
Abstract
Desalted Salicornia europaea L. (SE) inhibits acetylcholine esterase, attenuates oxidative stress and inflammatory cytokines, and activates neurotrophic pathway. We performed 12-week, randomized, double-blind, placebo-controlled study to evaluate the efficacy of PhytoMeal(a desalted SE)-ethanol extract (PM-EE), in improving the cognitive performance in patients with subjective memory impairment. 63 participants complaining memory dysfunction without dementia (Korean Mini-Mental State Examination [K-MMSE] score ≥ 23) were assigned to PM-EE 600 mg/day or placebo. The cognitive domain of the Alzheimer's disease assessment scale-Korean version (ADAS-K) was set as the primary outcome. After 12 weeks, there was no differences in the changes in the primary outcome or the frequency of adverse events between the groups. In the subgroup analysis for the 30 subjects with mild cognitive impairment (MCI, baseline K-MMSE scores ≤ 28), PM-EE significantly improved the color-reading score of the Korean color-word stroop test (8.2 ± 25.0 vs. − 4.7 ± 13.2, P = 0.018). Our findings suggest that PM-EE is safe but might not be effective in this setting of this study. However, PM-EE may improve the frontal executive function in the patients with MCI. Further large-sized studies with longer follow-up period is warranted (trial registration number KCT0003418).
Similar content being viewed by others
Introduction
Dementia is a very prevalent geriatric disorder with a substantial clinical impact1. Due to the increasing life expectancy, the global incidence and the socioeconomic burden of dementia is rapidly expanding2. Although acetylcholine esterase (AchE) inhibitors such as donepezil and galantamine and N-methyl-D-aspartate receptor blockers such as memantine have been established to be effective in improving the symptoms of dementia, they have not been shown to change the long-term outcome of the disease3,4,5,6. Recent studies have demonstrated that pathological, functional, and structural changes in the brain begin to develop as early as twenty years before the clinical manifestation of dementia becomes evident7. Thus, the early recognition and management of the disease is increasingly being recognized to be crucial to improve the clinical course of dementia8,9. In this regard, the clinical attention of the pre-dementia disease stages such as mild cognitive impairment (MCI), a status with objective deficit in cognitive function but without impairment of daily activities10,11, or subjective memory impairment (SMI), the pre-MCI status that presents with a subjective complaint of memory dysfunction with no clinical evidence of deficit in memory or other cognitive functions18, are rapidly increasing. Numerous investigations evaluating the candidate medications to treat MCI or SMI have been performed, but no drug has been established as effective12,13,14,15,16,17.
The two major types of dementia, Alzheimer’s disease (AD) and vascular dementia (VD), share common disease pathophysiology consisting of oxidative stress, chronic neuronal inflammation, and dysfunction in the cholinergic system in the brain19,20,21,22,23. Therefore, a broad and robust mechanism of action that widely covers multiple pathological processes while maintaining sufficient safety might be required for a new therapeutic candidate.
Salicornia europaea L. (SE) is a halophytic plant species that has recently been recognized for its substantial neuroprotective effects24,25,26. Recently, our study group demonstrated an anti-amnesic effect of the desalted and enzyme-digested SE ethanol extract (SE-EE) using a scopolamine-administered amnestic mouse model27. A subsequent study also isolated and identified Acanthoside B from SE-EE as a potential candidate substance and reported that Acanthoside B improved cognitive function in the amnestic mouse model with negligible toxicity28. We also demonstrated that Acanthoside B regulates cholinergic function by enhancing AchE inhibitory activity, attenuates oxidative stress and inflammatory cytokines, and activates the neurotrophic tropomyosin receptor kinase B/cAMP response element binding/brain-derived neurotrophic factor (TrkB/CREB/BDNF) pathway28, which are all processes involved in the key pathomechanism of both AD and VD.
In this study, we hypothesized that the use of PhytoMeal(a desalted SE)-ethanol extract (PM-EE), would be safe and more effective than placebo in improving cognitive performance in patients who are complaining memory impairment but without dementia and performed a randomized placebo controlled clinical trial to evaluate the efficacy and safety of PM-EE.
Materials and methods
Study subjects
In this study, we recruited patients who (1) complained subjective feeling of decrement in memory function and visited the outpatient clinic of the Neurology Department of the Seoul National University Hospital (SNUH), (2) were aged between 50 and 85 years, and (3) were without dementia according to the baseline Mini-Mental State Examination-Korean version (K-MMSE) scores (scores of 23 or higher). We excluded patients who (1) had been admitted to hospital within 3 months of the inclusion time with a diagnosis including malignant neoplasm, acute intracerebral haemorrhage, acute cerebral infarction, acute myocardial infarction, unstable angina, or arrhythmia with congestive heart failure; (2) had a current or past history of schizophrenia or major depressive disorder; (3) had a chronic brain disease that affects cognitive function, such as cerebral infarction, dementia, or parkinsonism; (4) had taken medications such as antipsychotics, tricyclic antidepressants, or other neuroprotective agents within 4 weeks of the time of inclusion; (5) were currently taking vitamin E with a dosage higher than 400 international units per day; (6) had undergone estrogen replacement therapy (excluding topical estrogen) within 2 months of the time of inclusion (due to the potential protective effect of estrogen on the cognitive function29); (7) had ingested any food products known to be related to cognitive function within 2 weeks of the time of inclusion; (8) were alcohol abusers or dependent on alcohol; (9) had laboratory abnormalities such as a thyroid stimulating hormone level of ≤ 0.1 uIU/ml or ≥ 10 uIU/ml, serum creatinine level ≥ 2 times higher than the upper normal limit, or serum aspartate aminotransferase or alanine aminotransferse levels ≥ 3 times higher than the upper normal limit; (10) had uncontrolled hypertension with resting blood pressure higher than 160/110 mmHg; (11) had uncontrolled diabetes with a fasting blood glucose level of ≥ 180 mg/dl; or (12) had participated in other clinical trials within 3 months of the time of inclusion or were planning to participate in other clinical trials while participating in the current study. This study was registered at Clinical Research Information Service; Korea Centers for Disease Control and Prevention, Ministry of Health and Welfare, Republic of Korea (KCT0003418, date of registration:10/01/2019). This study protocol and supporting documentation were approved by Institutional Review Board (IRB) of SNUH (H-1711-092-901) and the study was performed in compliance with the SNUH IRB regulations and the International Conference on Harmonisation guideline for Good Clinical Practice. Written informed consent to participate was obtained from all enrolled patients.
Study design and procedures
The current study was designed as a randomized, double-blind, placebo-controlled experiment. Due to the lack of previous clinical study data that evaluated the effect of PM-EE on cognition, we used a study data that evaluated the effect of using Panax Ginseng for 12 weeks on ADAS score changes to estimate the size of the study population30. Parameters for the estimation was as follows: allocation ratio 1:1, power 0.8, and α error probability 0.05. The estimation returned 26 participants required in each groups, and study population was set as 30 considering a predicted drop-out rate of 10%. At baseline, we obtained participants’ medical histories, performed physical and laboratory examinations, and administered baseline scores for cognitive evaluations. Participants who met the inclusion criteria were randomized into two groups in a 1 to 1 ratio using a simple randomization method by utilizing SAS randomization program. The random allocation sequence was generated by M-H.K and the details of randomization table were unknown to all researchers who enrolled the participants and the clinicians who evaluated the patients. The groups were assigned to receive PM-EE 600 mg/day (two 150 mg tablets, twice daily) or placebo (two placebo tablets, twice daily) for 12 weeks. In the placebo tablets, desalted SE extract substituted with the equal mass of crystalline cellulose and the PM-EE and the placebo tablets were identical in appearance. The details of the contents of raw materials in PM-EE and Placebo tablets are described in Supplemental Table 1. Randomization was performed at the SNUH Clinical Research Unit using a list of computer-generated random numbers. At 6 weeks after treatment, participants were followed up with physical and laboratory examinations and were checked for the development of any adverse events. At 12 weeks after treatment, participants were followed up again with physical and laboratory examinations, were checked for the development of any adverse events, and performed the follow-up cognitive evaluations. The use of medications included in the exclusion criteria was not allowed during the trial.
Measurements
The K-MMSE was evaluated for baseline cognitive function. At baseline and at 12 weeks after treatment, scores on the Korean version of the Alzheimer's Disease Assessment Scale (ADAS-K) were used to evaluate the global function31; scores on the cognitive domain of the ADAS-K (ADAS-cog) the Korean version of the Consortium to Establish a Registry for Alzheimer's Disease (CERAD-K) assessment packet32, and the Korean version of the Color-Word Stroop Test (K-CWST) were used to evaluate cognitive function33,34; and scores on the Alzheimer Disease Cooperative Study assessment for the Activities of Daily Living (ADCS-ADL) to evaluate the ability to perform daily activities35, and the Short-form Geriatric Depression Scale (SGDS) were used to measure the severity of depression36. Before K-CWST, subject were asked whether he or she had discomfort in reading words or colors, and performed a pre-test using 5–6 words using the same setting that the test was performed, to check subjects’ capacity of performing the test. Treatment-emergent adverse events were recorded using the preferred term of the medical dictionary for regulatory activities (MedDRA)37. Laboratory measurements included complete blood count and serum panels including electrolytes, creatinine levels, liver enzymes, glucose level, and cholesterol profiles, routine urinalysis results, systolic/diastolic blood pressure and pulse rate, and routine 12-lead electrocardiography results. Serum samples for laboratory evaluations were collected after 8 h of fasting.
End points
The primary endpoint was set as the change in the total score of the ADAS-cog at 12 weeks from the total score at baseline5,13,17. The secondary endpoints were changes in the ADAS-cog subdomain, total ADAS-K, K-CWST, ADCS-ADL, SGDS, and CERAD-K scores.
Statistical analysis
SAS (Version 9.4, SAS Institute, Cary, North Carolina, USA) was used for the statistical analyses. All analyses to evaluate the drug efficacy were carried out according to the per-protocol principle, and safety analysis was performed using the safety set. Baseline characteristics were compared using Fisher’s exact test for categorical variables or t-tests or Wilcoxon rank sum tests for continuous variables. The change in the scores from baseline to 12 weeks after treatment was evaluated using paired t-tests or Wilcoxon rank sum tests, and inter-group comparisons were performed using analysis of covariance (ANCOVA) and two-sample t-tests. The frequency of adverse events was compared between the groups using chi-square tests or Fisher’s exact test. Post-hoc analysis were performed for the subgroup of 30 subjects with MCI (baseline K-MMSE scores of ≤ 28)38. All statistical evaluations were two-tailed, and P values of < 0.05 were set as statistically significant. Power analyses for the minimal sample size to reach a statistical significance were estimated for all of the outcome parameters, using allocation ratio of 1:1, power of 0.8, and α error probability of 0.05. Effect sized was measured using the mean values and the standard deviation of the score changes in the groups.
Results
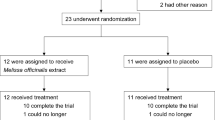
Between February 2018 and December 2018, a total of 63 subjects were included in this study. Thirty-two participants were randomized into the PM-EE group, and the remaining 31 were randomized into the placebo group (safety set). During follow-up, 4 subjects (3 in the PM-EE group and 1 in the placebo group) were excluded for violating the exclusion criteria and 1 subject in the placebo group for withdrawing consent for study participation. Four subjects (3 in the PM-EE group and 1 in the placebo group) were excluded from the analysis for low (< 70%) drug compliance and 1 subject for withdrawing consent for study participation (Fig. 1). Finally, 53 participants (26 in the PM-EE group and 27 in the placebo group) were included in the analysis. Demographic and baseline clinical characteristics were comparable between the study groups (Table 1).

A flow chart illustrating the study process.
After 12 weeks, there were no significant differences in the changes in ADAS-cog scores between the groups (P = 0.9858). None of the secondary outcome parameters, such as changes in the ADAS-cog subdomain, total ADAS-K, K-CWST, ADCS-ADL, SGDS, or CERAD-K scores, showed a statistically significant difference between the groups (Table 2). In the power analysis, the total number of study subjects required to reach a statistical significance for the effect of PM-EE on the changes in the ADAS-cog score was 8262. However, when single items of the ADAS-cog were compared, a significant improvement in the comprehension of spoken language items was observed in the PM-EE group (− 0.04 ± 0.20 vs. 0.19 ± 0.56, P = 0.0498, Table 3).
For the safety analysis, 4 subjects (12.5%, number of events = 7) in the PM-EE group and 4 subjects (12.9%, number of events = 6) in the placebo group reported an adverse event (P = 1.000). All of the reported adverse events were of mild severity and were not relevant to the use of the drugs (Table 4). There was no significant difference in the changes in the laboratory parameters from baseline to follow-up between the groups.
In the subgroup analysis for the 30 subjects with baseline K-MMSE scores of ≤ 28 (17 in the PM-EE group and 13 in the placebo group), the baseline profiles of cognitive evaluation scores were comparable between the groups (Table 5). However, the PM-EE group showed higher interval improvement in the color-reading score of the K-CWST (8.2 ± 25.0 vs. − 4.7 ± 13.2, P = 0.018). Other parameters, such as ADAS-cog, total ADAS-K, ADCS-ADL, SGDS, or CERAD-K scores, did not exhibit significantly different score changes from baseline between the groups (Table 5). In the power analysis for this subgroup, the total number of study subjects required to reach a statistical significance for the effect of PM-EE on the changes in the ADAS-cog score was 566.
Discussion
This is the first randomized controlled study to investigate the effect of PM-EE in subjects complaining memory dysfunction without overt dementia. The results found that PM-EE did not significantly improve the total score or subdomain scores of the ADAS-cog, K-CWST, ADCS-ADL, SGDS, or CERAD-K. However, in the subpopulation with baseline K-MMSE scores of ≤ 28, PM-EE was associated with significantly more improvement in the color-reading score of the K-CWST. PM-EE also showed significantly improved the comprehension of spoken language items in the language domain of the ADAS-cog. Additionally, PM-EE was safe, without provoking a higher frequency of adverse events, and all of the reported adverse events were of mild severity and were not relevant to the use of PM-EE.
The primary outcome analysis returned a negative result. This finding is in accordance with those from the previous studies that evaluated the disease modifying effects of various candidates in MCI12,13,14,15,16,17. Donepezil, the most established medication for treating AD and other forms of dementia, was not significantly effective in treating MCI and was also associated with a higher frequency of adverse events and a higher rate of drug discontinuation12,13,17. Accordingly, this study did not observe a certain trend favouring the effects of PM-EE in the change in the ADAS-cog scores or other outcome parameters. The power analyses also returned very high numbers of participants required to observe a statistically significant effect of PM-EE. This might be due to the participants’ relatively mild or minimal degree of the cognitive dysfunction included in this study, which might have been insufficient to properly evaluate the effect of the drug. However, the number of participants required for a statistical significance of PM-EE in the MCI subgroup (baseline K-MMSE scores of ≤ 28) was comparable to those of the previous studies (250-2100) which evaluated the long-term effects of the candidate drugs in patients with MCI12,13,14,16,17. This implies that in further large-sized studies, PM-EE might be established to be effective in patients with objective cognitive deficits. The properties of the ADAS-cog, the primary outcome parameter of those previous studies and the present study, might also have at least partially contributed to the negative result. A previous pooled analysis of three clinical trials using donepezil in mild-to-moderate AD reported that ADAS-cog components have a substantial ceiling effect (7 out of 11 components)39. Therefore, ADAS-cog might be too simple to reflect the expected depth and range of cognitive performance39, and might not have been an optimal measurement to evaluate the effects of PM-EE in this clinical setting.
However, we observed a beneficial effect of PM-EE over placebo in improving the comprehension of spoken language function. Language comprehension is a complex cognitive task that requires the simultaneous activation of perception, attention, and language processes and, therefore, is highly implicated in overall cognitive function40. A recent study demonstrated that a wide range of brain structures, including the left inferior and middle frontal gyri, left inferior parietal cortex, and left middle temporal gyrus, is activated during the comprehension of spoken language, and their level of activation and functional connectivity decreases with ageing41. Therefore, improvement in spoken language comprehension function due to PM-EE might provide a beneficial effect on overall cognitive function.
Notably, PM-EE significantly improved stroop test results in patients with K-MMSE scores compatible with the diagnosis of MCI38. The sroop test is a sensitive measurement of the frontal executive function which includes attention, flexibility, cognitive inhibition, and working memory42. Frontal executive function is generally diminished in both amnestic and non-amnestic MCI patients, and alterations in executive function is associated with the response to AChE inhibitors43,44,45,46. A previous study reported that AChE inhibitor activity was up-regulated in the frontal areas of patients with MCI, and the authors assumed this upregulation to be a compensatory mechanism47. Additionally, executive dysfunction in MCI and AD has been recognized as a key contributor to patients’ impairment in activities of daily living48.
Taken together with the modest improvement in the ADAS-cog in the subgroup with MCI, the effects of PM-EE in improving stroop test results in patients with MCI implies that this drug might be potentially effective in improving the core symptoms of MCI. As disease progression from MCI to dementia takes several years or even a decade8,10,11, candidate medications to treat MCI should be safe for long-term use without provoking any harmful effect. Considering the broad mechanism of PM-EE that both enhances AChE inhibitors and has anti-inflammatory, antioxidative, and neuroprotective effects with a favourable safety profile28, PM-EE might serve as a potential candidate drug for the long-term treatment of MCI.
In addition to the small number of study population, this study has some limitations to be addressed. First, in previous studies evaluating the effects of various medical candidates in patients with MCI, the change in ADAS-cog scores over time was very slow, and the follow-up duration was 3 years to evaluate the long-term effects of the medication12,13,14,15,16,17. As the current study evaluated the effects of PM-EE after using the drug for only for 12 weeks, a longer duration of follow-up is warranted to properly evaluate the disease-modifying effects of PM-EE in MCI. Second, this study only used a single dose of PM-EE. Because PM-EE was safe without provoking adverse events related to drug usage, future studies might include multiple dosage regimens with higher doses of PM-EE. Additionally, the present study also has limitations as a single-centre study. These limitations might be overcome by further randomized controlled studies with a larger number of patients, a longer follow-up evaluation period, and multiple dosage regimens to demonstrate the long-term clinical effects of PM-EE.
References
Prince, M. et al. The global prevalence of dementia: a systematic review and metaanalysis. Alzheimer’s Dementia 9, 63-75.e62 (2013).
Brookmeyer, R., Johnson, E., Ziegler-Graham, K. & Arrighi, H. M. Forecasting the global burden of Alzheimer’s disease. Alzheimer’s Dementia 3, 186–191 (2007).
Birks, J. & Craig, D. Galantamine for vascular cognitive impairment. Cochrane Database Syst. Rev. (2006).
Reisberg, B. et al. Memantine in moderate-to-severe Alzheimer’s disease. N. Engl. J. Med. 348, 1333–1341 (2003).
Erkinjuntti, T. et al. Efficacy of galantamine in probable vascular dementia and Alzheimer’s disease combined with cerebrovascular disease: a randomised trial. Lancet 359, 1283–1290 (2002).
Courtney, C. et al. Long-term donepezil treatment in 565 patients with Alzheimer’s disease (AD2000): randomised double-blind trial. Lancet 363, 2105–2115 (2004).
Sperling, R. A. et al. Toward defining the preclinical stages of Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimer’s Dementia 7, 280–292 (2011).
Livingston, G. et al. Dementia prevention, intervention, and care. Lancet 390, 2673–2734 (2017).
Weimer, D. L. & Sager, M. A. Early identification and treatment of Alzheimer’s disease: social and fiscal outcomes. Alzheimer’s Dementia 5, 215–226 (2009).
Gauthier, S. et al. Mild cognitive impairment. Lancet 367, 1262–1270 (2006).
Morris, J. C. et al. Mild cognitive impairment represents early-stage Alzheimer disease. Arch. Neurol. 58, 397–405 (2001).
Tricco, A. C. et al. Efficacy and safety of cognitive enhancers for patients with mild cognitive impairment: a systematic review and meta-analysis. CMAJ 185, 1393–1401 (2013).
Doody, R. et al. Donepezil treatment of patients with MCI: a 48-week randomized, placebo-controlled trial. Neurology 72, 1555–1561 (2009).
Winblad, B. et al. Safety and efficacy of galantamine in subjects with mild cognitive impairment. Neurology 70, 2024–2035 (2008).
Loy, C. & Schneider, L. Galantamine for Alzheimer's disease and mild cognitive impairment. Cochrane Database of Systematic Reviews (2006).
Petersen, R. C. et al. Vitamin E and donepezil for the treatment of mild cognitive impairment. N. Engl. J. Med. 352, 2379–2388 (2005).
Salloway, S. et al. Efficacy of donepezil in mild cognitive impairment: a randomized placebo-controlled trial. Neurology 63, 651–657 (2004).
Jessen, F. et al. The characterisation of subjective cognitive decline. Lancet Neurol. (2020).
Heneka, M. T. et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 14, 388–405 (2015).
Iadecola, C. The overlap between neurodegenerative and vascular factors in the pathogenesis of dementia. Acta Neuropathol. 120, 287–296 (2010).
Craft, S. The role of metabolic disorders in Alzheimer disease and vascular dementia: two roads converged. Arch. Neurol. 66, 300–305 (2009).
Bennett, S., Grant, M. M. & Aldred, S. Oxidative stress in vascular dementia and Alzheimer’s disease: a common pathology. J. Alzheimer’s Disease 17, 245–257 (2009).
Román, G. C. & Kalaria, R. N. Vascular determinants of cholinergic deficits in Alzheimer disease and vascular dementia. Neurobiol. Aging 27, 1769–1785 (2006).
Patel, S. Salicornia: evaluating the halophytic extremophile as a food and a pharmaceutical candidate. 3 Biotech 6, 104 (2016).
Min, A. Y. et al. N-palmitoyl serotonin alleviates scopolamine-induced memory impairment via regulation of cholinergic and antioxidant systems, and expression of BDNF and p-CREB in mice. Chem. Biol. Interact. 242, 153–162 (2015).
Kim, Y. A. et al. Evaluation of Salicornia herbacea as a potential antioxidant and anti-inflammatory agent. J. Med. Food 12, 661–668 (2009).
Karthivashan, G. et al. Ameliorative potential of desalted Salicornia europaea L. extract in multifaceted Alzheimer’s-like scopolamine-induced amnesic mice model. Sci. Rep. 8, 7174 (2018).
Karthivashan, G. et al. Cognitive-enhancing and ameliorative effects of acanthoside B in a scopolamine-induced amnesic mouse model through regulation of oxidative/inflammatory/cholinergic systems and activation of the TrkB/CREB/BDNF pathway. Food Chem. Toxicol. 129, 444–457 (2019).
Sherwin, B. B. Estrogen effects on cognition in menopausal women. Neurology 48, 21S-26S (1997).
Lee, S.-T., Chu, K., Sim, J.-Y., Heo, J.-H. & Kim, M. Panax ginseng enhances cognitive performance in Alzheimer disease. Alzheimer Dis. Assoc. Disord. 22, 222–226 (2008).
Suh, G. H. & Mohs, R. C. Development of the Korean version of Alzheimers Disease Assessment Scale (ADAS-K) to assess cognition in dementia. J. Korean Geriatrics Soc. 7, 269–277 (2003).
Lee, J. H. et al. Development of the Korean Version of the Consortium to Establish a Registry for Alzheimer’s Disease Assessment Packet (CERAD-K) clinical and neuropsychological assessment batteries. J. Gerontol. Ser. B Psychol. Sci. 57, P47–P53 (2002).
Kim, T. Y. et al. Development of the Korean stroop test and study of the validity and the reliability. J. Korean Geriatrics Soc. 8, 233–240 (2004).
Fisher, L. M., Freed, D. M. & Corkin, S. StroopColor-Word Test performance in patients with Alzheimer’s disease. J. Clin. Exp. Neuropsychol. 12, 745–758 (1990).
Galasko, D. et al. An inventory to assess activities of daily living for clinical trials in Alzheimer's disease. Alzheimer diseaseassociated disorders (1997).
Cho, M. J. et al. Validation of geriatric depression scale, Korean version (GDS) in the assessment of DSM-III-R major depression. J. Korean Neuropsychiatric Assoc. 38, 48–63 (1999).
Brown, E. G., Wood, L. & Wood, S. The medical dictionary for regulatory activities (MedDRA). Drug Saf. 20, 109–117 (1999).
Lin, J. S. et al. Screening for cognitive impairment in older adults: an evidence update for the US Preventive Services Task Force. Rockville, MD: Agency for Healthcare Research and Quality Report No: 14-05198-EF-1 (2013).
Cano, S. J. et al. The ADAS-cog in Alzheimer’s disease clinical trials: psychometric evaluation of the sum and its parts. J. Neurol. Neurosurg. Psychiatry 81, 1363–1368 (2010).
Wingfield, A. & Tun, P. A. Cognitive supports and cognitive constraints on comprehension of spoken language. J. Am. Acad. Audiol. 18, 548–558 (2007).
Peelle, J. E., Troiani, V., Wingfield, A. & Grossman, M. Neural processing during older adults’ comprehension of spoken sentences: age differences in resource allocation and connectivity. Cereb. Cortex 20, 773–782 (2009).
Demakis, G. J. Frontal lobe damage and tests of executive processing: a meta-analysis of the category test, stroop test, and trail-making test. J. Clin. Exp. Neuropsychol. 26, 441–450 (2004).
Traykov, L. et al. Executive functions deficit in mild cognitive impairment. Cognit. Behav. Neurol. 20, 219–224 (2007).
Mega, M. S. et al. Orbital and dorsolateral frontal perfusion defect associated with behavioral response to cholinesterase inhibitor therapy in Alzheimer’s disease. J. Neuropsychiatry Clin. Neurosci. 12, 209–218 (2000).
Connelly, P., Prentice, N. & Fowler, K. Predicting the outcome of cholinesterase inhibitor treatment in Alzheimer’s disease. J. Neurol. Neurosurg. Psychiatry 76, 320–324 (2005).
Brandt, J. et al. Selectivity of executive function deficits in mild cognitive impairment. Neuropsychology 23, 607 (2009).
DeKosky, S. T. et al. Upregulation of choline acetyltransferase activity in hippocampus and frontal cortex of elderly subjects with mild cognitive impairment. Ann Neurol 51, 145–155 (2002).
Marshall, G. A. et al. Executive function and instrumental activities of daily living in mild cognitive impairment and Alzheimer’s disease. Alzheimer’s Dementia 7, 300–308 (2011).
Acknowledgements
This research was supported by the Korea Institute of Planning and Evaluation for Technology in Food, Agriculture, Forestry and Fisheries (IPET) through High Value-added Food Technology Development Program, funded by Ministry of Agriculture, Food and Rural Affairs (MAFRA) (116018-03-3-HD040). This research was supported by the Brain Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Science, ICT & Future Planning (2016M3C7A1914002).
Author information
Authors and Affiliations
Contributions
W-J.L. analyzed the data, interpreted study results, and drafted the manuscript. Y-W.S. designed and performed the study and analyzed the data. D-E.K. participated in the conceptualization and planning of the study, managed the study procedures, and managed the study data. M-H.K. conceptualized the study, managed the study procedures, analyzed the data, conducted statistical analysis, and revised the manuscript. M.K. designed and conceptualized the study, supervised all study procedures, analyzed the data, interpreted study results, and revised the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Lee, WJ., Shin, YW., Kim, DE. et al. Effect of desalted Salicornia europaea L. ethanol extract (PM-EE) on the subjects complaining memory dysfunction without dementia: a 12 week, randomized, double-blind, placebo-controlled clinical trial. Sci Rep 10, 19914 (2020). https://doi.org/10.1038/s41598-020-76938-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-76938-x
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
