
Abstract
Forkhead box F1 (FOXF1) is a lung embryonic mesenchyme-associated transcription factor that demonstrates persistent expression into adulthood in mesenchymal stromal cells. However, its biologic function in human adult lung-resident mesenchymal stromal cells (LR-MSCs) remain to be elucidated. Here, we demonstrate that FOXF1 expression acts as a restraint on the migratory function of LR-MSCs via its role as a novel transcriptional repressor of autocrine motility-stimulating factor Autotaxin (ATX). Fibrotic human LR-MSCs demonstrated lower expression of FOXF1 mRNA and protein, compared to non-fibrotic controls. RNAi-mediated FOXF1 silencing in LR-MSCs was associated with upregulation of key genes regulating proliferation, migration, and inflammatory responses and significantly higher migration were confirmed in FOXF1-silenced LR-MSCs by Boyden chamber. ATX is a secreted lysophospholipase D largely responsible for extracellular lysophosphatidic acid (LPA) production, and was among the top ten upregulated genes upon Affymetrix analysis. FOXF1-silenced LR-MSCs demonstrated increased ATX activity, while mFoxf1 overexpression diminished ATX expression and activity. The FOXF1 silencing-induced increase in LR-MSC migration was abrogated by genetic and pharmacologic targeting of ATX and LPA1 receptor. Chromatin immunoprecipitation analyses identified three putative FOXF1 binding sites in the 1.5 kb ATX promoter which demonstrated transcriptional repression of ATX expression. Together these findings identify FOXF1 as a novel transcriptional repressor of ATX and demonstrate that loss of FOXF1 promotes LR-MSC migration via the ATX/LPA/LPA1 signaling axis.
Similar content being viewed by others


Introduction
Mesenchymal cells are an important component of cellular niches in adult organs and are being increasingly recognized to display tissue-specific transcriptome and functions. We have previously demonstrated that human adult lung contains a population of resident, long-lived mesenchymal stromal cells (MSC) with clonal multi-lineage differentiation potential1. MSCs derived from adult lungs exhibit unique mesenchymal transcriptional signature suggesting their lung-specificity and origin from embryonic lung mesenchyme1,2. Expansion and mobilization of lung-resident mesenchymal stem cells (LR-MSCs) is noted during conditions of lung injury and fibrosis2,3, and the lipid mediator lysophosphatidic acid (LPA) has been identified as a key inducer of LR-MSC migration4. LR-MSCs can regulate LPA expression in an autocrine manner by secretion of Autotaxin (ATX), a lysophospholipase D that enzymatically produces LPA from extracellular lysophosphatidylcholine5. We have recently demonstrated that ATX expression is upregulated in LR-MSCs derived from diseased lungs and can drive β-catenin activation via downstream LPA/LPA1 signaling5. While these data shed light on mechanisms of MSC mobilization and activation, transcriptional regulation of MSCs under homeostatic conditions remain to be identified.
The Forkhead Box (Fox) family of transcription factors is a group of proteins that share a common DNA binding domain termed a winged-helix or forkhead domain, with FOXF1 being a mesenchyme-specific, putative transcription factor6,7, which plays a critical role in lung development. In mice, FOXF1 expression is noted in the lateral mesenchyme at embryonic day 9.5 and its deficiency is associated with defects in branching morphogenesis of the lung8,9. FOXF1 is unique among the embryonic lung mesenchyme-associated transcription factors in that it demonstrates persistent expression in the mesenchymal cells of an adult lung, and we have reported that LR-MSCs derived from human adult lungs express ~ 35,000-fold higher FOXF1 mRNA than bone-marrow MSCs5. However, the significance of FOXF1 expression in adult human LR-MSCs remains to be elucidated.
In this study, we investigated the mechanistic role of FOXF1 in regulating the biologic functions of lung-resident mesenchymal stem cells. Our investigations identify FOXF1 as a transcriptional repressor, with loss of FOXF1 promoting an activated mesenchymal phenotype accompanied by intense mitogenic activity, higher rates of cellular migration, proliferation, and the secretion of pro-inflammatory chemokines and cytokines. Importantly, our data demonstrates that FOXF1 regulates the migration of LR-MSCs via its novel role as a transcriptional repressor of ATX.
Materials and methods
Human subjects and ethics statement
Informed consent was obtained directly from all human subjects after a full explanation of the study objectives and procedures. The study was carried out in accordance with relevant guidelines and regulations using a protocol for human studies approved by the University of Michigan Institutional Review Board (approval number HUM00042443) and was in compliance with the Helsinki Declaration.
Isolation and culture conditions of LR-MSCs
LR-MSCs were isolated and characterized as previously described from bronchoalveolar lavage derived from lung transplant recipients with or without chronic lung allograft rejection1,2,3,4,5,10,11. LR-MSCs cultured in high-glucose DMEM (11965-118, Gibco) supplemented with 10% FBS, 100 U/ml penicillin/streptomycin, and 0.5% fungizone were utilized at passages 3–6. All methods were carried out in accordance with relevant guidelines and regulations.
RNA interference
At 60–70% confluence, LR-MSCs were transfected with 100 nM FOXF1-specific (Stealth RNAi HSS142031, Invitrogen) or scrambled control siRNA (sc-37007, Santa Cruz), using Oligofectamine (12252-011, Invitrogen) and Opti-MEM I reduced serum medium. For double gene silencing, LR-MSCs were transfected with FOXF1-specific siRNA or scrambled control (100 nM each). 24 h later, LR-MSCs were transfected again with 100 nM of ATX-specific siRNA or scrambled control, incubated overnight, and then maintained in serum-free DMEM. Cells were subsequently assayed for migration rates after 48 h. RNA and protein lysates were harvested after 72 h for real-time PCR and immunoblotting analysis.
Lentiviral transduction of LPAR1 short hairpin RNA
For lentivirus transduction, LR-MSCs were transfected with FOXF1-specific siRNA or scrambled control (100 nM each) and incubated for 48 h. Subsequently, the cells were infected with lentiviral vectors that contain either a control transduction particles-shRNA (Mission pLKO.1-puro, Sigma) or LPAR1-specific transduction particles-shRNA (Mission: Clone #: NM_057179, Sigma) in serum-free growth medium with 2.5 multiplicities of infection (MOI) using protamine sulphate as linker. After incubating for a period of 48 h, the cells were subsequently assayed for migration rates.
Affymetrix analysis
Total RNA extracted from LR-MSCs transfected with FOXF1-specific or scrambled control siRNA were used to synthesize cDNA, followed by Affymetrix GeneChip expression profiling (U133 Plus 2.0 Array) at the University of Michigan DNA Sequencing Core. The affy and limma packages of bioconductor implemented in the R statistical environment were used12,13,14. The GO analysis was performed using conditional hypergeometric tests from the GO stats package of Bioconductor15. A cutoff of p values < 0.0001 was utilized to determine GO terms that were relevant. The microarray data has been deposited in GEO under the Accession number GSE161903.
RNA isolation and quantitative PCR
Total RNA was isolated from LR-MSCs using the RNeasy mini kit (74104, Qiagen) and cDNA synthesized utilizing the High Capacity cDNA reverse transcription kit (4368814, Applied Biosystems). Real-time PCR for FOXF1, ATX and β-actin were conducted with probes Hs00230962_m1, Hs00905125_m1, and 4310881E (Applied Biosystems), respectively in 1× Taqman gene expression master mix (4369016, Applied Biosystems). The forward and reverse primer sequences for the genes analyzed using SYBR Green PCR Master Mix (4,309,155, Applied Biosystems) are listed in Table 1. Relative mRNA expression for target genes was calculated as 2 log− (ΔCt target mRNA − ΔCt β-actin).
Immunoblotting and autotaxin activity assay
Whole cell lysates of LR-MSCs were extracted as reported previously4. Western blot was performed using primary antibodies against FOXF1 (AF4798, R&D, 1:1000), ATX (10005375, Cayman Chemical, 1:100), PCNA (2586, Cell Signaling Technologies, 1:1000), Cyclin D1 (sc-718, Santa Cruz Biotechnology, 1:1000), phospho-Histone H3 Ser10 (PA5-17869, Thermo Fisher, 1:500) and GAPDH (MAB374, Millipore, 1:5000). HRP-conjugated secondary antibodies used in this study included A5420 (anti-goat, Sigma, 1:5,000), A8924 (anti-mouse, Sigma, 1:20,000) and A0545 (anti-rabbit, Sigma, 1:10,000), respectively. ATX activity in the conditioned media was measured with a fluorogenic ATX substrate FS-3 as previously described5.
Proliferation assay
LR-MSCs were transfected with FOXF1 siRNA or scrambled control (100 nM each), and 24 h post-transfection, the mesenchymal cells were trypsinized and seeded at 5000 cells/well in 96 well plates. Cells were cultured in medium for 72 h and assayed with a CyQUANT NF Cell Proliferation Assay Kit (C35006, ThermoFisher Scientific) as per manufacturer´s instructions.
Protein measurement in cell supernatant
Mesenchymal cells were transfected with FOXF1-specific or the scrambled siRNA in Opti-MEM I reduced serum medium. After overnight incubation, media was exchanged for serum-free DMEM for 48 h and the conditioned media was measured for CCL5 and CCL7 by ELISA according to the manufacturer’s protocols: R&D systems (Minneapolis, MN): Human CCL5/RANTES Quantikine ELISA Kit (DRN00B), Human CCL7/MCP-3 Quantikine ELISA Kit (DCC700). Absorbance was read with a SpectraMax M3 multi-mode microplate reader (Molecular Devices).
Migration assay
Migration rates of LR-MSCs was measured in matrigel-coated transwells as previously described4. Briefly, transwells were freshly coated with matrigel overnight at 37 °C. LR-MSCs were transfected with FOXF1-specific or the scrambled siRNA for three days, trypsinized, and re-suspended in serum-free DMEM. 1 × 105 cells were seeded into each upper chamber of the transwells, inserted into a 24-well plate containing DMEM supplemented with 5% FBS. Each condition was performed in triplicates. For migration assay involving PF-8380 treatment, LR-MSCs transfected with FOXF1-specific or scrambled siRNA were pre-treated with medium containing 1 µM PF-8380 (HY-13344, MedChem Express) for 30 min. They were then seeded into the transwell setup with 1 µM PF-8380 added to both the upper and lower chambers. 18 h later, LR-MSCs on the transwells were fixed and stained by Hema 3 staining kit (Fisher Scientific, Kalamazoo, MI). Finally, after the removal of the cells on the upper surface of the transwells using cotton swabs, the cells on the surface underneath were counted in five 200 × microscopic fields to quantify amount of cellular migration.
Murine Foxf1 overexpression
Mouse Foxf1 was overexpressed in LR-MSCs by transfection of pShuttle A-CMV-mFoxf1 utilizing Lipofectamine transfection reagent in Opti-MEM I reduced serum medium. The mFoxf1 overexpression plasmids were a generous gift by Dr. Vladimir V. Kalinichenko, MD, PhD, Cincinnati Children’s Hospital Medical Center, OH. It should be noted that the human FOXF1 (NCBI: NP_001442, 379 amino acid) is 94% homologous to murine FOXF1a (NCBI: NP_034556.1, 353 amino acid)16. Dr. Kalinichenko’s lab analyzed human and mouse FOXF1 sequences and identified two highly conserved homologous regions containing potential transcription factor binding sites for the following families: basic leucine zipper CCAAT/enhancer binding protein β, winged helix/Forkhead Box A, zinc finger GATA, homeodomain Nkx2.5, and cut-homeodomain HNF-6 transcription factors17. Since Dr. Kalinichenko’s lab has well-documented evidence of the efficacy of this plasmid16,18,19, we have chosen to utilize this plasmid to overexpress FOXF1 in our LR-MSCs. Empty pShuttle A was used as control. Transfection efficacy was confirmed by immunoblotting techniques.
Luciferase reporter assay
To examine the transcriptional functionality of 3 potential binding sites of FOXF1 in ATX promoter, 1.5 kb ATX promoter plus 150 bp downstream of the transcriptional start site was cloned into pGAL4.23 vector to drive luc2 luciferase gene expression. Three truncated versions of the ATX promoter (pATX1, pATX2 and pATX3) lacking one, two or all three potential FOXF1 binding sites were generated to drive luc2 luciferase gene expression in pGAL4.23. Plasmids were co-transfected with Renilla luciferase reporter pRL-TK (E224A, Promega) in LR-MSCs, and the luciferase activity was assayed 24 h post-transfection using Dual-Luciferase Reporter Assay System kit (E1960, Promega) by a Promega GloMax Explorer System Multimode Reader. The luc2 luciferase activity was normalized to the control Renilla luciferase activity to represent the transcriptional activity of the promoters.
Chromatin immunoprecipitation (ChIP) assay
ChIP assay was performed utilizing EZ-ChIP kit (17–371, Millipore) according to the manufacturer´s protocol. Briefly, LR-MSCs cultured in three 10 cm dishes at 80% confluence were cross-linked by 1% formaldehyde (10 min × RT), then quenched with 1 mM Glycine. Cells were pelleted and resuspended in SDS lysis buffer, and DNA sheared by sonication to reach ~ 200–1000 base pairs in length. Total fragmented DNA aliquots were incubated with anti-FOXF1 antibody (AF4798, R&D) or goat IgG (I5256, Sigma) as the background binding control at 4 °C overnight. Samples were subsequently incubated with Protein G agarose (4 °C × 1 h) followed by elution of protein/DNA complexes and heated at 65 °C × 5 h to reverse cross-linked complexes. Finally, DNA were purified and subjected to real-time PCR analysis using the following primers to detect binding at putative sites: CHIPSite1F, ACTAGATTCTAAGAATCTGTAATGAA; CHIPSite1R, GTAACAGAGTAGTGGCTCT; CHIPSite2F, AGAGCCACTACTCTGTTAC; CHIPSite2R, TTTTTGGCCTCTTCCTCAGCA; CHIPSite3F, ATGTGATACTAGGGACAGG; CHIPSite3R, AATGGCGTCAACCTCACCA.
Statistical analysis
All data are presented as Means ± SEM. Statistical significance was assessed with Student’s paired two-tailed t test for comparing scrambled and FOXF1-silenced groups, or with one-way ANOVA and a post hoc Bonferroni test for 3 or more groups, unless specified otherwise using GraphPad Prism 8 software (La Jolla, CA). p < 0.05 was considered significant.
Results
FOXF1 as a transcriptional regulator of key functional pathways in LR-MSCs
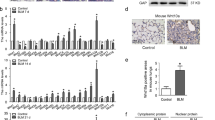
We have previously demonstrated that lung mesenchyme associated embryonic transcription factor Foxf1 is uniquely expressed in human lung allograft-derived MSCs. Comparison of the MSCs derived from patients with and without chronic allograft rejection demonstrated lower expression of FOXF1 transcripts (Fig. 1A) and protein (Fig. 1B) in fibrotic mesenchymal cells. In order to gain insight into the functional role of FOXF1 in human LR-MSCs, we used RNAi-mediated FOXF1 silencing and Affymetrix whole transcriptome array analyses. LR-MSCs silenced for FOXF1 demonstrated a 55% decrease in FOXF1 mRNA expression (Fig. 1C) and a concomitant downregulation in FOXF1 protein (Fig. 1D). Affymetrix analyses of the RNA isolated from LR-MSCs transfected with FOXF1-specific or scrambled siRNA revealed that 98 probe sets had a fold change ≥ 1.5 and an adjusted p-value below 0.01. Of these probesets, 69 genes were upregulated and 29 genes were downregulated as a result of FOXF1 silencing, thus suggesting that FOXF1 predominantly functions as a transcriptional repressor in LR-MSCs (Fig. 1E). We further filtered the dataset and focused on genes with a fold change above 2 or below − 2, and an adjusted p value below 0.05. This process revealed the top 35 differentially expressed genes, which were subsequently used to construct a gene–gene interaction network (STRING) to predict associations due to FOXF1-silencing (Fig. 1F). Gene ontology (GO) analysis was performed using conditional hypergeometric tests from the GO stats package of bioconductor15,20. Top ten GO terms in biological processes were widely modulated by FOXF1 silencing including proliferation, inflammatory responses, and migration (Table 2). Proliferation/cell cycle GO analyses revealed predominant upregulation of genes involved in positive regulation of proliferation and the downregulation of genes implicated in cell cycle arrest (Fig. 1G). Loss of FOXF1 in LR-MSCs induced an inflammatory response in these cells with upregulation of 34 of 44 genes associated with the inflammatory response GO term (GO:0006954) (Fig. 1H). A similar trend was noted with differentially expressed genes in positive regulation of cell proliferation and regulation of cell migration (GO:0008284 and GO:0030334, respectively) (Fig. 1I). Taken together, these data suggest a role for FOXF1 in fundamental cellular process of LR-MSCs including proliferation, cell cycle progression, inflammation, and migration.

Loss of FOXF1 promotes fundamental cellular processes in LR-MSCs. (A) mRNA was isolated from fibrotic and non-fibrotic LR-MSCs derived from bronchoalveolar lavage fluid of transplant patients, and FOXF1 expression was analyzed by real-time PCR. Values: Means ± SEM; n = 9 (non-Fib-MSCs); n = 8 (Fib-MSCs); **p < 0.0034. (B) Protein lysates from fibrotic and non-fibrotic LR-MSCs were analyzed for FOXF1 and GAPDH by western blotting. Graph shows densitometry analyses of these immunoblots. Values: Means ± SEM; n = 16; **p < 0.0086. (C) LR-MSCs were transfected with scrambled or FOXF1-specific siRNA and confirmed by real-time PCR. Values: Means ± SEM; n = 7; ***p < 0.0005. (D) Protein lysates from (A) were subjected to immunoblotting with antibodies against FOXF1 and GAPDH. (E–I) Gene regulation due to FOXF1 silencing was analyzed by Affymetrix gene array in 3 lines of LR-MSCs. Data reflects fold changes ≥ 1.5, and an adjusted p < 0.01. (E) Diagram showing the number of upregulated and downregulated genes. (F) Gene–gene interaction network (using STRING database) showing associations due to FOXF1-silencing. (G–I) Heatmaps showing two-fold Log changes are presented for positive regulation of cell cycle ((G) GO:0045787), inflammatory response ((H) GO:0006954), and regulation of cell migration ((I) GO:0030334). Note: Full length blots for Fig. 1B and Fig. 1D are provided in Supplementary Fig. S1 and S2.
Loss of FOXF1 induces migration and the expression and activity of ATX in human LR-MSCs
In order to ascertain the role of FOXF1 in cellular migration, a functional in vitro assay using a modified Boyden chamber was utilized. Comparision of LR-MSCs transfected with scrambled control or FOXF1-specific siRNA demonstrated an approximate 2.5-fold increase in cell migration following FOXF1-silencing, suggesting that the loss of FOXF1 imparts LR-MSCs with a robust migratory phenotype (Fig. 2A,B). ATX-LPA axis is key regulator of cellular migration and Autotaxin-encoding gene—ENPP2 was noted to be among the top ten upregulated genes in FOXF1-silenced cells (Fig. 1I). Increased ATX expression in response to FOXF1-silencing was confirmed at mRNA and protein level by real-time PCR and western blotting respectively (Fig. 2C,D). Furthermore, supernatant from FOXF1-silenced LR-MSCs demonstrated significantly higher ATX activity utilizing a fluorimetric substrate, FS-3, compared to scrambled siRNA controls (Fig. 2E). We next overexpressed mFoxf1 in LR-MSCs and assessed ATX protein levels and activity by immunoblotting and fluorimetry, respectively. Efficacy of mFoxf1 overexpression was confirmed by immunoblotting for HA as shown in Fig. 2F. A 40% decrease in ATX protein expression was noted in LR-MSCs overexpressing mFoxf1 (Fig. 2F), with a concordant decrease in ATX activity compared to control vector (Fig. 2G). Together these findings demonstrated that decreased FOXF1 leads to increased LR-MSC migration and ATX secretion and activity.

FOXF1 silencing increases migration rates, and the expression and activity of ATX in LR-MSCs. (A) LR-MSCs invasion was analyzed using matrigel-coated transwells with 8 μm pore size. (B) Quantification of data in (A). Values: Means ± SEM; n = 9; **p < 0.0045. (C,D) FOXF1 silencing upregulated the expression of ATX mRNA ((C); n = 6; ***p < 0.0003) and protein ((D); n = 11; **p < 0.0337) expressions. (E) The activity of ATX in the cell supernatant was assayed using the fluorogenic phospholipid substrate, FS-3. RFU: relative fluorescent unit, Values: Means ± SEM. n = 9, *p < 0.05. (F,G) DNA from pShuttle A-pCMV-HA-mFoxf1 was utilized to overexpress mFoxf1 in LR-MSCs, and the backbone vector pShuttle A was used as control. Immunoblotting analyses was utilized to confirm overexpression of FOXF1, and regulation of ATX and GAPDH ((F); n = 7; *p < 0.01). (G) ATX activity in cell supernatant is shown (n = 5; **p < 0.01). Values: Means ± SEM. Note: Full length blots for Fig. 2D and Fig. 2F are provided in Supplementary Fig. S3 and S4.
ATX/LPA/LPA1 signaling axis mediates increased migration rates in FOXF1-silenced LR-MSCs
To further ascertain if increased ATX expression mediates the increase in migration induced by loss of FOXF1, LR-MSCs transfected with FOXF1 siRNA were subjected to subsequent transfection with siRNA specific to ATX or scrambled control (Fig. 3A). Migration was compared between LR-MSCs silenced for FOXF1 alone or in combination with ATX silencing in the modified Boyden chamber migration assay (Fig. 3B). Increased migration noted in response to FOXF1-silencing was abrogated in LR-MSCs subjected to gene silencing for both FOXF1 and ENPP2 (ATX) (Fig. 3C). That ATX is a key factor in mediating the pro-migratory effect of FOXF1 inhibition was further confirmed by using PF8380, a specific pharmacologic inhibitor of ATX. FOXF1-silenced LR-MSCs treated with PF8380 demonstrated significant reduction in migration rates with levels comparable to scrambled control siRNA transfected LR-MSCs (Fig. 3D).

ATX-dependent cell migration in FOXF1-silenced LR-MSCs. (A) LR-MSCs were transfected with scrambled or FOXF1-specific siRNA. 24 h later, these LR-MSCs were transfected with scrambled or ATX-specific siRNA. Immunoblotting was performed to confirm RNAi-mediated FOXF1 and ATX silencing efficacy. n = 5 per group. (B) Migration assay was conducted in LR-MSCs transfected with scrambled or siRNA specific to FOXF1, ATX, or both FOXF1- and ATX-specific siRNA. Representative images of cell migration are shown. (C) Quantification of (B), n = 5, ***p < 0.0003. (D) LR-MSCs transfected with scrambled or FOXF1-specific siRNA were treated with the ATX inhibitor, PF-8380 (1 μM) and migration assay was performed. Values: Means ± SEM. n = 5, **p < 0.0142, *p < 0.0325. (E,F) Migration assays are shown with LR-MSCs transfected with scrambled or FOXF1-specific siRNA, and then treated with the LPA1 inihibitor, VPC12249 (1 μM) (E), or subjected to lentivirus-mediated shRNA interference against LPAR1 (F). Values: Means ± SEM. n = 5. ***p < 0.0002, ****p < 0.0001. Note: Full length blots for Fig. 3A are provided in Supplementary Fig. S5.
ATX regulates cellular migration via its generation of lipid mediator LPA and downstream LPA receptor signaling. We have previously shown that LR-MSCs predominantly express LPA receptor isoform 1 (LPA1) and that migration of LR-MSCs in response to LPA is mediated via LPA1 receptor ligation4. To study the pharmacologic blockade of LPA signaling on migration of FOXF1-silenced LR-MSCs, we utilized VPC12249, an LPA1-specific antagonist. FOXF1-silenced LR-MSCs demonstrated a two-fold higher migration, which was significantly diminished by the pharmacologic blockade of LPA1 (Fig. 3E). Furthermore, lentiviral gene silencing of the LPA receptor—LPAR1, using short hairpin RNA (shRNA), was adopted as a complementary approach to determine the effects of LPA signaling on migration. FOXF1-silenced LR-MSCs demonstrated 1.5-fold higher migration, which was significantly mitigated by shRNA-mediated lentiviral repression of LPAR1 gene expression (Fig. 3F). Together, these data demonstrate that loss of FOXF1 promotes LR-MSC migration via ATX/LPA/LPA1 signaling pathway.
Identification of novel FOXF1 binding sites in the − 1.5 kb upstream region of the ATX promoter (− 1217/− 1127/− 458)
Silencing of FOXF1 in LR-MSCs resulted in a robust induction of ATX at mRNA level, so we next sought to investigate whether FOXF1 directly binds to regions of the ATX promoter to influence transcription. Utilizing JASPAR promoter analysis (www.jaspar.genereg.net), we observed three putative FOXF1/2 binding sites (RTAAAYA)21 in the − 1.5 kb upstream region of ATX promoter (Fig. 4A). To study the function of these three potential binding sites, we constructed luc2 luciferase expressing vectors driven by full length or truncated ATX promoters on the pGAL4.23 backbone (Fig. 4B). These reporter constructs were co-transfected with control Renilla vector pRL-TK into LR-MSCs and the luciferase activity was measured. As shown in Fig. 4B, deletion of the two most upstream putative FOXF1 binding sites (sites 1 and 2) increased luciferase expression two and fourfold, respectively. Promoter truncation omitting all 3 putative FOXF1 binding sites resulted in a sixfold induction of ATX transcription, suggesting a role for all three binding sites in repressing ATX transcription. Next, we utilized chromatin immunoprecipitation (ChIP) analysis to investigate if FOXF1 can bind these putative sites in the ATX promoter. FOXF1 antibody was used to pull down the FOXF1/chromosome complexes with goat IgG as the negative control. ChIP data demonstrated an over 20-fold increase in FOXF1 binding at all three sites of the ATX promoter compared to IgG control (Fig. 4C). Collectively, these results suggest that FOXF1 transcriptionally represses expression of ATX by directly binding to regions of the ATX promoter.

FOXF1 inhibits ATX transcription and directly binds to ATX promoter. (A) Three potential FOXF1 binding sites exist in the 1.5 kb ATX promoter and their locations upstream the ATX transcription initiation site were marked. (B) Three versions of truncated ATX promoter were utilized to drive luciferase expression in LR-MSCs and corresponding luciferase activities were measured, n = 5, *p < 0.05, **p < 0.01. (C) ChIP assay was performed to detect the direct binding of FOXF1 with its three potential binding sites in the ATX promoter. Values: Means ± SEM. ****p < 0.0001.
FOXF1-silencing induces proliferation and inflammatory responses in LR-MSCs
We also further investigated other biological functions which were identified to be significantly altered by FOXF1 silencing by affymetrix analyses (Table 2). Quantitative assessment of cellular proliferation by CyQUANT NF cell proliferation assay demonstrated approximately 75% higher proliferation in FOXF1-silenced LR-MSCs compared to that of the scrambled siRNA control (Fig. 5A). Real-time PCR analyses confirmed upregulation of genes involved in cell cycle progression upon FOXF1 silencing, such as cyclin D1 (CCND1), cyclin B1 (CCNB1), cyclin-dependent kinase 1 (CDK1) and phosphoprotein enriched in astrocytes 15 (PEA15) (Fig. 5B). Additionally, FOXF1-silencing in LR-MSCs demonstrated upregulation of proteins marking proliferation and cell cycle progression such as proliferating cell nuclear antigen (PCNA), phosphorylated histone H3 (Ser 10) and cyclin D1 (Fig. 5C,D).

FOXF1 silencing promotes cellular proliferation and secretion of inflammatory mediators in LR-MSCs. (A) Proliferation rate was analyzed using the CyQUANT NF Cell Proliferation Assay. n = 5; values: Means ± SEM. *p < 0.05. (B) Real-time PCR analyses of specific genes detected in the Affymetrix array analysis. Values: Means ± SEM; n = 7; **p < 0.01. (C) Protein lysates with equal concentrations (~ 10 µg) from LR-MSCs transfected with FOXF1-specific or scrambled siRNA were subjected to immunoblotting against proliferation markers—anti-PCNA, anti- phosphorylated histone H3 (Ser 10), anti-cyclin D1 and anti-GAPDH (loading control). (D) Quantification of data in (C) is shown as fold change over scrambled control. Values: Means ± SEM; n = 4; *p < 0.05. (E) LR-MSCs were transfected with scrambled or FOXF1 siRNA and subjected to real-time PCR analyses of key cytokines. Values: Means ± SEM. n = 7, ***p < 0.001. (F) Secreted CCL5 and CCL7, in the conditioned media from (E) were measured by ELISA. Values: Means ± SEM. n = 4 (CCL5) and n = 8 (CCL7). *p < 0.05. (G) Real-time PCR analyses of PTGS2. Values: Means ± SEM. n = 7, **p < 0.01. Note: Full length blots for Fig. 5C are provided in Supplementary Fig. S6.
The gene expression pattern demonstrating increased pro-inflammatory cytokines noted in FOXF1-silenced LR-MSCs by Affymetrix analyses was also further confirmed by real-time PCR. An approximately 100-fold increase in the gene expression of CCL5 and tenfold increase in the gene expression of CCL7, and a 150-fold increase in the gene expressions of CXCL10 and CXCL11 was found in FOXF1-silenced LR-MSCs relative to scrambled siRNA control (Fig. 5E). FOXF1-silencing induced increase in cytokine secretion by LR-MSCs was documented by ELISA where higher levels of CCL5 and CCL7 were noted in conditioned media collected from FOXF1-silenced LR-MSCs compared to the respective scrambled controls (Fig. 5F). Real-time PCR also confirmed that loss of FOXF1 induced expression of Prostaglandin-Endoperoxide Synthase 2 (PTGS2 or COX2) a key enzyme in prostaglandin biosynthesis (Fig. 5G).
Discussion
Mesenchymal cells are a critical component of cellular niches in all organs and play a key role in the pathogenesis of fibrotic diseases22. However, transcriptional networks and signaling mechanisms involved in regulating mesenchymal progenitor cells in homeostatic conditions are not well identified. Here, we identify a role for transcription factor forkhead protein FOXF1 as a master repressor of key cellular functions in human LR-MSCs. FOXF1 silencing was noted to promote proliferation, migration, and secretory function of LR-MSCs. Furthermore, FOXF1 was identified as a novel transcriptional repressor of ATX, a key enzyme largely responsible for the synthesis of extracellular pro-fibrotic mediator, LPA. Increased ATX secretion followed by subsequent LPA synthesis and autocrine LPA1 signaling, mediated LR-MSC migration in response to decreased FOXF1 expression. Together, these data shed light on novel restraining mechanisms in mesenchymal cells which limit their activation in homeostatic conditions. These findings have significant relevance to understanding both adaptive and mal-adaptive reparative processes in the lung.
Our studies provide first evidence for the role of FOXF1 as a transcriptional repressor of key enzyme ATX in human LR-MSCs. ATX, a secreted glycoprotein from the family of ectonucleotide pyrophosphatases/phosphodiesterases, is essential for development and is implicated in a variety of physiologic and pathologic processes23. ATX produces majority of the extracellular LPA and the ATX/LPA/LPA1 signaling axis has been shown to play a key role in fibrosis, inflammation, and cancer across various organs5,24,25,26,27,28,29,30,31. ATX-LPA signaling is implicated in fibrotic diseases of the lung5,30,32, and we have demonstrated stable increased expression of ATX in mesenchymal cells derived from fibrotic lung allografts5. In these studies, ATX mRNA expression was noted to be regulated by nuclear factor of activated T cells 2 (NFAT1). NFAT1 is a known enhancer of ATX transcription with NFAT binding sites described in the ATX promoter region in breast cancer cells33. Other transcription factors such as HOXA13, v-JUN, NF-κB and Stat3 have also been identified as transcriptional activators of ATX in various murine and human cellular conditions33,34,35,36, however, no ATX repressor has been reported to date. ATX as a target of FOXF1 was identified by global affymetrix analysis where ENPP2 was among the top differentially expressed genes in FOXF1-silenced LR-MSCs. We utilized both FOXF1 silencing and overexpression strategies to confirm regulation of ATX by FOXF1 in LR-MSCs. Silencing of FOXF1 resulted in robust increases in ATX at the transcriptional level as well as increased ATX expression and function—as indicated by increased ATX mRNA, protein, and activity. FOXF1 overexpression was associated with reduced ATX expression at both the RNA and protein level. We identified, previously uncharacterized, three putative FOXF1 binding sites on the ATX promoter. That FOXF1 binds to and is a repressor of the ATX gene ENPP2 was confirmed by its direct binding to the ATX promoter using ChIP analysis. Increases in ATX transcription was noted in luciferase assays with subsequent promoter truncations. Future studies will focus on identifying the exact binding site. Previous studies in NIH3T3 cells have identified FOXF1 as a repressor of the CDH11 gene37 and other members of the FOX family such as FOXP1 and FOXP2 which are expressed in the lung epithelium have also been characterized as transcriptional repressors38.
A key finding of our work is recognition of the role of transcriptional factor FOXF1 as a inhibitory regulator of LR-MSC migration. Downregulation of FOXF1 resulted in a robust migratory phenotype in LR-MSCs which was found to be dependent on ATX secretion and downstream LPA/LPA1 signaling. Mesenchymal cell migration is a key feature of its activated state and its positive regulation by growth factors and biological mediators is well studied in context of tissue repair and fibrosis. However, the fundamental question of what prevents activation of mesenchymal cell migration in a quiescent condition has not been previously explored. Our data demonstrating FOXF1 as a transcriptional repressor of ATX and its loss promoting ATX/LPA/LPA1 signaling axis mediated migration suggests that FOXF1 expression could be critical brake on cellular migration in homeostatic conditions by keeping autocrine ATX expression in check. Loss of FOXF1 has been linked to increased invasiness of hepatocellular cancer cells39. FOXF1 has also been identified as a target of p53 in a separate study of human cancer cell lines, with its ectopic expression inhibiting cancer cell invasion and migration and its inactivation of FOXF1 stimulating cell invasion and migration40.
MSCs are key components of cellular niches, and regulate biologic processes via their paracrine actions and locally generated ATX has been demonstrated to be important in cellular interactions within tissue microenvironment41. Further evidence for the role of FOXF1 in regulating the secretome of the LR-MSCs was provided by affymetrix analysis where a significant change in the cytokine transcriptome was noted with marked upregulation of key chemokines such as CCL5, CCL7, CXCL10 and CXCL11. PTGS2, the enzyme that regulates prostanoid synthesis, was also significantly upregulated in FOXF1-silenced LR-MSCs. This suggests that FOXF1 regulates multiple downstream pathways in human LR-MSCs, the mechanism of which remains to be elucidated. Future studies are needed to identify other transcriptional targets of FOXF1 in LR-MSCs.
Among top upregulated biological processes identified in FOXF1-silenced LR-MSCs by GO analysis were positive regulation of cell proliferation. Mesenchymal cells within the lungs have relatively low turnover42, but our previous longitudinal studies of human lung allografts have provided clues regarding conditions associated with LR-MSC proliferation and mobilization3. An increase in LR-MSC numbers were noted early post-transplant during an active repair phase and later post-transplant preceding development of allograft fibrosis3. Both these conditions are marked by significant epithelial injury and FOXF1 plays a key role in mesenchymal-epithelial interactions during lung development43. FOXF1 is a Shh target gene and loss of Shh signaling has been implicated in mesenchymal cell proliferation in murine models44,45. Our finding that loss of FOXF1 promotes cellular proliferation suggests that FOXF1 could be a key intermediary for the actions of Shh. That loss of FOXF1 can promote mesenchymal cell activation and contribute to fibrosis is suggested in studies of transgenic mice with myofibroblast-specific deletion of Foxf1, where worse fibrotic remodeling was noted in response to bleomycin37. Further investigations are needed to shed more light on the regulation of this novel regulatory mechanism of mesenchymal cell activation in normal reparative and aberrant fibrotic responses within tissue niches in a human lung.
In conclusion, our study elucidates a critical mechanistic role of transcription factor FOXF1 that acts as a master regulator of cellular functions and paracrine actions of resident MSCs in human adult lungs. Furthermore, these studies are novel in their elucidation of the first transcriptional repressor of ATX in any cell type, a finding that has significant implication across various organs and diseases.
Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Abbreviations
- FOXF1:
-
Forkhead box isoform 1
- ATX:
-
Autotaxin
- ENPP2 :
-
Ectonucleotide pyrophosphatase/phosphodiesterase 2 (encodes for ATX)
- LPA:
-
Lysophosphatidic acid
- LPA1:
-
LPA receptor isoform 1
- LR-MSCs:
-
Lung-resident mesenchymal cells
- mFoxf1 :
-
Murine FOXF1 overexpressing vector
- MOI:
-
Multiplicities of infection
- DMEM:
-
Dulbecco’s minimum essential medium
- FBS:
-
Fetal bovine serum
- PCNA:
-
Proliferating cell nuclear antigen
- HRP:
-
Horseradish peroxidase
- ChIP:
-
Chromatin immunoprecipitation
- CCL5/RANTES:
-
Chemokine ligand 5
- CCL7/MCP-3:
-
Chemokine ligand 7
- STRING:
-
Search tool for the retrieval of interacting genes/proteins
- GO:
-
Gene ontology
- CCND1:
-
Cyclin D1
- CCNB1:
-
Cyclin B1
- CDK1:
-
Cyclin dependent kinase 1
- PEA15:
-
Phosphoprotein enriched in astrocytes 15
- PTGS2:
-
Prostaglandin-endoperoxide synthase 2
- COX2:
-
Cyclooxygenase isoform 2
- PGE2:
-
Prostaglandin E2
- NFAT1:
-
Nuclear factor of activated T cells 2
- HOXA13:
-
Homeobox A13
- CDH11:
-
Cadherin 11 or osteoblast-cadherin
- CXCL10:
-
C-X-C Motif chemokine ligand 10
- CXCL11:
-
C-X-C Motif chemokine ligand 11
References
Lama, V. N. et al. Evidence for tissue-resident mesenchymal stem cells in human adult lung from studies of transplanted allografts. J. Clin. Investig. 117, 989–996. https://doi.org/10.1172/JCI29713 (2007).
Walker, N. et al. Resident tissue-specific mesenchymal progenitor cells contribute to fibrogenesis in human lung allografts. Am. J. Pathol. 178, 2461–2469. https://doi.org/10.1016/j.ajpath.2011.01.058 (2011).
Badri, L. et al. Mesenchymal stromal cells in bronchoalveolar lavage as predictors of bronchiolitis obliterans syndrome. Am. J. Respir. Crit. Care Med. 183, 1062–1070. https://doi.org/10.1164/rccm.201005-0742OC (2011).
Badri, L. & Lama, V. N. Lysophosphatidic acid induces migration of human lung-resident mesenchymal stem cells through the beta-catenin pathway. Stem Cells 30, 2010–2019. https://doi.org/10.1002/stem.1171 (2012).
Cao, P. et al. Autocrine lysophosphatidic acid signaling activates beta-catenin and promotes lung allograft fibrosis. J. Clin. Investig. 127, 1517–1530. https://doi.org/10.1172/JCI88896 (2017).
Mahlapuu, M., Pelto-Huikko, M., Aitola, M., Enerback, S. & Carlsson, P. FREAC-1 contains a cell-type-specific transcriptional activation domain and is expressed in epithelial-mesenchymal interfaces. Dev. Biol. 202, 183–195. https://doi.org/10.1006/dbio.1998.9010 (1998).
Hellqvist, M., Mahlapuu, M., Samuelsson, L., Enerback, S. & Carlsson, P. Differential activation of lung-specific genes by two forkhead proteins, FREAC-1 and FREAC-2. J. Biol. Chem. 271, 4482–4490 (1996).
Costa, R. H., Kalinichenko, V. V. & Lim, L. Transcription factors in mouse lung development and function. Am. J. Physiol. Lung Cell. Mol. Physiol. 280, L823-838 (2001).
Hoggatt, A. M. et al. The transcription factor Foxf1 binds to serum response factor and myocardin to regulate gene transcription in visceral smooth muscle cells. J. Biol. Chem. 288, 28477–28487. https://doi.org/10.1074/jbc.M113.478974 (2013).
Jarvinen, L. et al. Lung resident mesenchymal stem cells isolated from human lung allografts inhibit T cell proliferation via a soluble mediator. J. Immunol. 181, 4389–4396. https://doi.org/10.4049/jimmunol.181.6.4389 (2008).
Walker, N. M. et al. Prostaglandin E2 as an inhibitory modulator of fibrogenesis in human lung allografts. Am. J. Respir. Crit. Care Med. 185, 77–84. https://doi.org/10.1164/rccm.201105-0834OC (2012).
Smyth, G. K. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat. Appl. Genet. Mol. Biol. 3, 3. https://doi.org/10.2202/1544-6115.1027 (2004).
Ritchie, M. E. et al. Empirical array quality weights in the analysis of microarray data. BMC Bioinform. 7, 261. https://doi.org/10.1186/1471-2105-7-261 (2006).
Benjamini, Y., Drai, D., Elmer, G., Kafkafi, N. & Golani, I. Controlling the false discovery rate in behavior genetics research. Behav. Brain Res. 125, 279–284 (2001).
Falcon, S. & Gentleman, R. Using gostats to test gene lists for GO term association. Bioinformatics 23, 257–258. https://doi.org/10.1093/bioinformatics/btl567 (2007).
Pradhan, A., Ustiyan, V., Zhang, Y., Kalin, T. V. & Kalinichenko, V. V. Forkhead transcription factor FoxF1 interacts with Fanconi anemia protein complexes to promote DNA damage response. Oncotarget 7, 1912–1926. https://doi.org/10.18632/oncotarget.6422 (2016).
Fulford, L. et al. The transcription factor FOXF1 promotes prostate cancer by stimulating the mitogen-activated protein kinase ERK5. Sci. Signal 9, 48. https://doi.org/10.1126/scisignal.aad5582 (2016).
Milewski, D. et al. FoxF1 and FoxF2 transcription factors synergistically promote rhabdomyosarcoma carcinogenesis by repressing transcription of p21(Cip1) CDK inhibitor. Oncogene 36, 850–862. https://doi.org/10.1038/onc.2016.254 (2017).
Flood, H. M. et al. The Forkhead box F1 transcription factor inhibits collagen deposition and accumulation of myofibroblasts during liver fibrosis. Biol. Open https://doi.org/10.1242/bio.039800 (2019).
Alexa, A., Rahnenfuhrer, J. & Lengauer, T. Improved scoring of functional groups from gene expression data by decorrelating GO graph structure. Bioinformatics 22, 1600–1607. https://doi.org/10.1093/bioinformatics/btl140 (2006).
Pierrou, S., Hellqvist, M., Samuelsson, L., Enerback, S. & Carlsson, P. Cloning and characterization of seven human forkhead proteins: binding site specificity and DNA bending. EMBO J. 13, 5002–5012 (1994).
El Agha, E. et al. Mesenchymal stem cells in fibrotic disease. Cell Stem Cell 21, 166–177. https://doi.org/10.1016/j.stem.2017.07.011 (2017).
Perrakis, A. & Moolenaar, W. H. Autotaxin: structure-function and signaling. J. Lipid Res. 55, 1010–1018. https://doi.org/10.1194/jlr.R046391 (2014).
Knowlden, S. & Georas, S. N. The autotaxin-LPA axis emerges as a novel regulator of lymphocyte homing and inflammation. J. Immunol. 192, 851–857. https://doi.org/10.4049/jimmunol.1302831 (2014).
Liu, S., Murph, M., Panupinthu, N. & Mills, G. B. ATX-LPA receptor axis in inflammation and cancer. Cell Cycle 8, 3695–3701. https://doi.org/10.4161/cc.8.22.9937 (2009).
Nishioka, T. et al. ATX-LPA1 axis contributes to proliferation of chondrocytes by regulating fibronectin assembly leading to proper cartilage formation. Sci. Rep. 6, 23433. https://doi.org/10.1038/srep23433 (2016).
Leblanc, R. et al. Interaction of platelet-derived autotaxin with tumor integrin alphaVbeta3 controls metastasis of breast cancer cells to bone. Blood 124, 3141–3150. https://doi.org/10.1182/blood-2014-04-568683 (2014).
Erstad, D. J., Tager, A. M., Hoshida, Y. & Fuchs, B. C. The autotaxin-lysophosphatidic acid pathway emerges as a therapeutic target to prevent liver cancer. Mol. Cell. Oncol. 4, e1311827. https://doi.org/10.1080/23723556.2017.1311827 (2017).
Sakai, N. et al. The involvement of autotaxin in renal interstitial fibrosis through regulation of fibroblast functions and induction of vascular leakage. Sci. Rep. 9, 7414. https://doi.org/10.1038/s41598-019-43576-x (2019).
Ninou, I., Magkrioti, C. & Aidinis, V. Autotaxin in pathophysiology and pulmonary fibrosis. Front. Med. (Lausanne) 5, 180. https://doi.org/10.3389/fmed.2018.00180 (2018).
Valdes-Rives, S. A. & Gonzalez-Arenas, A. Autotaxin-lysophosphatidic acid: from inflammation to cancer development. Mediat. Inflamm. 2017, 9173090. https://doi.org/10.1155/2017/9173090 (2017).
Oikonomou, N. et al. Pulmonary autotaxin expression contributes to the pathogenesis of pulmonary fibrosis. Am. J. Respir. Cell Mol. Biol. 47, 566–574. https://doi.org/10.1165/rcmb.2012-0004OC (2012).
Chen, M. & O’Connor, K. L. Integrin alpha6beta4 promotes expression of autotaxin/ENPP2 autocrine motility factor in breast carcinoma cells. Oncogene 24, 5125–5130. https://doi.org/10.1038/sj.onc.1208729 (2005).
Black, E. J., Clair, T., Delrow, J., Neiman, P. & Gillespie, D. A. Microarray analysis identifies Autotaxin, a tumour cell motility and angiogenic factor with lysophospholipase D activity, as a specific target of cell transformation by v-Jun. Oncogene 23, 2357–2366. https://doi.org/10.1038/sj.onc.1207377 (2004).
McCabe, C. D. & Innis, J. W. A genomic approach to the identification and characterization of HOXA13 functional binding elements. Nucleic Acids Res. 33, 6782–6794. https://doi.org/10.1093/nar/gki979 (2005).
Williams, T. M. et al. Candidate downstream regulated genes of HOX group 13 transcription factors with and without monomeric DNA binding capability. Dev. Biol. 279, 462–480. https://doi.org/10.1016/j.ydbio.2004.12.015 (2005).
Black, M. et al. FOXF1 inhibits pulmonary fibrosis by preventing CDH2-CDH11 cadherin switch in myofibroblasts. Cell Rep. 23, 442–458. https://doi.org/10.1016/j.celrep.2018.03.067 (2018).
Shu, W., Yang, H., Zhang, L., Lu, M. M. & Morrisey, E. E. Characterization of a new subfamily of winged-helix/forkhead (Fox) genes that are expressed in the lung and act as transcriptional repressors. J. Biol. Chem. 276, 27488–27497. https://doi.org/10.1074/jbc.M100636200 (2001).
Zhao, Z. G., Wang, D. Q., Hu, D. F., Li, Y. S. & Liu, S. H. Decreased FOXF1 promotes hepatocellular carcinoma tumorigenesis, invasion, and stemness and is associated with poor clinical outcome. Onco Targets Ther. 9, 1743–1752. https://doi.org/10.2147/OTT.S95002 (2016).
Tamura, M. et al. Forkhead transcription factor FOXF1 is a novel target gene of the p53 family and regulates cancer cell migration and invasiveness. Oncogene 33, 4837–4846. https://doi.org/10.1038/onc.2013.427 (2014).
Nakasaki, T. et al. Involvement of the lysophosphatidic acid-generating enzyme autotaxin in lymphocyte-endothelial cell interactions. Am. J. Pathol. 173, 1566–1576. https://doi.org/10.2353/ajpath.2008.071153 (2008).
Hogan, B. L. et al. Repair and regeneration of the respiratory system: complexity, plasticity, and mechanisms of lung stem cell function. Cell Stem Cell 15, 123–138. https://doi.org/10.1016/j.stem.2014.07.012 (2014).
Mahlapuu, M., Enerback, S. & Carlsson, P. Haploinsufficiency of the forkhead gene Foxf1, a target for sonic hedgehog signaling, causes lung and foregut malformations. Development 128, 2397–2406 (2001).
Herriges, M. J. et al. Long noncoding RNAs are spatially correlated with transcription factors and regulate lung development. Genes Dev. 28, 1363–1379. https://doi.org/10.1101/gad.238782.114 (2014).
Bohnenpoll, T. et al. A SHH-FOXF1-BMP4 signaling axis regulating growth and differentiation of epithelial and mesenchymal tissues in ureter development. PLoS Genet. 13, e1006951. https://doi.org/10.1371/journal.pgen.1006951 (2017).
Acknowledgements
This work was supported by National Institutes of Health Grants R01 HL118017 and R01 HL094622 (VNL), and Cystic Fibrosis Foundation Grant LAMA16XX0 (VNL).
Author information
Authors and Affiliations
Contributions
Conceptualization and experiment design: P.C., N.M.W. and V.N.L.; Data acquisition, analysis, and interpretation: P.C., N.M.W., R.B., S.M.-P., Y.A., K.M., D.S.W., R.V. and V.N.L.; Drafting of the manuscript: R.V., P.C., N.M.W. and V.N.L.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Cao, P., Walker, N.M., Braeuer, R.R. et al. Loss of FOXF1 expression promotes human lung-resident mesenchymal stromal cell migration via ATX/LPA/LPA1 signaling axis. Sci Rep 10, 21231 (2020). https://doi.org/10.1038/s41598-020-77601-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-77601-1
This article is cited by
-
A combined opposite targeting of p110δ PI3K and RhoA abrogates skin cancer
Communications Biology (2024)
-
Gene expression profiles in mesenchymal stromal cells from bone marrow, adipose tissue and lung tissue of COPD patients and controls
Respiratory Research (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
