
Abstract
Neuroblastoma is the most common and deadly childhood tumor. Relapsed or refractory neuroblastoma has a very poor prognosis despite recent treatment advances. To investigate genomic alterations associated with relapse and therapy resistance, whole-genome sequencing was performed on diagnostic and relapsed lesions together with constitutional DNA from seven children. Sequencing of relapsed tumors indicates somatic alterations in diverse genes, including those involved in RAS-MAPK signaling, promoting cell cycle progression or function in telomere maintenance and immortalization. Among recurrent alterations, CCND1-gain, TERT-rearrangements, and point mutations in POLR2A, CDK5RAP, and MUC16 were shown in ≥ 2 individuals. Our cohort contained examples of converging genomic alterations in primary-relapse tumor pairs, indicating dependencies related to specific genetic lesions. We also detected rare genetic germline variants in DNA repair genes (e.g., BARD1, BRCA2, CHEK2, and WRN) that might cooperate with somatically acquired variants in these patients with highly aggressive recurrent neuroblastoma. Our data indicate the importance of monitoring recurrent neuroblastoma through sequential genomic characterization and that new therapeutic approaches combining the targeting of MAPK signaling, cell cycle progression, and telomere activity are required for this challenging patient group.
Similar content being viewed by others


Introduction
Neuroblastoma (NB) is an aggressive pediatric malignancy originating from the sympathetic nervous system. It is characterized by a diverse clinico-biological phenotype, with nearly half of all patients having a high-risk profile. Despite recent improvements in multimodal treatment, including high-dose induction chemotherapy, followed by surgery and myeloablative therapy with autologous stem cell rescue and local radiotherapy, maintenance treatment with retinoic acid, as well as immunotherapy using anti-GD2, the long-term survival of children diagnosed with high-risk NB remains below 50% at 5 years from treatment1,2. Although initial response to treatment is seen in most patients, a significant portion will subsequently progress with lesions resistant to standard therapy, and for NB patients with relapsed or refractory metastatic disease, survival is below 10%2. Genetic risk factors previously identified in primary NB include larger chromosomal aberrations, such as amplification of the MYCN oncogene, 11q-deletion, 1p-deletion, and 17q-gain3, as well as activating mutations in ALK encoding anaplastic lymphoma kinase4,5,6,7,8. Additional genomic alteration that recently have been associated to inferior outcome are distal 6q-deletion9,10, 19p-deletion and 1q-gain11,12 and aberrations connected to ATRX, TERT, TP53 or RAS13. Analysis of primary NB tumors indicates a relative paucity of recurrent somatic alterations, with mutations in ALK being most frequent14,15,16,17. However, treatment-resistant tumors will likely have novel molecular and genetic aberrations different from those present at time of diagnosis, either induced by or selected for during treatment. Previous studies of relapsed NB indicate that alterations associated with activation of the ALK-RAS-MAPK pathway18,19 or mesenchymal transition20 are enriched in relapsed samples. To further investigate the genetic mechanisms linked to recurrent disease, alterations between seven triplicate samples of primary tumor, relapsed or refractory tumor, and constitutional DNA were investigated using whole-genome sequencing (WGS).
Results
Clinical background and patient characteristics
We performed WGS on the genomes of triplicate samples of primary and relapsed/refractory NB tumors and lymphocytes from seven patients. All tumors were high-stage, high-risk tumors with metastatic disease at time of diagnosis. The median age at time of diagnosis was 50 months (range 17–118 months), while the median time from diagnosis to relapse or progression was 26 months (range 12–71 months). All subjects were diagnosed between 2012 and 2017 and initially treated according to the HR-NBL1/SIOPEN protocol and thus, underwent similar treatment consisting of induction chemotherapy (rapid COJEC ± TVD), local therapy (surgery and local irradiation), high-dose chemotherapy (BuMel) with autologous stem cell rescue, and maintenance therapy (retinoids). Six of the seven patients had recurrence after complete clinical remission after completed therapy and one patient experienced disease progression during treatment (Table 1). None of these patients had received any additional treatment using targeted therapy before the time of relapse/progression.
Constitutional alterations in cancer-predisposing genes
To detect possible genetic predisposition to NB, constitutional DNA was analyzed, focusing only on variants in 565 genes with established associations with sporadic and/or hereditary cancer (Supplemental Table 2). This analysis did not detect any activating point mutations in hot-spot regions of ALK or loss-of-function variants in PHOX2B, the two genes mainly connected to NB predisposition. However, a novel ALK frameshift variant (i.e., ALK p.V1471fs*6) of unknown significance was detected in one patient. This variant is predicted to result in a truncated protein that retains the kinase domain but lacks a portion of the C-terminal domain containing multiple phosphorylation sites crucial for ALK functions including tyrosine residue 1604. Thus, it is unlikely that this variant has the transforming potential seen for of the ALK hot-spot mutations common in NB. Three of the seven patients had germline variants likely to affect the function of DNA repair genes. These include a nonsense mutation in BARD1 (p.Q545), a missense variant in WRN (p.S1141L) affecting an ATM phosphorylation site important for WRN protein function, and two rare missense variants in CHEK2 (p.I157T) and BRCA2 (p.R2991C), respectively, detected in one patient. These variants and other variants associated with loss-of-function or predicted to be damaging by SIFT and PolyPhen2 are summarized in Table 2.
Overall comparison of somatic variants between time of diagnosis and time of recurrence
In total, 529 nonsynonymous somatic SNVs were detected in the analyzed tumors (Table 3 and Supplemental Table 3), with an average of 28 SNVs (range 8–63) in primary tumors, increasing to an average of 61 SNVs in relapsed tumors (range 51–76), corresponding to 0.7 and 2.1 variants per megabase of the coding genome, respectively. The mutational burden at time of relapse/progression was significantly higher than at time of diagnosis (p = 0.002) (Fig. 1A,B). Overall, the mutational load was in the range of that previously reported in NB. The median number of somatic SNVs shared by each primary and relapse/refractory pair was 14 (range 0–40), corresponding to 50% of all mutations detected in primary tumors and to 23% of all mutations detected in relapsed/refractory tumors. However, for patient NB60R6, for whom the investigated biopsy specimens were from a metastasis at time of diagnosis and a metastasis at time of relapse, no somatic nonsynonymous SNV shared by the two samples could be detected, not even after including mutations below the 10% variant allele fraction.

Mutational distribution of primary and relapsed NBs. (A) The number of non-silent somatic mutations per megabase identified in seven primary tumors and their corresponding relapse tumors. Mutations shared by primary and relapse tumors are shown in blue, mutations unique to primary tumors are shown in green, and mutations unique to relapse tumors are shown in red. (B) The total number of somatic structural variants identified in the seven primary tumors and their corresponding relapse tumors; color scheme as in (A). (C) Number of nonsynonymous somatic mutations per megabase in primary and relapse tumors in relation to age at time of diagnosis and age at time of relapse/progression. (D) Recurrent and other biological relevant copy number alterations and SNVs detected through WGS. (E) Case-by-case overview of somatic SNVs and SVs in primary and relapsed tumors with biologically relevant mutated genes and/or genomic alterations indicated.
The number of somatic mutations at time of diagnosis was correlated with patient age at time of sampling, at time of both diagnosis and relapse/progression. Pearson correlations were calculated after removing case NB67R9, which appears to have had a markedly higher mutation rate than the other samples (Fig. 1C). For the remaining tumor specimens, Pearson correlation coefficients indicate statistically significant correlation between the total number of non-synonymous SNVs and patient age at time of diagnosis or relapse occurrence (R = 0.915, p = 3.0 × 10–5) (Fig. 1C).
Copy number alterations and structural variants
Although some persistent copy number alterations (CNAs) were detected in matched primary and relapsed tumors, the relapsed tumors had increased numbers of structural variants (SVs) at time of relapse (Fig. 1B, Supplemental Fig. 1, and Supplemental Table 4). At time of diagnosis, a median of 38 somatic SVs (range 5–122) were detected, the number later increasing to a median of 49 SVs (range 14–89) at time of relapse when excluding the excessive number of SVs within highly amplified regions (i.e., the MYCN locus in NB63R4 and the 12q-amplicons in NB59R9). Persistent CNAs present in both diagnostic and relapsed/refractory tumors include: 17q-gain, detected in all tumors except one that instead displayed gain of whole chromosome 17; 11q-deletion, detected in four of six patients; MYCN amplification, detected in one patient; and CDK4/MDM2/FRS2 amplification, detected in one patient (Fig. 2 and Supplemental Fig. 1). Loss of chromosome 1p, also associated with poor prognosis in NB21, was seen in six patients: three cases (i.e., NB67R9, NB63R4, and NB69R8) in which 1p-deletion was detected in both primary and relapse tumors, one case (i.e., NB60R6) in which 1p-deletion was detected only in the diagnostic specimen, one case (i.e., NB53R2) in which 1p-deletion was detected only in the relapsed specimen, and one case (NB67R5) in which 1p-deletion was detected in both primary and relapsed tumors, albeit with different breakpoints (Fig. 2 and Supplemental Table 4). On the genomic level, additional recurrent CNAs and structural variants were seen in association with TERT (in three patients, one relapse specific), CCND1 (in two patients, one relapse specific), and LSAMP (in two patients, both relapse specific) (Fig. 1D,E). Interestingly, both TERT SVs in patient NB67R5 and the CCND1-gains in patient NB60R6 appear to have emerged on separate occasions as parallel events, given the difference in translocation partners and breakpoints relating to these genomic alterations (Supplemental Table 4). In patient NB60R6 we also detected CNAs in both primary and relapse tumors, resulting in gain of chromosomal regions 7q and 6p; however, this also emerged through parallel events, as the two tumors have different breakpoints for these CNAs and also gains of different parental chromosomes as judged by the allele fraction (Figs. 1D and 2). The paired primary and relapse tumors of patient NB60R6 shared only one common SV, a translocation between chromosomes 11 and 17 that ultimately resulted in 11q-deletion and 17q-gain. However, as the position of the translocation was identical in both samples, this indicates that both analyzed specimens share a common descent. Patient NB59R9 had the highest number of SVs due to high genomic instability, with a cataclysmic event resulting in extensive focal genomic shattering and rejoining. This affected regions in chromosome 5 and amplified regions in chromosome 12, connected to MDM2 and CDK4, were present in both samples for this patient, while similar focal genomic shattering was seen in chromosomes 7, 18, and 21 in primary tumors and in chromosome 4 in relapse tumors (Supplemental Table 4).

Copy number aberrations. Coverage-based plots displaying segmental alterations in primary and relapsed tumors.
Recurrent alterations and mutations in cancer-associated pathways
Telomere maintenance is crucial for sustained proliferative capacity and is associated with poor prognosis in NB. In our cohort, tumors from three patients had TERT-associated SVs and tumors from two patients had tandem duplications causing disruption of ATRX (Fig. 1D,E). All three patients with TERT alterations had genomic profiles that included 11q deletions, though in two patients we did not detect any alterations in genes directly connected to telomere maintenance. Both these patients displayed high levels of genomic amplification: patient NB59R9 for several 12q loci, including CKD4, MDM2, and FRS2, and patient NB63R4 for the MYCN locus. Interestingly, patient NB67R5, with two different TERT-associated structural variants in primary and relapsed tumors, also had two mutations in the chromatin-associated genes ARID1A and ARID1B. The primary tumor of NB67R5 had a structural variant causing disruption of ARID1B, a variant that was lost in the relapse sample, which instead carried a missense mutation in ARID1A (Fig. 1D, and Supplemental Tables 2 and 3).
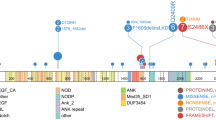
Previous studies by us and others have indicated that mutations in ALK and other genes of the RAS-MAPK pathway are connected to relapsed NB18,19. In our cohort, an activating somatic ALK mutation was seen in the tumors from one patient (i.e., NB69R8). This is an ALK Q1275R alteration in which the tumor displayed increased variant allele frequency from time of diagnosis to time of relapse. This increase was likely due to a new copy-neutral loss of heterozygosity (CN-LOH) of chromosome 2 that preserves the mutated allele, as judged by B-allele frequency. In addition to ALK, somatic variants were also detected in other genes associated with RAS-MAPK signaling. This includes mutations in KRAS, FGFR1, and MAP3K14 (NIK), the first two genes being detected only in the relapsed samples of two different patients, while the last was shared by the primary and relapse tumors in one patient (Fig. 1D). We also detected a somatic frameshift mutation in PHOX2B, a gene important for proper neural crest development and present in both the primary and recurrent tumors of patient NB67R5. Somatic alterations were seen in several genes connected to cell cycle progression and/or DNA repair, such as CCND1, CDK4, MDM2, BRCA1, BRINP1, and WRN (Fig. 1D).
Besides CCND1 and TERT, 30 additional genes were altered in at least two tumor samples in our cohort, either through SNV or SV. Recurrent mutated genes present only in tumors at time of diagnosis were limited to ABCA5, CNTNAP2, CTNND2, and PTPRM, while we detected recurrent mutations in relapsed samples of 16 genes either already present in primary tumors or acquired in relapse. Of these 16 genes, eight were found to be relapse specific (i.e., CDK5RAP1, CFAP44, LSAMP, MUC16, NUBPL, POLR2A, PRR12, and SLC6A18) (Fig. 1D and Table 3). Two tumors carried missense mutations predicted to be damaging by SIFT and PolyPhen2 in the tumor-suppressor gene FAT1 (Fig. 1D). The FAT1 mutation in NB63R4 was acquired during relapse, while the FAT1 mutation in NB67R5 was present at the subclonal level with a 2% variant allele fraction in the primary tumor but increasing in relapse (i.e., a 15% variant allele fraction), indicating clonal expansion (Supplemental Table 3 and Supplemental Fig. 2).
Pathway-centered gene set enrichment of relapsed and primary neuroblastoma
Pathway analyses were performed on somatically mutated genes detected among all relapsed and primary tumors, respectively, to detect differences in pathway signature. Gene set analyses in KEGG for all mutations present in relapse tumors identified pathways connected to the glutamatergic synapse, PI3K-Akt signaling, longevity regulation, and Rap1 signaling among the highest ranked enrichment although not at a statistically significant level after correction for multiple testing (Supplemental Fig. 3 and Supplemental Table 5). Gene set analyses also indicated the prior enrichment of genes involved in PI3K-Akt signaling and in cancer pathways in the diagnostic samples (Supplemental Fig. 3 and Supplemental Table 5).

Broad patient-specific tumor heterogeneity with converging genomic alterations. (A) Striking tumor heterogeneity is seen between tumor material from time of diagnosis and time of relapse in patient NB60R6. Only one SV, an unbalanced translocation between chromosomes 11 and 17, is shared by the two samples. (B) The two samples display converging genomic events, such as gain of CCND1, 7q, and 6p, albeit with different breakpoints and different parental alleles.
Discussion
In attempting to identify molecular features related to disease progression in NB, we have characterized the genomes of seven relapsed/progressive NB tumors and corresponding tumor specimens from the time of diagnosis. In this study, we show that relapsed tumors generally have a higher mutational load and have acquired additional segmental variants (Fig. 1A,B, and Supplemental Fig. 1), in line with previous studies of relapsed NB19,20. We also show that the number of mutations present in primary tumors and the number of acquired mutations in relapsed tumors correlates well with patient age at diagnosis and recurrence, respectively (Fig. 1C). The coverage depth used for WGS in this study limits the ability to analyze whether relapse-specific mutations were acquired de novo or emerged from subclonal events in the primary tumor. However, we do detect examples of the clonal expansion of specific mutations, for example, as seen for FAT1 (Supplemental Fig. 2), as well as the presence of clonal eradication, as only a mean of 36% of all mutations detected in primary tumors are also present in relapse tumors. For some samples, such as NB60R6 and NB67R9, there is striking in-patient heterogeneity, with very few somatic SNVs shared by different tumor samples (Fig. 1E). This indicates that the longitudinal monitoring of disease burden and treatment response using specific genetic alterations could be challenging for a subgroup of NB patients.
Recurrent alterations were seen in 32 genes in tumors from at least two patients; eight of these genes were relapse specific and include MUC16 and LSAMP (Fig. 1D). Tumors from three patients also display alterations connected to cell cycle genes. These include tumors from two patients with CCND1 gain and tumors from one patient with CDK4/MDM2 amplification (Fig. 2).
Mutations in ALK have previously been shown to be enriched in relapsed NB18, as have other mutations that activate RAS-MAPK signaling, such as RAS, NF1, BRAF, and FGFR119. Tumors from four of our seven patients carry somatic ALK-MAPK-RAS signaling pathway alterations. These include somatic mutations in ALK and MAP3K14 that were already present at time of diagnosis and mutations in KRAS and FGFR1 that are relapse specific (Fig. 1D). The KRAS K117N alteration, detected in patient NB67R5, has been shown to decrease GTPase activity22,23 although studies from colorectal cancer indicate this alteration to be less potent than mutations in codons 12 and 1322. The tumor with KRAS K117N also carried a FAT1 mutation present at the subclonal level in the primary tumor and later found to be enriched at time of relapse. FAT1 is a tumor-suppressor gene that restrains the oncogenic activity of the Yes-associated protein (YAP)24, and studies of other malignancies show that YAP is a critical effector of oncogenic RAS25,26. Therefore, it is possible that the malignant capacity of KRAS K117N is further potentiated through increased YAP signaling in relapsed tumors.
Both PHOX2B and ALK have previously displayed recurrent mutations in both familial and sporadic NB4,5,27, as they are crucial for proper development of the sympathetic nervous system. In our cohort, the tumors from one patient (i.e., NB67R5) shared a somatic frameshift mutation of PHOX2B (p.G230fs). Truncating PHOX2B mutations have been shown to possess a dominant-negative effect resulting in halted sympathetic nervous system differentiation and increased proliferation, making cells susceptible to additional transforming events28. Regarding PHOX2B and ALK alterations in constitutional DNA, we detected a novel ALK frameshift variant already present in the germline (p.V1471fs*6) in one investigated patient. Although ALK is of great interest in NB etiology, it is difficult to envision the role of this particular variant in the oncogenic process, as it is expected to lack phosphorylation sites essential for ALK activation.
However, it has been shown that children and adolescents with cancer have a higher incidence of pathogenic or likely pathogenic germline mutations than do healthy controls29. When expanding our investigation of other cancer predisposition genes besides ALK and PHOX2B, we detect rare variants such as a nonsense mutation in BARD1 as well as missense variants in CHEK2, BRCA2, and WRN (Table 2). Germline variants in BARD1 and CHEK2 have indeed been shown to be enriched in NB patients16,30, while BRCA2 has been shown to be enriched in pediatric cancer patients29. The gene WRN encodes a RecQ helicase important for maintaining genome stability through resolving stalled replication forks in association with double-strand breaks. The missense variant detected in WRN affects an ATM phosphorylation site required for correctly handling stalled forks31. The tumors of patients who are heterozygous for the WRN variant display high genomic instability, with extensive focal genomic shattering and rejoining that might be attributed to deficient double-strand break repair.
For one patient (i.e., NB60R6), we analyzed a metastasis at time of diagnosis and a metastasis at time of relapse that provided indications of branched evolution. These two tumor specimens displayed no common nonsynonymous SNVs and only one shared structural variation, a translocation between chromosomes 11 and 17 resulting in 11q-deletion and 17q-gain (Fig. 3A). That the fusion point was exactly the same in both samples indicates that they emerged from a common ancestral tumor cell progenitor and not through multifocal events. However, both lesions acquired 6p-gain, 7q-gain, and CCND1-gain, albeit with different breakpoints and gains of different parental alleles (Figs. 2 and 3B). Additional parallel events between primary and relapse tumors were seen in case NB67R5, which displayed two different SVs close to the TERT locus, and in case NB67R9, which displayed two different mutations in ARID1B and ARID1A, two members of the SWI/SNF chromatin-remodeling complex, in tumors at time of diagnosis and time of relapse, respectively (Fig. 1D). The primary and relapsed tumors from patient NB67R9 also had a common TERT rearrangement that might be further potentiated through the ARID1B and ARID1A mutations, as loss of ARID1A has been shown to enhance TERT transcription32.
The findings of alterations of TERT, ARID1A, and ARID1B are related to telomere maintenance, which is a strong predictor of poor outcome in NB13,33. Besides the MYCN-amplified NB case, in which sustained telomere length might be achieved through elevated TERT expression driven by MYCN, all six non-MYCN-amplified cases in our cohort displayed genomic aberrations associated with telomere maintenance or chromatin remodeling; three of these cases had TERT rearrangements, two had ATRX alterations, and one had a mutation in ARID2, which is also part of the SWI/SNF chromatin-remodeling complex. This further supports the finding that telomere maintenance is associated with an unfavorable clinical course in NB13,33, as most tumors in this study were characterized by different means of sustaining telomeres.
In this study we have identified a diverse set of gene mutations in tumors from time of diagnosis and time of recurrence in this challenging patient group. This set includes mutations in genes promoting cell cycle progression, activating ALK-RAS-MAPK signaling, or important for telomere maintenance and immortalization. Our findings in combination with previous studies of relapsed NB provide a rationale for the comprehensive genomic characterization of recurrent NB in order to optimize treatment and aim for prolonged survival. This information could be used to spur novel combination therapies using compounds abrogating MAPK signaling, cell cycle progression, and telomere activity.
Material and methods
Patient material and ethics
NB tumors were collected from Swedish patients after either written or verbal informed consent was obtained from parents/guardians according to ethical permits approved by the local ethics committee (Karolinska Institutet and Karolinska University Hospital, registration numbers 03-736 and 2009/1369-31/1) in accordance with the Declaration of Helsinki. Clinical data on all patients were obtained from hospital records and/or the Swedish Childhood Cancer Registry.
Whole-genome sequencing
DNA was extracted from frozen tumors or blood using the DNeasy blood and tissue kit (Qiagen, Hilden, Germany) according to the manufacturer’s protocol and evaluated using absorbance measurements, fluorometric quantitation, and DNA integrity assessment on an Agilent TapeStation (Agilent, Santa Clara, CA) before sequencing. WGS was performed on triplicate samples of DNA from tumor material at time of diagnosis, tumor material at time of relapse/refractory disease, and corresponding constitutional DNA extracted from the blood of seven NB patients. For TruSeq DNA PCR-Free library preparation, 1 µg of DNA was used for all samples except for the primary tumor from NB59R9, for which a modified low-input protocol using 100 ng of DNA was required due to limited material. WGS was performed aiming for an average coverage of at least 60X for tumor libraries prepared according to the PCR-free library protocol and 30X for constitutional DNA and tumor samples prepared according to the low-input protocol using Illumina xTen instrumentation (Illumina, San Diego, CA, USA) located at Clinical Genomics, SciLife Laboratories, Stockholm, Sweden. Details of sequencing coverage are given in Supplemental Table 1. Mapping to the human reference genome hg19, removal of read duplicates, realignment around InDels, and base-score recalibration and variant calling were carried out using the Sentieon suite of bioinformatics tools (Sentieon Inc., Mountain View, CA, USA). This included the variant calling of germline single nucleotide variants (SNVs) and small InDels (up to about 80 bp in size) using Sentieon DNAscope software, while Sentieon TNscope was used for the calling of corresponding somatic variants, supplying the normal sample to filter out germline variants. In both DNAscope and TNscope variant calling, the corresponding machine learning models developed by Sentieon were used to filter out likely artifacts (Sentieon version v201808.03). Only high-quality called variants with a minimum 10% variant allele frequency and a total read coverage of ten were considered for further analysis. All synonymous variants or variants in non-coding regions were excluded, except those affecting canonical splice sites and remaining variants were assessed manually using the Integrative Genomics Viewer (IGV)34 to remove calls due to mapping artifacts and paralogs. The germline variants were analyzed using a more stringent filtering approach excluding all variants with a population allele frequency above 1.0%, in either gnomAD v2.1.1 (https://gnomad.broadinstitute.org/), 1000 genomes, the Exome Aggregation Consortium (ExAC) (Cambridge, MA, USA, http://exac.broadinstitute.org), or the NHLBI Exome Sequencing Project (http://evs.gs.washington.edu/EVS/), unless being an already established pathogenic variant, and keeping only nonsynonymous variants of genes selected based on established connections to hereditary and/or sporadic cancer genes, as listed in Supplemental Table 2. The filtering of somatic and germline variants was done using the ingenuity variant analysis software v5.6 (Qiagen, Hilden, Germany).
The Canvas tool (version 1.38.0.1554)35 was used to call copy number alterations (CNAs), with gains and losses of genomic regions as well as LOH regions being identified using read depth coverage in combination with SNV b-allele frequencies. For the calling of germline and somatic CNVs, the SmallPedigree-WGS and Somatic-WGS algorithms of Canvas were used, respectively. Somatic structural variants (SVs) were called using the Manta tool (version 1.1.1)36, which applies information about soft-clipped read ends, disjointed read pairs, and read-pair orientation to identify larger structural variations (e.g., deletions, duplications, inversions, and translocations). Calls from the constitutional DNA were used to filter out germline variation and artifacts caused by problematic regions. In addition, further filtering and removal were done based on presence in the SweGen Variant Frequency dataset (https://swefreq.nbis.se/) or in our in-house set of normal controls. Genes affected either directly by SVs or through smaller focal gain/loss were also noted for further genomic analysis.
To verify same individual origin for matched tumor-normal pair, we ran a Python script developed in-house (available on request) that calculated the fraction of shared single nucleotide polymorphisms (SNPs) relative the total number of SNPs using 400,000 SNPs (all present in dbSNP; version 138) from chromosomes 1–20. Using this method, the fraction reliably ends up in the 0.97–1 range for verified tumor-normal matched pairs, whereas unrelated randomly selected samples have a fraction between 0.6 and 0.7, while a parent and child-level of relatedness ends up close to 0.8. All paired samples in this study showed a tumor-normal matched fraction above 0.97 and thus, authenticated sample identity.
Statistics
IBM SPSS software was used to calculate Pearson correlations between mutational loads at diagnosis and relapse, respectively, relative to age at time of sampling and to perform independent t-testing for differences in the number of somatic SNVs between primary and relapsed tumors. Gene set analysis for the enrichment of somatically mutated genes was conducted using the curated Kyoto Encyclopedia of Genes and Genomes (KEGG) cell signaling pathway database37 (www.kegg.jp/kegg/kegg1.html) through the Enrichr web server38.
References
Valteau-Couanet, D. et al. Long-term results of the combination of the N7 induction chemotherapy and the busulfan-melphalan high dose chemotherapy. Pediatr. Blood Cancer 61, 977–981. https://doi.org/10.1002/pbc.24713 (2014).
Ladenstein, R. et al. 28 years of high-dose therapy and SCT for neuroblastoma in Europe: lessons from more than 4000 procedures. Bone Marrow Transplant. 41(Suppl 2), S118-127. https://doi.org/10.1038/bmt.2008.69 (2008).
Caren, H. et al. High-risk neuroblastoma tumors with 11q-deletion display a poor prognostic, chromosome instability phenotype with later onset. Proc. Natl. Acad. Sci. USA 107, 4323–4328. https://doi.org/10.1073/pnas.0910684107 (2010).
Mosse, Y. P. et al. Identification of ALK as a major familial neuroblastoma predisposition gene. Nature 455, 930–935. https://doi.org/10.1038/nature07261 (2008).
Janoueix-Lerosey, I. et al. Somatic and germline activating mutations of the ALK kinase receptor in neuroblastoma. Nature 455, 967–970. https://doi.org/10.1038/nature07398 (2008).
Caren, H., Abel, F., Kogner, P. & Martinsson, T. High incidence of DNA mutations and gene amplifications of the ALK gene in advanced sporadic neuroblastoma tumours. Biochem. J. 416, 153–159 (2008).
George, R. E. et al. Activating mutations in ALK provide a therapeutic target in neuroblastoma. Nature 455, 975–978. https://doi.org/10.1038/nature07397 (2008).
Chen, Y. et al. Oncogenic mutations of ALK kinase in neuroblastoma. Nature 455, 971–974. https://doi.org/10.1038/nature07399 (2008).
Ognibene, M., Morini, M., Garaventa, A., Podesta, M. & Pezzolo, A. Identification of a minimal region of loss on chromosome 6q27 associated with poor survival of high-risk neuroblastoma patients. Cancer Biol. Ther. 21, 391–399. https://doi.org/10.1080/15384047.2019.1704122 (2020).
Depuydt, P. et al. Genomic amplifications and distal 6q loss: novel markers for poor survival in high-risk neuroblastoma patients. J. Natl Cancer Inst. 110, 1084–1093. https://doi.org/10.1093/jnci/djy022 (2018).
Abbasi, M. R. et al. Impact of disseminated neuroblastoma cells on the identification of the relapse-seeding clone. Clin. Cancer Res. 23, 4224–4232. https://doi.org/10.1158/1078-0432.CCR-16-2082 (2017).
Lasorsa, V. A. et al. 19p loss is significantly enriched in older age neuroblastoma patients and correlates with poor prognosis. NPJ Genom. Med. 5, 18. https://doi.org/10.1038/s41525-020-0125-4 (2020).
Ackermann, S. et al. A mechanistic classification of clinical phenotypes in neuroblastoma. Science 362, 1165–1170. https://doi.org/10.1126/science.aat6768 (2018).
Pandey, G. K. et al. The risk-associated long noncoding RNA NBAT-1 controls neuroblastoma progression by regulating cell proliferation and neuronal differentiation. Cancer Cell 26, 722–737. https://doi.org/10.1016/j.ccell.2014.09.014 (2014).
Molenaar, J. J. et al. Sequencing of neuroblastoma identifies chromothripsis and defects in neuritogenesis genes. Nature 483, 589–593. https://doi.org/10.1038/nature10910 (2012).
Pugh, T. J. et al. The genetic landscape of high-risk neuroblastoma. Nat. Genet. 45, 279–284. https://doi.org/10.1038/ng.2529 (2013).
Sausen, M. et al. Integrated genomic analyses identify ARID1A and ARID1B alterations in the childhood cancer neuroblastoma. Nat. Genet. 45, 12–17. https://doi.org/10.1038/ng.2493 (2013).
Schleiermacher, G. et al. Emergence of new ALK mutations at relapse of neuroblastoma. J. Clin. Oncol. 32, 2727–2734. https://doi.org/10.1200/JCO.2013.54.0674 (2014).
Eleveld, T. F. et al. Relapsed neuroblastomas show frequent RAS-MAPK pathway mutations. Nat. Genet. 47, 864–871. https://doi.org/10.1038/ng.3333 (2015).
Schramm, A. et al. Mutational dynamics between primary and relapse neuroblastomas. Nat. Genet. 47, 872–877. https://doi.org/10.1038/ng.3349 (2015).
Caron, H. et al. Allelic loss of chromosome 1p as a predictor of unfavorable outcome in patients with neuroblastoma. N. Engl. J. Med. 334, 225–230. https://doi.org/10.1056/NEJM199601253340404 (1996).
Janakiraman, M. et al. Genomic and biological characterization of exon 4 KRAS mutations in human cancer. Can. Res. 70, 5901–5911. https://doi.org/10.1158/0008-5472.CAN-10-0192 (2010).
Stolze, B., Reinhart, S., Bulllinger, L., Frohling, S. & Scholl, C. Comparative analysis of KRAS codon 12, 13, 18, 61, and 117 mutations using human MCF10A isogenic cell lines. Sci. Rep. 5, 8535. https://doi.org/10.1038/srep08535 (2015).
Martin, D. et al. Assembly and activation of the Hippo signalome by FAT1 tumor suppressor. Nat. Commun. 9, 2372. https://doi.org/10.1038/s41467-018-04590-1 (2018).
Zhang, W. et al. Downstream of mutant KRAS, the transcription regulator YAP is essential for neoplastic progression to pancreatic ductal adenocarcinoma. Sci. Signal. 7, ra42. https://doi.org/10.1126/scisignal.2005049 (2014).
Slemmons, K. K., Crose, L. E., Rudzinski, E., Bentley, R. C. & Linardic, C. M. Role of the YAP oncoprotein in priming RAS-driven rhabdomyosarcoma. PLoS ONE 10, e0140781. https://doi.org/10.1371/journal.pone.0140781 (2015).
Mosse, Y. P. et al. Germline PHOX2B mutation in hereditary neuroblastoma. Am. J. Hum. Genet. 75, 727–730. https://doi.org/10.1086/424530 (2004).
Pei, D. et al. Distinct neuroblastoma-associated alterations of PHOX2B impair sympathetic neuronal differentiation in zebrafish models. PLoS Genet. 9, e1003533. https://doi.org/10.1371/journal.pgen.1003533 (2013).
Zhang, J. et al. Germline mutations in predisposition genes in pediatric cancer. N. Engl. J. Med. 373, 2336–2346. https://doi.org/10.1056/NEJMoa1508054 (2015).
Lasorsa, V. A. et al. Exome and deep sequencing of clinically aggressive neuroblastoma reveal somatic mutations that affect key pathways involved in cancer progression. Oncotarget 7, 21840–21852. https://doi.org/10.18632/oncotarget.8187 (2016).
Ammazzalorso, F., Pirzio, L. M., Bignami, M., Franchitto, A. & Pichierri, P. ATR and ATM differently regulate WRN to prevent DSBs at stalled replication forks and promote replication fork recovery. EMBO J. 29, 3156–3169. https://doi.org/10.1038/emboj.2010.205 (2010).
Suryo Rahmanto, Y. et al. Inactivating ARID1A tumor suppressor enhances TERT transcription and maintains telomere length in cancer cells. J Biol Chem 291, 9690–9699. https://doi.org/10.1074/jbc.M115.707612 (2016).
Peifer, M. et al. Telomerase activation by genomic rearrangements in high-risk neuroblastoma. Nature 526, 700–704. https://doi.org/10.1038/nature14980 (2015).
Thorvaldsdottir, H., Robinson, J. T. & Mesirov, J. P. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief. Bioinform. 14, 178–192. https://doi.org/10.1093/bib/bbs017 (2013).
Roller, E., Ivakhno, S., Lee, S., Royce, T. & Tanner, S. Canvas: versatile and scalable detection of copy number variants. Bioinformatics 32, 2375–2377. https://doi.org/10.1093/bioinformatics/btw163 (2016).
Chen, X. et al. Manta: rapid detection of structural variants and indels for germline and cancer sequencing applications. Bioinformatics 32, 1220–1222. https://doi.org/10.1093/bioinformatics/btv710 (2016).
Kanehisa, M., Goto, S., Furumichi, M., Tanabe, M. & Hirakawa, M. KEGG for representation and analysis of molecular networks involving diseases and drugs. Nucleic Acids Res. 38, D355-360. https://doi.org/10.1093/nar/gkp896 (2010).
Kuleshov, M. V. et al. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 44, W90-97. https://doi.org/10.1093/nar/gkw377 (2016).
Acknowledgements
The authors wish to acknowledge the Science for Life Laboratory/Clinical Genomics, Gothenburg University/Sahlgrenska University Hospital and the Science for Life Laboratory/Clinical Genomics, Stockholm for performing WGS, for help with sequencing analysis, and for help with computational infrastructure. We also thank Vanja Börjesson from the Bioinformatics Core facility at Gothenburg University for the generation of circos plots.
Funding
Open Access funding provided by Gothenburg University Library. Open Access funding provided by Gothenburg University Library. Open Access funding provided by Gothenburg University Library. Open Access funding provided by Gothenburg University Library. This work was supported by grants from the Swedish Childhood Cancer Foundation (SF: PR2018-099, NCP2015-0061, and TJ2014-0064; TM: PR2017-0166, PK: PR2017-0122), the Swedish Cancer Foundation (TM: 18-0711, PK: 19-0566), the Swedish state under the LUA/ALF agreement (ALFGBG-447171), the Swedish Research Council (TM/SF: 2014-3031), and the Swedish Foundation for Strategic Research (TM: RB13-0204).
Author information
Authors and Affiliations
Contributions
M.J. performed computational data handling, A.M.M. analyzed the data, and R.M. prepared the samples. C.B., G.L., T.E., and P.K. helped with patient care, tissue sampling, and obtaining clinical information, while T.M. and P.K. formulated strategy and directed the research. S.F. designed the research, analyzed and interpreted the data, performed statistical analyses, and drafted the manuscript; all authors approved the final version of the paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Fransson, S., Martinez-Monleon, A., Johansson, M. et al. Whole-genome sequencing of recurrent neuroblastoma reveals somatic mutations that affect key players in cancer progression and telomere maintenance. Sci Rep 10, 22432 (2020). https://doi.org/10.1038/s41598-020-78370-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-78370-7
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
