the response to induction chemotherapy, in future TPOG ALL protocols. In addition, further investigation of the mutations in the JAK-STAT pathway could facilitate possible target-based therapies. These approaches might increase the survival of childhood T-ALL in Taiwan. Currently, the sequencing of these genes might require NGS with targeted sequencing or whole exome sequencing (WES) if it is available.
The genetic frequency differed slightly from previous reports22,30; for example, NOTCH1 was around 50% in most studies that used the methodology of Sanger sequencing8,23,26,28,29,47. When Liu et al. used WES and/or WGS to profile the genetic landscape of T-ALL, NOTCH1 was found to be the most frequently mutated gene, with 264 sequence mutations, identified in 196 cases, and most mutations were in the heterodimerization domain (62.9%; 166/264) and the PEST domain (31.4%; 83/264). Of the 264 mutations, 116 (43.9%) were subclonal (AF < 30%)20. Our target sequencing data are similar to this, with a higher somatic mutational rate of NOTCH1 due to subclonal mutations. Zhang et al. observed a high frequency of mutations resulting in aberrant cytokine receptor and RAS signaling, and alterations of genes with roles in hematopoietic and lymphoid development in ETP-ALL19. However, due to the smaller sample size of this cohort and the lack of adequate immunophenotypes to determine ETP-ALL in some of the patients, we were only able to conclude that alterations of the RAS pathway were higher in ETP-ALL patients in this study. In addition, we had ten patients with paired diagnostic and relapsed samples. One of the notable findings in our study was that four patients had acquired TP53 mutations, which is compatible with our previous study53.
There are some limitations of this study. The first is that the cohort is relatively small, and complete genetic profiling might need a larger cohort. Second, some novel genetic pathways related to T-ALL were not designed in this target-seq panel, such as the MYC and FA pathways20,54. Therefore, we did not have a comprehensive genetic picture of our Taiwanese cohort; in the future, we intend to use WES. Targeted sequencing might also miss some other genetic alterations in T-ALL cases. Finally, most of the patients in this cohort lacked MRD data, following induction chemotherapy; therefore, we are not able to correlate the genetic alterations with MRD. To assess the prognostic value of mutations to NOTCH1/FBXW7 and the RAS/PTEN germline, a larger prospective cohort is needed, to validate their association with MRD in TPOG protocols.
In conclusion, this study showed that amplicon-based next generation sequencing is able to identify the common genetic alterations in childhood T-ALL. Some of the identified genetic lesions may be suitable therapeutic targets and some might have prognostic value. The identification of JAK-STAT pathway alterations may also be useful additions for targeted therapy after induction in future TPOG ALL protocols39. A larger, prospective clinical trial is needed, coordinated with MRD data, to validate the clinical significance of key mutations in Taiwanese patients treated with TPOG protocols.
Data availability
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
Pui, C. H. & Evans, W. E. Treatment of acute lymphoblastic leukemia. N. Engl. J. Med. 354, 166–178 (2006).
Pui, C. H., Nichols, K. E. & Yang, J. J. Somatic and germline genomics in paediatric acute lymphoblastic leukaemia. Nat. Rev. Clin. Oncol. 16, 227–240 (2019).
Pui, C. H. et al. Childhood acute lymphoblastic leukemia: progress through collaboration. J. Clin. Oncol. 33, 2938–2948 (2015).
Ferrando, A. A. et al. Gene expression signatures define novel oncogenic pathways in T cell acute lymphoblastic leukemia. Cancer Cell 1, 75–87 (2002).
Homminga, I. et al. Integrated transcript and genome analyses reveal NKX2-1 and MEF2C as potential oncogenes in T cell acute lymphoblastic leukemia. Cancer Cell 19, 484–497 (2011).
Soulier, J. et al. HOXA genes are included in genetic and biologic networks defining human acute T-cell leukemia (T-ALL). Blood 106, 274–286 (2005).
Hebert, J., Cayuela, J. M., Berkeley, J. & Sigaux, F. Candidate tumor-suppressor genes MTS1 (p16INK4A) and MTS2 (p15INK4B) display frequent homozygous deletions in primary cells from T- but not from B-cell lineage acute lymphoblastic leukemias. Blood 84, 4038–4044 (1994).
Weng, A. P. Activating mutations of NOTCH1 in human T cell acute lymphoblastic leukemia. Science 306, 269–271 (2004).
Flex, E. et al. Somatically acquired JAK1 mutations in adult acute lymphoblastic leukemia. J. Exp. Med. 205, 751–758 (2008).
Zenatti, P. P. et al. Oncogenic IL7R gain-of-function mutations in childhood T-cell acute lymphoblastic leukemia. Nat. Genet. 43, 932–939 (2011).
Shochat, C. et al. Gain-of-function mutations in interleukin-7 receptor-α (IL7R) in childhood acute lymphoblastic leukemias. J. Exp. Med. 208, 901–908 (2011).
Van Vlierberghe, P. et al. ETV6 mutations in early immature human T cell leukemias. J. Exp. Med. 208, 2571–2579 (2011).
Della Gatta, G. et al. Reverse engineering of TLX oncogenic transcriptional networks identifies RUNX1 as tumor suppressor in T-ALL. Nat. Med. 18, 436–440 (2012).
Gutierrez, A. et al. The BCL11B tumor suppressor is mutated across the major molecular subtypes of T-cell acute lymphoblastic leukemia. Blood 118, 4169–4173 (2011).
De Keersmaecker, K. et al. The TLX1 oncogene drives aneuploidy in T cell transformation. Nat. Med. 16, 1321–1327 (2010).
Gutierrez, A. et al. Inactivation of LEF1 in T-cell acute lymphoblastic leukemia. Blood 115, 2845–2851 (2010).
Van Vlierberghe, P. et al. PHF6 mutations in T-cell acute lymphoblastic leukemia. Nat. Genet. 42, 338–342 (2010).
Tosello, V. et al. WT1 mutations in T-ALL. Blood 114, 1038–1045 (2009).
Zhang, J. et al. The genetic basis of early T-cell precursor acute lymphoblastic leukaemia. Nature 481, 157–163 (2012).
Liu, Y. et al. The genomic landscape of pediatric and young adult T-lineage acute lymphoblastic leukemia. Nat. Genet. 49, 1211–1218 (2017).
Belver, L. & Ferrando, A. The genetics and mechanisms of T cell acute lymphoblastic leukaemia. Nat. Rev. Cancer 16, 494–507 (2016).
Girardi, T., Vicente, C., Cools, J. & De Keersmaecker, K. The genetics and molecular biology of T-ALL. Blood 129, 1113–1123 (2017).
Breit, S. Activating NOTCH1 mutations predict favorable early treatment response and long-term outcome in childhood precursor T-cell lymphoblastic leukemia. Blood 108, 1151–1157 (2006).
Fogelstrand, L. et al. Prognostic implications of mutations in NOTCH1 and FBXW7 in childhood T-all treated according to the NOPHO ALL-1992 and ALL-2000 protocols. Pediatr. Blood Cancer 61, 424–430 (2014).
Jenkinson, S. et al. Impact of PTEN abnormalities on outcome in pediatric patients with T-cell acute lymphoblastic leukemia treated on the MRC UKALL2003 trial. Leukemia 30, 39–47 (2016).
Larson Gedman, A. et al. The impact of NOTCH1, FBW7 and PTEN mutations on prognosis and downstream signaling in pediatric T-cell acute lymphoblastic leukemia: A report from the Children’s Oncology Group. Leukemia 23, 1417–1425 (2009).
Mansour, M. R. et al. Prognostic implications of NOTCH1 and FBXW7 mutations in adults with T-Cell acute lymphoblastic leukemia treated on the MRC UKALLXII/ECOG E2993 protocol. J. Clin. Oncol. 27, 4352–4356 (2009).
Park, M.-J. et al. FBXW7andNOTCH1mutations in childhood T cell acute lymphoblastic leukaemia and T cell non-Hodgkin lymphoma. Br. J. Haematol. 145, 198–206 (2009).
Zhu, Y. M. NOTCH1 mutations in T-cell acute lymphoblastic leukemia: Prognostic significance and implication in multifactorial leukemogenesis. Clin. Cancer Res. 12, 3043–3049 (2006).
Belver, L. & Ferrando, A. The genetics and mechanisms of T cell acute lymphoblastic leukaemia. Nat. Rev. 16, 494–507 (2016).
Van Vlierberghe, P. & Ferrando, A. The molecular basis of T cell acute lymphoblastic leukemia. J. Clin. Investig. 122, 3398–3406 (2012).
Vicente, C. et al. Targeted sequencing identifies associations between IL7R-JAK mutations and epigenetic modulators in T-cell acute lymphoblastic leukemia. Haematologica 100, 1301–1310 (2015).
Gutierrez, A. et al. Absence of biallelic TCR deletion predicts early treatment failure in pediatric T-cell acute lymphoblastic leukemia. J. Clin. Oncol. 28, 3816–3823 (2010).
Gutierrez, A. et al. High frequency of PTEN, PI3K, and AKT abnormalities in T-cell acute lymphoblastic leukemia. Blood 114, 647–650 (2009).
De Keersmaecker, K. et al. Exome sequencing identifies mutation in CNOT3 and ribosomal genes RPL5 and RPL10 in T-cell acute lymphoblastic leukemia. Nat. Genet. 45, 186–190 (2013).
Pui, C. H. et al. Clinical impact of minimal residual disease in children with different subtypes of acute lymphoblastic leukemia treated with response-adapted therapy. Leukemia 31, 333–339 (2017).
Beldjord, K. et al. Oncogenetics and minimal residual disease are independent outcome predictors in adult patients with acute lymphoblastic leukemia. Blood 123, 3739–3749 (2014).
Petit, A. et al. Oncogenetic mutations combined with MRD improve outcome prediction in pediatric T-cell acute lymphoblastic leukemia. Blood 131, 289–300 (2018).
Inaba, H., Azzato, E. M. & Mullighan, C. G. Integration of next-generation sequencing to treat acute lymphoblastic leukemia with targetable lesions: The St. Jude children’s research hospital approach. Front Pediatr 5, 258 (2017).
Coustan-Smith, E. et al. Early T-cell precursor leukaemia: A subtype of very high-risk acute lymphoblastic leukaemia. Lancet Oncol. 10, 147–156 (2009).
Li, M.-J. et al. Treatment for childhood acute lymphoblastic leukemia in Taiwan: Taiwan Pediatric Oncology Group ALL-2002 study emphasizing optimal reinduction therapy and central nervous system preventive therapy without cranial radiation. Pediatr. Blood Cancer 64, 234–241 (2017).
Yang, Y. L. et al. IKZF1 deletions predict a poor prognosis in children with B-cell progenitor acute lymphoblastic leukemia: A multicenter analysis in Taiwan. Cancer Sci. 102, 1874–1881 (2011).
Bergeron, J. et al. Prognostic and oncogenic relevance of TLX1/HOX11 expression level in T-ALLs. Blood 110, 2324–2330. https://doi.org/10.1182/blood-2007-04-079988 (2007).
Seki, M. et al. Recurrent SPI1 (PU.1) fusions in high-risk pediatric T cell acute lymphoblastic leukemia. Nat. Genet. 49, 1274–1281 (2017).
van Grotel, M. et al. Prognostic significance of molecular-cytogenetic abnormalities in pediatric T-ALL is not explained by immunophenotypic differences. Leukemia 22, 124–131 (2007).
Trinquand, A. et al. Toward a NOTCH1/FBXW7/RAS/PTEN–based oncogenetic risk classification of adult t-cell acute lymphoblastic leukemia: A group for research in adult acute lymphoblastic leukemia study. J. Clin. Oncol. 31, 4333–4342 (2013).
Yeh, T.-C. et al. Clinical and biological relevance of genetic alterations in pediatric T-cell acute lymphoblastic leukemia in Taiwan. Pediatr. Blood Cancer 66, e274962 (2019).
Marchesini, M. et al. Blockade of oncogenic NOTCH1 with the SERCA inhibitor CAD204520 in T cell acute lymphoblastic leukemia. Cell Chem. Biol. 27, 678-697.e613 (2020).
Sanchez-Martin, M. et al. Synergistic antileukemic therapies in NOTCH1-induced T-ALL. Proc. Natl. Acad. Sci. 114, 2006–2011 (2017).
Papayannidis, C. et al. A Phase 1 study of the novel gamma-secretase inhibitor PF-03084014 in patients with T-cell acute lymphoblastic leukemia and T-cell lymphoblastic lymphoma. Blood Cancer J. 5, e350–e350 (2015).
Aste-Amézaga, M. et al. Characterization of notch1 antibodies that inhibit signaling of both normal and mutated notch1 receptors. PLoS ONE 5, e9094 (2010).
Wu, Y. et al. Therapeutic antibody targeting of individual Notch receptors. Nature 464, 1052–1057 (2010).
Yu, C.-H. et al. TP53 alterations in relapsed childhood acute lymphoblastic leukemia. Cancer Sci. 111, 229–238 (2020).
Pouliot, G. P. et al. Fanconi-BRCA pathway mutations in childhood T-cell acute lymphoblastic leukemia. PLoS ONE 14, e0221288 (2019).
Acknowledgements
The authors express their gratitude to all of the patients who participated in this study, and their parents. The authors also acknowledge the efforts of the TPOG and the Childhood Cancer Foundation in Taiwan. This work was supported by grants from the Ministry of Science and Technology, Taiwan (MOST-106-2314-B-002-199- and MOST-107-2314-B-002-173-MY2 to YLY).
Author information
Authors and Affiliations
Contributions
Y.-H.C., C.-H.Y., H.-Y.C., and Y.-L.Y. designed the study, analyzed the data and wrote the manuscript. C.-H.Y. and S.-W.L. acquired, processed patient specimens and performed experiments. Y.-H.C., C.-Y.L., H.-Y.C. performed the analysis. K.-H.L., S.-T.J., M.-Y.L., K.-H.W., H.-H.C. and D.-T.L. provided clinical samples and data. The manuscript was written by Y.-L.Y, and H.-Y.C. and was reviewed and edited by all authors.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Chang, YH., Yu, CH., Jou, ST. et al. Targeted sequencing to identify genetic alterations and prognostic markers in pediatric T-cell acute lymphoblastic leukemia. Sci Rep 11, 769 (2021). https://doi.org/10.1038/s41598-020-80613-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-80613-6
This article is cited by
-
Integrated analysis of transcriptome and genome variations in pediatric T cell acute lymphoblastic leukemia: data from north Indian tertiary care center
BMC Cancer (2024)
-
Relapsed/Refractory T- Acute Lymphoblastic Leukemia — Current Options and Future Directions
Indian Journal of Pediatrics (2024)
-
Revealing key lncRNAs in cytogenetically normal acute myeloid leukemia by reconstruction of the lncRNA–miRNA–mRNA network
Scientific Reports (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
