
Abstract
A new flavanol derivative, (2R,3R)-3-acetoxy-7-hydroxy-3′,4′-methylenedioxyflavan (1), was co-isolated from the rhizomes of Zephyranthes ajax Hort. with the following seven known compounds: 7-hydroxyflavan (2), 7,4′-dihydroxyflavan (3), 7,4′-dihydroxy-8-methylflavan (4), 7,3′-dihydroxy-4′-methoxyflavan (5), 5,4′-dihydroxy-7-methoxy-6-methylflavan (6), 7-hydroxy-3′,4′-methylenedioxyflavanone (7) and haemanthamine (8). Their structures were elucidated by combining 1D-/2D-NMR, CD, UV and HRESIMS data, and comparisons with reported data in literature were made. Among these known compounds, 2, 3, 4, 6 and 7 were isolated from the genus Zephyranthes for the first time. In addition, the cytotoxicity assay indicated that compound 8 has potent cytotoxic activity against human hepatocellular carcinoma (the HepG2 cell line), human lung carcinoma (the SK-LU-1 cell line), human carcinoma in the mouth (the KB cell line), human colon carcinoma (the SW480 cell line) and human stomach gastric adenocarcinoma (the AGS cell line), with IC50 values ranging from 4.4 to 11.3 µM. This is the first study reporting the cytotoxicity of compound 8 against the SK-LU-1 cancer cell lines.
Similar content being viewed by others
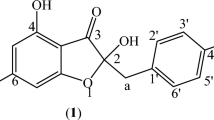

Introduction
The plants of the family Amaryllidaceae consist of ca. 85 genera and 1100 species widely distributed in the temperate and tropical regions of the globe1. Many species of this family have been used as remedies for inflammation, circulatory and neurological diseases2. Amaryllidaceae species contain numerous alkaloids with proven significant medical value. According to literature, there are over 500 Amaryllidaceae alkaloids possessing acetylcholinesterase inhibitory, cytotoxic, antitumor, analgesic, antifungal, antimalarial, antiviral and antibacterial activities2,3.
Zephyranthes ajax Hort. is an amaryllidaceous bulbous perennial. Bulbs are around 1 cm in diameter. Leaves are green, smooth and have linear blades which are around 30 cm long and 0.4–0.5 cm wide. Flowers are yellow and they have six petals (each 2 cm long) and equal-length stamens (around half of the petal length). Z. ajax is mainly used as an ornamental plant in Vietnam4. To date, however, the knowledge on the chemical constituents of Z. ajax is still limited. Phytochemical studies have revealed that this genus contains many alkaloids with cytotoxic, acetylcholinesterase inhibition, antiviral and antibacterial activity5,6,7,8. In addition, the genus contains flavonoids, flavans, gibberllins, phospholipids, sterols, lectins and terpenoids7. In this paper, we describe the isolation, structure elucidation and cytotoxic activities of a new flavanol derivative (1) and seven known compounds (2–8) found from Z. ajax collected in Vietnam.
Results and discussion
The structures of known compounds were established by means of spectroscopic (1H-, 13C-NMR and MS) and these results were in good agreement with the previous studies, including 7-hydroxyflavan (2)9, 7,4′-dihydroxyflavan (3)10, 7,4′-dihydroxy-8-methylflavan (4)11, 7,3′-dihydroxy-4′-methoxyflavan (5)12, 5,4′-dihydroxy-7-methoxy-6-methylflavan (6)13, 7-hydroxy-3′,4′-methylenedioxyflavanone (7)14 and haemanthamine (8)15,16 (Fig. 1). To our best knowledge, this is the first study reporting the presence and isolation of compounds 2, 3, 4, 6 and 7 in the genus Zephyranthes.

Structures of 1–8 isolated from Zephyranthes ajax Hort.
Compound 1 was obtained as a colourless powder. The high-resolution electron spray ionization mass spectrometry (HRESIMS) of 1 showed a quasi-molecular ion peak at m/z 329.1021 [M+H]+. Based on the HRESIMS and NMR data, its molecular formula is evidently C18H16O6, requiring eleven degrees of unsaturation. The 1H NMR spectrum of 1 exhibited the characteristic signals corresponding to two sets of ABX patterns at δH 6.90 (d, J = 8.5 Hz, H-5), 6.40 (dd, J = 8.5, 2.5 Hz, H-6), 6.37 (d, J = 2.5 Hz, H-8) and δH 6.98 (d, J = 1.0 Hz, H-2′), 6.82 (d, J = 8.0 Hz, H-5′), 6.95 (dd, J = 8.0, 1.0 Hz, H-6′), belonging to two 1,3,4-trisubstituted benzene rings. In addition, the signals of a dioxygenated methylene group at δH 5.97 (s), two oxygenated methine groups at δH 5.11 (s, H-2), 5.37 (m, H-3) and an acetoxy group at δH 1.89 (s) were observed. The acetoxy methyl protons resonate at higher field due to the anisotropic effect of two aromatic rings. This observation is in accordance with previous studies on 3-acetoxyflavanol derivatives17,18. The 13C NMR, DEPT and HSQC spectra of 1 revealed 18 signals including a carbonyl carbon (δC 171.9), twelve aromatic carbons (δC 158.0, 156.2, 149.0, 148.7, 133.7, 131.3, 121.0, 110.8, 110.0, 108.8, 108.1, 104.0), a dioxygenated methylene carbon (δC 102.4), two oxygenated methine carbons (δC 78.4, 70.2), a methylene carbon (δC 31.1) and a methyl carbon (δC 20.8) (Table 1). The present results suggest that compound 1 is a flavanol derivative. The planar structure of 1 was established by detailed HMBC analysis (Fig. 2). Notably, the HMBC cross-peak from H-3 (δH 5.37) to carbonyl carbon (δC 171.9) led to the introduction of acetoxy group at C-3. This was supported by the strong downfield shifts of C-3 (δC 70.2), H-3 (δH 5.37) compared to those of (2R,3R)-3,7-dihydroxy-3,4-methylenedioxyflavan (δC 67.1, δH 4.21)19.

Key HMBC (1H → 13C, arrows) correlations of 1.
The CD spectrum of 1 indicated a negative Cotton effect at λ 288 nm (1Lb band) which was associated with P-helicity of heterocyclic C ring. Therefore, the absolute configuration of C-2 was determined as R form20. Furthermore, the small vicinal coupling constant values (JH-2/H-3 = 0 Hz, JH-3/H-4a = 2.0 Hz, JH-3/H-4b = 4.0 Hz) were indicative of the cis relationship between H-2 and H-321. Based on the above evidences, compound 1 was elucidated to be (2R,3R)-3-acetoxy-7-hydroxy-3′,4′-methylenedioxyflavan.
The cytotoxicity of all isolates was initially evaluated against human hepatocellular carcinoma (the HepG2 cell line) and human lung carcinoma (the SK-LU-1 cell line). As shown in Table 2, compounds 1–7 did not show any cytotoxicity against the two tested cancer cells. Compound 8 exhibited potent activity against the HepG2 and SK-LU-1 cell lines, with IC50 values of 9.7 and 5.4 µM, respectively. Particularly, the cytotoxic activity against the SK-LU-1 cancer cell line of 8 has not been reported previously. Based on the initial results, compound 8 was selected for the further investigation against human carcinoma in the mouth (the KB cell line), human colon carcinoma (the SW480 cell line) and human stomach gastric adenocarcinoma (the AGS cell line). As expected, compound 8 showed a strong inhibitory effect towards the KB, SW480 and AGS cell lines, with IC50 values of 11.3, 4.4 and 6.5 µM, respectively. These findings are in agreement with those reported in literature22,23. According to Havelek et al., compound 8 acts as a potential anticancer agent owing to the decrease in cell viability and induction of apoptosis accelerating the caspases activation. With treatment of 8 in Jurkat cells, caspase-9 and caspase-3/7 activation are more intense and caspase-8 activation was less intense, in addition to the arrest of the cell cycle and the decrease in mitochondrial membrane potential which plays a crucial step in the apoptotic process. Furthermore, the agent is able to stimulate DNA damage checkpoint kinase Chk1 and the p16INK4a cyclin-dependent kinase inhibitor24. Pellegrino et al. reported that compound 8 is capable to form complex with the Saccharomyces cerevisiae 80S ribosome through binding at the A-site cleft of the peptidyl tranferase centre on the large ribosomal subunit, thus generating specific molecular interactions with the 25S rRNA25. The authors found that that ribosomes are bound by one molecule of compound 8 at a time, thus enabling highly specific drug targeting. Moreover, compound 8 was found to form a sandwich-like structure between the two 25S rRNA redidues (U2875 and C2821). The accommodation of compound 8 in the present structure is also stabilized by the formation of two additional hydrogen bonds associated with the C11-hydroxyl group25. These result in the halting of the elongation phase of eukaryotic translation. Consequently, haemanthamine treatment specifically restrains ribosome biogenesis, activates nucleolar stress response and stabilises p53 in cancer cells, which are responsible for the preferential killing of cancer cells25. Compound 8 has been proven to have a cytotoxic activity, but it is very poorly soluble in water. Therefore, we recently developed novel haemanthamine (compound 8)-loaded amphiphilic nanofibers to overcome these formulation challenges26. Noticeably, secondary metabolites from natural products have been the most successful leads in the discovery and development of novel drugs27,28. However, these plant-origin compounds possess a number of disadvantages associated with their large molecule size, poor water solubility, poor oral bioavailability, limitations in target-specific drug delivery, and in-vivo instability29,30. Hence, the development of novel drug delivery systems for natural compounds could resolve these critical issues, thus leading to novel and versatile applications of natural products in medicine. In our recent study, we succeeded to yield (through extraction and crystallization) haemanthamine (compound 8) in the amounts enough to fabricate the active-loaded amphiphilic nanofibers26. The results suggested that the formulation of such amphiphilic nanofibers for haemanthamine could provide a promising drug delivery system for the present large-molecule and poorly water-soluble active agent.
Materials and methods
General experimental procedures
A JASCO P-2000 polarimeter (Hachioji, Tokyo, Japan) was used to record optical rotation. UV spectra were measured using a Shimadzu UV-1800 spectrophotometer (Shimadzu, Kyoto, Japan). Circular dichroism spectra were studied with a Chirascan CD spectrometer (Applied Photophysics Ltd., Surrey, United Kingdom). A Bruker Avance 500 spectrometer (Bruker, MA, USA) was exploited for investigating NMR spectra and TMS as an internal reference. HRESIMS data were performed using an Agilent 6530 Accurate-Mass spectrometer (Agilent, CA, USA). The column chromatography studies were conducted with YMC RP-18 (Fuji Silysia Chemical Ltd, Kasugai, Aichi, Japan), Cosmosil 75C18-OPN (Nacalai Tesque Inc., Kyoto, Japan), silica gel (40–50 µm, Kanto Chemical Co., Tokyo, Japan), Diaion HP-20 (Mitsubishi Chem. Co., Tokyo, Japan) and Sephadex LH-20 (Dowex 50WX2-100, Sigma–Aldrich, USA). Pre-coated silica gel 60F254 and RP-18 F254 plates (0.25 or 0.50 mm thickness, Merck, Germany) were exploited to investigate analytical TLC. Preparative HPLC with a DAD detector applied was Agilent 1260 Infinity II system (Agilent, CA, USA) and using a Zorbax SB–C18 column (5 µm particle size, 9.4 × 250 mm).
Plant material
The whole plants of Z. ajax were collected in Hue city, Vietnam (geographical coordinates: 16°27′43.8″N; 107°33′55.4″E) in May 2017. The plant material was authenticated by Dr Chinh Tien Vu (Vietnam National Museum of Nature, VAST, Vietnam). A voucher specimen has been stored at the Faculty of Pharmacy, Hue University of Medicine and Pharmacy, Hue University, Vietnam.
Extraction and isolation
The powdered bulbs (cut from the ground parts) of Z. ajax (5.5 kg) were extracted three time with MeOH (10.0 L each, at room temperature) to obtain 467 g extract. After suspension of the extract using water (2.0 L), partition was conducted with dichloromethane, ethyl acetate, n-butanol (3 times, 2.0 L each). The solvents were removed in vacuo to yield the dichloromethane (D, 120.3 g), ethyl acetate (E, 126.8 g), n-butanol (43.5 g) and water (140.3 g)-soluble portions.
The D extract was loaded onto a silica gel column, eluted with a n-hexane–acetone gradient solvent system (100:0, 95:5, 90:10, 50:10, 10:10, 0:100 v/v, each 1.5 L to get 6 fractions D1–D6). Fraction D3 (20.5 g) was subjected to a silica gel column using n-hexane–ethyl acetate (8:1, v/v) as an eluent to give 8 sub-fractions (D3.1–D3.8). Fraction D3.2 (2.3 g) was chromatographed with a RP-18 column chromatography eluting with MeOH–water (4:1, v/v) to give 10 fractions (D3.2.1–D3.2.10). Fraction D3.2.5 (212 mg) was then applied to a Sephadex LH-20 column by eluting with MeOH to give 3 fractions (D3.2.5.1–D3.2.5.3). Fraction D3.2.5.2 (78 mg) was separated by preparative reversed-phase HPLC using acetonitrile–water (75:25, flow rate 2.0 mL/min) as an eluent to obtain 1 (4.2 mg) and 2 (3.7 mg). Fraction D3.4 gave a precipitate. After filtering and drying, the precipitate was dissolved in CHCl3–MeOH (1:1, v/v), and then recrystallised to yield 8 (210 mg) as a white precipitate.
The E extract was chromatographed on a silica gel column using n-hexane–acetone gradient solvent system (100:0, 75:10, 50:10, 25:10, 10:10, 0:100 v/v, each 1.5 L to get 6 fractions E1–E6) as the eluent. Fraction E4 (18.5 g) was further fractionated on a silica gel column by eluting with n-hexane–ethyl acetate–acetone (10:1:1, v/v) to yield 7 fractions (E4.1–E4.7). Fraction E4.2 (2.1 g) was subsequently purified with a RP-18 column by eluting with MeOH–water (3:1, v/v) to get 6 fractions (E4.2.1–E4.2.6). Fraction E4.2.4 (281 mg) was further separated by a Sephadex LH-20 column by eluting with MeOH–water (4:1, v/v) to give 4 sub-fractions (E4.2.4.1–E4.2.4.4). Fraction E4.2.4.2 (94 mg) was purified by preparative reversed-phase HPLC using acetonitrile–TFA in water 0.05% (65:35, flow rate 2.0 mL/min) as an eluent to obtain 3 (3.0 mg), 4 (3.8 mg). Fraction E4.4 (2.1 g) was further applied to a silica gel column and eluted with n-hexane–CH2Cl2–MeOH (5:1:1, v/v) to get 5 fractions (E4.4.1–E4.4.5). Fraction E4.4.3 (451 mg) was further fractionated with a Sephadex LH-20 column by eluting with MeOH to yield 4 fractions (E4.4.3.1–E4.4.3.4). Fraction E4.4.3.3 (143 mg) was further purified by preparative reversed-phase HPLC and eluted with MeOH–water (73:27, flow rate 2.0 mL/min) to produce 5 (4.1 mg), 6 (2.9 mg) and 7 (4.4 mg).
(2R,3R)-3-Acetoxy-7-hydroxy-3′,4′-methylenedioxyflavan (1): Colourless powder; \({[}\alpha {]}_{D}^{25}\) − 18.0 (c 0.1, MeOH); UV (MeOH) λmax (nm): 203, 284, 288; CD (MeOH) λmax (nm) (Δε, mdeg.): 206 (− 4.07), 224 (− 0.52), 231 (− 0.73), 252 (+ 0.05), 288 (− 0.45), 341 (− 0.15); 1H and 13C NMR: see Table 1; HRESIMS found: m/z 329.1021 [M+H]+ (calcd. for C18H17O6, 329.1025).
Cytotoxicity assay
The effects of compounds 1–8 on the growth of human cancer cells, including HepG2 and SK-LU-1, were tested using a sulforhodamine B assay31,32. Two human cancer cell lines were grown in a Dulbecco’s modified eagle medium (DMEM) consisting of 2.0 mM l-glutamine, 10.0 mM HEPES and 1.0 mM sodium pyruvate. The DMEM was supplemented with 10% foetal bovine serum, FBS (GIBCO). The cells were sub-cultured every 3–5 days and kept in a humidified atmosphere containing 5% CO2 at 37 °C. Subsequently, the cells were detached using 0.05% Trypsin–EDTA. A sulforhodamine B (SRB) method based on the determination of cellular protein content was exploited to assess the proportion of viable cells in a cell population. In 96-well microplates, viable cells were cultured overnight (4 × 104 cells/well) in 180 μL of growth medium. The cells were then treated with tested samples at various concentrations of 100, 20, 4 and 0.8 μg/mL and maintained under the same conditions for 3 days. After removing the medium, cold 20% (w/v) trichloroacetic acid was used to fix the remaining cell monolayers for 1 h at 4 °C. A 1X SRB staining solution was applied to stain the fixed cells for 30 min. Subsequently, 1% (v/v) acetic acid was applied three times to remove the unbound dye. ELISA Plate Reader (Bio-Rad) was used for the absorbance measurement (at 515 nm) of the protein-bound dye dissolved in a 10-mM Tris base solution. DMSO 10% and ellipticine were used as a negative control and positive control, respectively. The half maximal inhibitory concentration (IC50) was determined using TableCurve Version 4.0 (Systat Software, Inc., USA)33,34. All experiments were performed in triplicate.
Supporting information
HRESIMS, UV, CD and NMR spectra (1H and 13C NMR, DEPT, HSQC, HMBC) for the new compound 1 (Supplementary Information).
References
Willis, J. C. Amaryllidaceae. In A Dictionary of the flowering plants & ferns 8th edn (ed. Shaw, A. H. K.) (Cambridge University Press, Cambridge, 1988).
Ding, Y. et al. Phytochemical and biological investigations of Amaryllidaceae alkaloids: A review. J. Asian Nat. Prod. Res. 19, 53–100 (2017).
Meerow, A. W. & Snijman, D. A. Amaryllidaceae. In The Families and Genera of Vascular Plants Vol. 3 (ed. Kubitzki, K.) 83–110 (Springer, Berlin, 1998).
Ho, P. H. An Illustrated Flora of Vietnam Vol. 3, 496–503 (Youth Publishing House, Ho Chi Minh City, 2003).
Jin, Z. Amaryllidaceae and Sceletium alkaloids. Nat. Prod. Rep. 30, 849–868 (2013).
Bastida, J. et al. Chemical and biological aspects of Amaryllidaceae alkaloids. In Recent Advances in Pharmaceutical Sciences (ed. Munoz-Torrero, D.) 65–100 (Transworld Research Network, Trivandrum, 2011).
Katoch, D. & Singh, B. Phytochemistry and pharmacology of genus Zephyranthes. Med. Aromat. Plants. https://doi.org/10.4172/2167-0412.1000212 (2015).
Zhan, G. et al. Zephycandidine A, the first naturally occurring imidazo[1,2-f]phenanthridine alkaloid from Zephyranthes candida, exhibits significant anti-tumor and anti-acetylcholinesterase activities. Sci. Rep. 6, 33990. https://doi.org/10.1038/srep33990 (2016).
Moodley, N. The chemical investigation of the Amaryllidaceae and Hyacinthaceae. Doctor of Philosophy in the School of Pure and Applied Chemistry, University of KwaZulu-Natal, Durban, South Africa 41–42 (2004).
Meksuriyen, D. & Cordell, G. A. Traditional medicinal plants of Thailand XIII. Flavonoid derivatives from Dracaena loureiri (Agavaceae). ScienceAsia. 14, 3–24 (1988).
Ioset, J. R., Marston, A., Gupta, M. P. & Hostettmann, K. A methylflavan with free radical scavenging properties from Pancratium littorale. Fitoterapia 72, 35–39 (2001).
Sun, Q. et al. Flavans with cytotoxic activity from the stem and root bark of Daphne giraldii. RSC Adv. 6, 55919–55929 (2016).
Zheng, Q. A., Li, H. Z., Zhang, Y. J. & Yang, C. R. Flavonoids from the resin of Dracaena cochinchinensis. Helv. Chim. Acta. 87, 1167–1171 (2004).
Ali, R., Rahim, A. & Islam, A. Synthesis and antimicrobial activity of 7-hydroxy-3′,4′-methylenedioxy- and 7-benzyloxy-3′,4′-methylenedioxy flavanones. J. Sci. Res. 9, 297–306 (2017).
Bastida, J. et al. Alkaloids from Narcissus confusus. Phytochemistry 26, 1519–1524 (1987).
Bohno, M. et al. Total synthesis of Amaryllidaceae alkaloids, (+)-vittatine and (+)-haemanthamine, starting from d-glucose. Tetrahedron 63, 6977–6989 (2007).
Meng, D., Wu, J. & Zhao, W. Glycosides from Breynia fruticosa and Breynia rostrata. Phytochemistry 71, 325–331 (2010).
Li, D. L., Li, X. M., Peng, Z. Y. & Wang, B. G. Flavanol derivatives from Rhizophora stylosa and their DPPH radical scavenging activity. Molecules 12(5), 1163–1169 (2007).
Jitsuno, M., Yokosuka, A., Sakagami, H. & Mimaki, Y. Chemical constituents of the bulbs of Habranthus brachyandrus and their cytotoxic activities. Chem. Pharm. Bull. 57, 1153–1157 (2009).
Slade, D., Ferreira, D. & Marais, J. P. J. Circular dichroism, a powerful tool for the assessment of absolute configuration of flavonoids. Phytochemistry 18, 2177–2215 (2005).
Vdovin, A. D., Kuliev, Z. A. & Abdullaev, N. D. 1H and13C spectroscopy in the study of flavan-3-ols, proanthocyanidins, and their derivatives. Chem. Nat. Compd. 33, 11–24 (1997).
Nair, J. J., Bastida, J., Viladomat, F. & van Staden, J. Cytotoxic agents of the crinane series of Amaryllidaceae alkaloids. Nat. Prod. Commun. 7, 1677–1688 (2012).
Havelek, R. et al. Anticancer potential of Amaryllidaceae alkaloids evaluated by screening with a panel of human cells, real-time cellular analysis and Ehrlich tumor-bearing mice. Chem. Biol. Interact. 275, 121–132 (2017).
Havelek, R. et al. The effect of Amaryllidaceae alkaloids haemanthamine and haemanthidine on cell cycle progression and apoptosis in p53-negative human leukemic Jurkat cells. Phytomedicine 21, 479–490 (2014).
Pellegrino, S. et al. The Amaryllidaceae alkaloid haemanthamine binds the eukaryotic ribosome to repress cancer cell growth. Structure. 26, 416-425.e4 (2018).
Nguyen, K. V. et al. Preformulation study of electrospun haemanthamine-loaded amphiphilic nanofibers intended for a solid template for self-assembled liposomes. Pharmaceutics. 11(10), 499. https://doi.org/10.3390/pharmaceutics11100499 (2019).
Mishra, B. B. & Tiwari, V. K. Natural products: An evolving role in future drug discovery. Eur. J. Med. Chem. 46, 4769–4807 (2011).
Rey-Ladino, J., Ross, A. G., Cripps, A. W., McManus, D. P. & Quinn, R. Natural products and the search for novel vaccine adjuvants. Vaccine. 29, 6464–6471 (2011).
Bonifácio, B. V. et al. Nanotechnology-based drug delivery systems and herbal medicines: A review. Int. J. Nanomed. 9, 1 (2014).
Patra, J. K. et al. Nano based drug delivery systems: Recent developments and future prospects. J. Nanobiotechnol. 16(1), 71 (2018).
Monks, A. et al. Feasibility of a high-flux anticancer drug screen using a diverse panel of cultured human tumor cell lines. J. Natl. Cancer Inst. 83, 757–766 (1991).
Wu, T. Y., Cho, T. Y., Lu, C. K., Liou, J. P. & Chen, M. C. Identification of 7-(4′-cyanophenyl)indoline-1-benzenesulfonamide as a mitotic inhibitor to induce apoptotic cell death and inhibit autophagy in human colorectal cancer cells. Sci. Rep. 7, 12406 (2017).
Nguyen, K. V. et al. Secondary metabolites from Alphonsea tonkinensis A.DC. showing inhibition of nitric oxide production and cytotoxic activity. J. Pharm. Pharmacogn. Res. 9(1), 24–32 (2021).
Ho, D. V. et al. A new triterpene ester and other chemical constituents from the aerial parts of Anodendron paniculatum and their cytotoxic activity. J. Asian Nat. Prod. Res. 20, 188–194 (2018).
Author information
Authors and Affiliations
Contributions
H.T.N., A.R. and J.H. contributed to the conception or design of the work. K.V.N., D.V.H., N.T.L., and K.V.P. performed experimental part of the study. K.V.N., H.T.N., D.V.H., K.V.P., A.R. and J.H. wrote the main manuscript text. K.V.N. prepared Figs. 1, 2. All authors analysed of data and accepted the submission.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Nguyen, K.V., Ho, D.V., Le, N.T. et al. Flavonoids and alkaloids from the rhizomes of Zephyranthes ajax Hort. and their cytotoxicity. Sci Rep 10, 22193 (2020). https://doi.org/10.1038/s41598-020-78785-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-78785-2
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
