
Abstract
Phosphorescence lifetime imaging microscopy (PLIM) combined with an oxygen (O2)-sensitive luminescent probe allows for high-resolution O2 imaging of living tissues. Herein, we present phosphorescent Ir(III) complexes, (btp)2Ir(acac-DM) (Ir-1) and (btp-OH)3Ir (Ir-2), as useful O2 probes for PLIM measurement. These small-molecule probes were efficiently taken up into cultured cells and accumulated in specific organelles. Their excellent cell-permeable properties allowed for efficient staining of three-dimensional cell spheroids, and thereby phosphorescence lifetime measurements enabled the evaluation of the O2 level and distribution in spheroids, including the detection of alterations in O2 levels by metabolic stimulation with an effector. We took PLIM images of hepatic tissues of living mice by intravenously administrating these probes. The PLIM images clearly visualized the O2 gradient in hepatic lobules with cellular-level resolution, and the O2 levels were derived based on calibration using cultured cells; the phosphorescence lifetime of Ir-1 gave reasonable O2 levels, whereas Ir-2 exhibited much lower O2 levels. Intravenous administration of NH4Cl to mice caused the hepatic tissues to experience hypoxia, presumably due to O2 consumption to produce ATP required for ammonia detoxification, suggesting that the metabolism of the probe molecule might affect liver O2 levels.
Similar content being viewed by others


Introduction
Molecular oxygen (O2) is essential for cellular function and is continuously supplied to whole tissues in the body to maintain homeostasis. Oxygen deprivation (hypoxia) in tissues is associated with the pathophysiology of various diseases such as cerebral infarction, fatty liver disease, chronic kidney disease, diabetic retinopathy, and cancer1,2,3. Understanding the cellular and molecular mechanisms underlying hypoxia-associated diseases requires O2 imaging technology capable of detecting the tissue oxygen status in real time and with high resolution. In vivo O2 detection methods that have been developed so far include oxygen electrodes4,5, blood oxygenation level dependent magnetic resonance imaging (Bold MRI)6,7, positron emission tomography (PET) with a hypoxia tracer8, electron paramagnetic resonance (EPR) oximetry9,10, hypoxia markers11,12,13, and optical imaging14,15,16. These methods can work in vivo, but they have advantages and limitations in terms of applicable targets, spatial resolution, tissue permeability, convenience, reversibility, etc. Of these known methods, optical imaging utilizing O2 quenching of probe phosphorescence has great advantages in that high-resolution O2 images can be obtained in a reversible manner, although it is limited to the detection of a penetration depth through tissues of ~ 1 cm even under near-infrared excitation17.
To visualize tissue O2 levels with high sensitivity and high stability, various phosphorescent metal complexes have been developed over the past few decades14,15,16,18,19. The most common compounds used as luminescent O2 probes include Pt(II) and Pd(II) porphyrins, Ru(II) complexes, Pt(II) complexes, and Ir(III) complexes, which give intense phosphorescence in the visible to near-infrared wavelength regions with reasonably long lifetimes (> 1.0 μs). Oxygen probes targeting various biological tissues of interest have been developed by modifying these O2-sensing luminophores: cell-penetrating conjugates of Pt(II)-porphyrins20,21, dendritic Pt(II)- and Pd(II)-porphyrins22,23,24, Ru(II) complex derivatives targeting the cell nucleus25, ratiometric O2 probes based on Ir(III) complexes26,27, etc.
With the use of phosphorescent probes, wide-field luminescence lifetime measurements using a gated CCD camera have enabled whole-body O2 imaging including tumor tissues, ocular fundus, and specific organs28,29,30,31. In contrast, phosphorescence lifetime imaging microscopy (PLIM)32,33 allows high-resolution O2 imaging at the cellular level, providing precise information on microenvironment along with real-time changes in O2 levels. The PLIM method has been applied to O2 imaging of giant cells34, cell spheroids20,24,35,36, neurospheres37,38 and the epithelium of rat and human colon tissues38 using cell-permeable small-molecule and nano-particle probes. By using dendrimerized Pt-porphyrin probes, the PLIM method has been successfully applied to microvascular and interstitial O2 imaging of the brain22,39,40, bone marrow41,42, and retinal tissue43 in vivo.
In contrast, few studies have reported tissue oxygen distributions in vivo using an intracellular O2 probe44. We have recently developed small-molecule O2 probes based on Ir(III) complexes18,19. Among them, BTPDM1 ((btp)2Ir(acac-DM); Ir-1) (Fig. 1A) allows for the measurement of O2 levels in hypoxic tumors45,46 and high-resolution O2 imaging in renal cortex in vivo47,48. To improve the reliability of the O2 level as measured with a small-molecule probe, it is necessary to quantify O2 levels in different tissues in vivo and investigate various issues: localization and stability of probes in tissues, calibration of emission lifetime in tissues (cell-specific calibration), the influence of probe administration on the target tissues, etc.

(A) Chemical structures of BTPDM1 (Ir-1) and (btp-OH)3Ir (Ir-2). Absorption and phosphorescence spectra of Ir-1 and Ir-2 in MeCN at room temperature are shown. The phosphorescence spectra were taken in both degassed (solid line) and aerated (dashed line) solutions. (B) Schematic views of hepatic lobules. CV central vein, PV portal vein.
In this study, we established an in vivo O2 imaging method using small-molecule probes by investigating the O2 distribution in the hepatic lobules of living mice by PLIM measurements with two structurally related Ir(III) complexes, Ir-1 and (btp-OH)3Ir (Ir-2) (Fig. 1A). The hepatic lobule is a fundamental structural unit of the liver with a hexagonal shape (Fig. 1B)49,50. In the lobule, blood runs unidirectionally from the vertices of the hexagon called portal triads, through microcirculatory networks known as sinusoids, to a central vein (CV) in the middle of the lobule. The portal triad contains two distinct inlets for blood flow: a well-oxygenated hepatic artery and a poorly-oxygenated portal vein, the latter of which constitutes ~ 70% of total liver blood flow. Therefore, even under physiological conditions, liver parenchymal cells and hepatocytes surrounding the portal triad are exposed to relatively low oxygen concentrations, and pericentral hepatocytes experience much lower oxygen concentrations, forming a zonal heterogenous distribution of oxygen along the sinusoids in the hepatic lobule. These intralobular oxygen distributions, however, are considered to be prerequisite for normal liver metabolic functions in a zone-specific manner. In addition, this metabolic zonation is often disrupted in disease conditions with aberrant oxygen distribution in the hepatic lobules. To deepen our knowledge about the dynamic alterations in liver metabolism in both physiological and pathophysiological conditions, real-time imaging of intracellular oxygen tension in situ is a promising method with the development of oxygen-sensitive probes.
We first compared the photophysical properties and oxygen sensitivity of Ir-1 and Ir-2 in solution and cultured cells. Next, we investigated the feasibility of these probes for the measurements of O2 distribution in cell spheroids. Based on these results, we performed PLIM measurements of hepatic tissues of mice using Ir-1 and Ir-2 to reveal the probe performances.
Results
Photophysical properties
Emission brightness and O2 sensitivity are basic characteristics required for an optical O2 probe. The emission brightness of the probes under the same concentration can be evaluated by the product (εΦp0) of the molar absorption coefficient (ε) at the excitation wavelength and the phosphorescence quantum yield (Φp0) under the absence of oxygen, and the O2 sensitivity by kqτp0, where kq and τp0 are the bimolecular quenching rate constant by O2 and the phosphorescence lifetime under the absence of oxygen. The first absorption band of Ir-1 appears at around 476 nm in MeCN (Fig. 1A), which originates from singlet metal-to-ligand-charge-transfer (1MLCT) transition. This band is slightly blue-shifted in Ir-2, giving a smaller ε value (2800 dm3mol−1 cm−1) compared with that (5400 dm3mol−1 cm−1) of Ir-1 at the excitation wavelength (488 nm) in our PLIM measurements. Ir-1 and Ir-2 exhibit red emission extending to the near infrared region and give vibrational structures with maximum wavelengths at 612 and 601 nm, respectively. Both complexes are highly emissive in MeCN (Φp0 = 0.26 for Ir-1 and 0.28 for Ir-2). Judging from the ε values at 488 nm and the almost equal Φp0, Ir-1 is superior to Ir-2 in brightness for excitation at 488 nm.
The O2 sensitivity of a probe in solution can be evaluated experimentally from the ratio (τp0/τp) of the phosphorescence lifetimes under deaerated and aerated conditions. The obtained τp0/τp values of Ir-1 and Ir-2 in MeCN were 72 and 97, and the kq values were calculated to be 7.59 × 104 mmHg−1 s−1 for Ir-1 and 8.22 × 104 mmHg−1 s−1 for Ir-2 (Table 1). Using the solubility of oxygen in MeCN at an oxygen partial pressure (pO2) of 0.21 atm51, the quenching rate constant can be converted from pressure units to concentration units. The converted kq values (6.38 × 109 M−1 s−1 for Ir-1 and 6.91 × 109 M−1 s−1 for Ir-2) were close to the diffusion-controlled rate of bimolecular reactions in MeCN51, indicating that oxygen has very high quenching ability to the excited triplet state of both complexes. We recently revealed that the phosphorescence quenching of Ir(III) complexes by molecular oxygen occurs not only by energy transfer but also by charge transfer from the triplet Ir(III) complex to O252. As a result, Ir(III) complexes show much larger kq values compared with metalloporphyrins and Ru(II) complexes. The O2 sensitivity is slightly higher in Ir-2 than Ir-1 owing to the longer intrinsic lifetime and larger kq value of Ir-2. The intrinsic lifetimes (5–10 μs) of Ir-1 and Ir-2 are shorter than those (10–100 μs) of Pt-porphyrins, but longer than those of typical Ru(II) complexes used as O2 probes18, and thus the O2 sensitivity of Ir-1 and Ir-2 is considered to be comparable with Pt-porphyrins and higher than Ru(II) complexes.
Characterization of intracellular O2 probes
To evaluate the cellular uptake efficiencies of Ir-1 and Ir-2, we compared the emission intensities of human colorectal adenocarcinoma (HT-29) cells incubated with these complexes (5 μM, incubation time: 2 h) using a plate reader. Here, we used their prototype complex BTP ((btp)2Ir(acac); btp = benzothienylpyridine, acac = acetylacetone)45 as a reference compound. The emission intensities of each complex under 21% and 2.5% O2 conditions were corrected for the number of HT-29 cells in each well and the ε values (4700, 5400, and 2800 M−1 cm−1 for BTP, Ir-1 and Ir-2) at 488 nm. The relative emission intensities after this correction (Fig. S1) suggest that the cellular uptake efficiency was greatly improved with Ir-1 and Ir-2 compared with BTP by introducing a hydrophilic dimethylamino and hydroxyl group, respectively.
The subcellular localization of Ir-1 and Ir-2 investigated by costaining experiments with organelle-specific trackers (LysoTracker Green and ERTracker Green) using HeLa cells showed preferential accumulation to lysosomes for Ir-1 and endoplasmic reticulum (ER) for Ir-2 (Fig. S2). Both complexes taken up into cells are presumed to accumulate in the organelle membrane because of their lipophilicity. The cytotoxicity of Ir-1 and Ir-2 evaluated with WST assay (incubation time: 24 h under the presence of the probe) revealed that the median lethal doses (LC50) of Ir-1 and Ir-2 for HT-29 cells were 10–15 μM and ~ 30 μM (Fig. S3A). In addition, analysis of mitochondrial membrane potential using JC-1 dye (Fig. S3B) showed that the membrane potential was almost unaffected by Ir-1 or Ir-2 loading (1 μM, 2 h). In typical experiments in this study, the cells were incubated with the probes (500 nM or 1 μM) for 2 h prior to microscopic measurements. Therefore, under these experimental conditions, the effect of the probes on cell activity is considered to be sufficiently small.
Photostability is another important characteristic required for an intracellular O2 probe. We investigated the photostability of Ir-1 and Ir-2 in cells by taking emission images of HT-29 cells under continuous 488 nm laser pulse irradiation with our PLIM system. The cells were incubated with Ir-1 or Ir-2 (1 μM) for 2 h under 21% or 2.5% O2 conditions, and PLIM images of the HT-29 cells were taken at every 50 scans until 550 scans (Fig. S4). During this irradiation time, the phosphorescence lifetimes of Ir-1 and Ir-2 were almost constant, although the phosphorescence intensity changed slightly by 488 nm light irradiation. Also, no significant change was observed in the cell morphology. In this study, the signals accumulated with 50 scans were usually averaged to obtain a single PLIM image. Therefore, Ir-1 and Ir-2 were found to have sufficient photostability for obtaining clear PLIM images.
Quantification of O2 levels
To quantify the oxygen levels in tissues based on lifetime measurements, we need a calibration curve that represents the relationship between lifetime and pO2. Since Ir-1 and Ir-2 are likely to accumulate in specific organelle membranes, the pO2 dependence of the phosphorescence lifetime in cells is expected to be different from that in solution. Therefore, we calibrated the lifetime by measuring PLIM images of HT-29 cells incubated with Ir-1 or Ir-2 under 21, 15, 10, 5, and 0% O2 conditions at 37 °C (Fig. 2A,B); we added 10 µM antimycin A (AntA) to the medium prior to the PLIM measurements to suppress the oxygen consumption by cellular respiration, and in the experiments under N2 saturated conditions we added Na2SO3 (500 mM) into the medium to remove oxygen remaining in the culture. The average lifetime of an entire image was plotted against the pO2 according to the Stern–Volmer equation, τp0/τp = 1 + kq τp0pO2. A linear relationship was obtained for both complexes (Fig. 2C), and the kq values for Ir-1 and Ir-2 were derived to be 4.22 × 103 mmHg−1 s−1 and 5.36 × 103 mmHg−1 s−1 along with the τp0, 5.20 µs and 6.18 µs, respectively. These values were used to quantify oxygen levels in cell spheroids from phosphorescence lifetimes (τp).

Intensity (upper) and PLIM (lower) images of HT-29 cells stained with (A) Ir-1 and (B) Ir-2 under different pO2 in an incubator. The average phosphorescence lifetimes are shown below each image. Cells were treated with Ant A (5–21% O2) and Na2SO3 (0% O2). Scale bar: 50 µm. (C) Stern–Volmer plots of \(\tau_{{\text{p}}}^{0}\)/\(\tau_{{\text{p}}}\) as a function of pO2 for Ir-1 (red) and Ir-2 (blue) partitioned into HT-29 cells under different pO2 in an incubator. Error bars: S.D.
Evaluation of O2 distribution in cell spheroids
In the past decade, three-dimensional (3D) spheroid systems have received much attention in fields such as drug discovery, cancer research, and toxicology. They provide a more physiologically-relevant environment and organ-like microarchitecture compared with conventional 2D cell cultures and better mimic the crucial tumor tissue properties and microenvironment. The excellent cell-permeable properties of Ir-1 and Ir-2 suggested their potential for in-depth staining of 3D cell spheroids and thus the potential of these probes for visualization of the O2 distribution within spheroids and living tissues.
So, we first acquired the PLIM images of HT-29 cell spheroids that were incubated with Ir-1 or Ir-2. Here, each probe (500 nM) was added to the medium after the spheroids were formed, and the spheroids were further incubated with the probe for 24 h prior to PLIM measurements. The bright-field images taken on different planes in the z direction (Fig. 3) show that spheroids with a diameter of 150–200 μm were almost uniformly stained, including the core, by Ir-1 and Ir-2. We found from the Z-stack phosphorescence lifetime images of an HT-29 spheroid stained with Ir-1 (Fig. 3B) and Ir-2 (Fig. 3C) that the cells closer to the center of the spheroid gave longer lifetimes, i.e. lower O2 levels compared with those in the peripheral region. For quantitative evaluation of the O2 gradient in a spheroid, we investigated the line profiles of the average phosphorescence lifetime on the plane approximately 20 μm from the bottom (Fig. 3D). We also measured the lifetimes along the central area in the z-direction (Fig. 3E), and derived the pO2 distribution (Fig. 3D,E) based on the τp0 and kq obtained for HT-29 cultured cells. Both probes showed that the pO2 is reduced near the center and bottom surface of the spheroids, because the oxygen supply from the bottom direction is cut off by the bottom glass, and also the peripheral cells consume oxygen carried by diffusion from the culture medium. The degree of hypoxia in the core depended on the spheroid size (Fig. S5) and the respiratory activity as will be described below.

(A) Bright-field image (left) and schematic view (right) of an HT-29 cell spheroid. (B, C) Z-stacked phosphorescence intensity (left) and lifetime (right) images of an HT-29 cell spheroid stained with (B) Ir-1 and (C) Ir-2. Each image corresponds to cross-section from the bottom to the upper part at an interval of 10 µm along the z-axis. Scale bar: 100 µm. (D) Line profiles of phosphorescence lifetime (blue) and pO2 (red) along the arrows shown in B and C. (E) Average phosphorescence lifetime (blue) and pO2 (red) of the square region along the z-axis in an HT-29 cell spheroid stained with probes.
We next investigated whether Ir-1 and Ir-2 can image changes in the oxygen status of spheroids caused by metabolic stimulus. We used FCCP (carbonyl cyanide p-(trifluoromethoxy)phenylhydrazone) and AntA to alter the oxygen status of HT-29 spheroids by stimulating metabolic processes; FCCP is an uncoupler of oxidative phosphorylation in mitochondria that disrupts ATP synthesis by transporting protons across the membrane and thus increases the O2 consumption rate. AntA is known to inhibit the mitochondrial electron transport chain from cytochrome b to cytochrome c1 and suppress O2 consumption. Metabolic stimulation of HT-29 cell spheroids by FCCP and AntA significantly changed the PLIM images (Fig. 4). The lifetime images of the spheroid plane ~ 30 μm from the bottom (Fig. 4B,C) demonstrated that upon addition of FCCP to the medium, the lifetimes of both probes quickly increased within ~ 10 min, especially in the core. The oxygen partial pressures calculated from the lifetimes based on the calibration in Fig. 2 indicate that the pO2 decreased sharply to less than 20 mmHg by increased O2 consumption, and then it slowly recovered. Conversely, the lifetimes of both probes rapidly dropped within 10 min upon metabolic stimulation using AntA (Fig. 4D,E), corresponding to the increase in pO2 by suppression of O2 consumption. The pO2 changes of spheroids reflected the metabolic effects of FCCP and AntA, demonstrating that Ir-1 and Ir-2 can be used to track the oxygen status of cell spheroids. These results confirmed the high penetrating ability of Ir-1 and Ir-2 into spheroids and their feasibility as tissue O2 probes.

Imaging of the oxygen status of an HT-29 cell spheroid upon metabolic stimulation with FCCP or AntA. (A) Schematic representation of a spheroid in media containing FCCP or AntA. PLIM images were taken at 30 μm from the bottom after oil sealing. (B,C) Variation in PLIM images (left), and their average phosphorescence lifetime and pO2 (right) of an HT-29 cell spheroid stained with (B) Ir-1 and (C) Ir-2 by metabolic stimulation with FCCP (1 μM) at 5 min and 6 min, respectively, and (D) Ir-1 and (E) Ir-2 by metabolic stimulation with AntA (1 μM) at 5 min. Scale bar: 100 µm.
In vivo O2 imaging of hepatic tissues
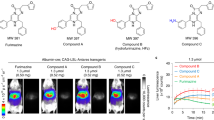
Since we confirmed that Ir-1 and Ir-2 can be efficiently taken up into spheroids and evaluate the oxygen status, we next attempted to visualize the oxygen gradient of hepatic lobules by in vivo PLIM measurements with Ir-1 or Ir-2 as a probe. Each probe (250 nmol in 25 μL dimethyl sulfoxide, diluted using PBS to 175 μL) was administered intravenously to anesthetized mice. Approximately 10 min after probe administration, the abdomen was opened to expose the liver, and the phosphorescence lifetime images of hepatic tissues (~ 10 μm from the surface of the liver) were measured around the central vein (CV) using different magnifications. Both probes gave clear PLIM images of hepatic tissues with cellular-level resolution (Fig. 5). Here, the areas that appear black are the regions where the emission intensity from the probe is extremely low, and these mainly correspond to the nucleus and sinusoid that have low probe uptake. It can be seen from the PLIM images measured with a 40 × objective lens that Ir-1 and Ir-2 are internalized into hepatocytes that line up along sinusoids. The arrangement of hepatocytes and lifetime distribution indicate that these images correspond to a hexagonal hepatic lobule (Fig. 1B) and that CV exists in the area with the longest lifetime (displayed in orange). The region surrounding the CV is considered to be the portal vein (PV) because it has a much shorter lifetime, i.e. much higher oxygen level. The direction of blood flow seen in the video image of hepatic tissues (Video S1) also indicates that the phosphorescence lifetime in the vicinity of CV is longer than that in the surrounding PV. PLIM images obtained with Ir-1 and Ir-2 show a similar lifetime gradient that increases from the PV to CV.

PLIM images of the hepatic surface of a mouse administered (A) Ir-1 and (B) Ir-2. The color bar indicates phosphorescence lifetime in μs. Left images: 10 × objective lens, center images: 20 × objective lens, right images: 40 × objective lens. Scale bar: 200 µm (left images), 100 µm (center images), and 50 µm (right images). CV: central vein, PV: portal vein. (C, D) Phosphorescence lifetime of Ir-1 and Ir-2 and pO2 in hepatic lobules. Average of phosphorescence lifetime and pO2 are shown. N = 23 ROIs for CV, 23 for IR (intermediate region between CV and PV), and 24 for PV in 5 mice administered Ir-1. N = 21 ROIs for CV, 23 for MR, and 27 for PV in 5 mice administered Ir-2. *p value < 0.01 by 2-tailed unpaired t test. Error bar: S.D.
To quantify the O2 levels in specific locations in the lobules from the phosphorescence lifetimes, we performed calibration of the phosphorescence lifetimes using an AML 12 (alpha mouse liver 12) cell line that was established from mouse hepatocytes (Fig. S6). Using the τp0 and kq of Ir-1 and Ir-2 as determined in a monolayer of AML 12 cells, we evaluated the average pO2 of the regions of interest (ROIs) that can be attributed to CV, PV, and the intermediate region (IR); the phosphorescence lifetime of Ir-1 gave reasonable O2 levels (24 ± 6.1 mmHg for CV, 32 ± 4.9 mmHg for IR, and 39 ± 4.2 mmHg for PV), whereas Ir-2 exhibited much lower O2 levels (3 ± 1.8 mmHg for CV, 5 ± 1.8 mmHg for IR, and 7 ± 2.7 mmHg for PV) (Table 2). These results indicated that PLIM images obtained with Ir-1 and Ir-2 clearly visualized the oxygen gradient from PV to CV. However, the O2 levels derived from the phosphorescence lifetimes differed between Ir-1 and Ir-2.
To confirm that Ir-1 and Ir-2 retain their spectral properties in hepatic tissues after systemic administration, we measured the emission spectra of the livers of living mice. Each probe molecule (250 nmol) was administered through the tail vein, and the emission images and spectra of livers were taken using a fluorescence microscope with an excitation filter of 450–500 nm and emission filter of > 532 nm (Fig. S7). The observed emission spectra with maxima at 619 nm for Ir-1 and 607 nm for Ir-2 were slightly red-shifted compared with those in MeCN, but they were in good agreement with those in AML 12 cells, demonstrating that these probes maintain their spectral properties in hepatic tissue.
The reason why Ir-2 gave an abnormally low O2 level seems to be related to the function of the liver to metabolize xenobiotics. So, to clarify how clearance of harmful metabolic byproducts and detoxification of xenobiotics affect hepatic O2 levels, we investigated the detoxification of ammonia in the liver, which has the function of converting toxic ammonia to urea by the urea cycle or to glutamine by glutamine synthesis in hepatocytes. With increased metabolic activity of the liver, oxygen consumption should be enhanced to produce ATP. We intravenously administered NH4Cl (0.13 g/kg) to anesthetized mice at ~ 30 min after 250 nmol Ir-1 injection into the tail vein. PLIM images of hepatic lobules measured 10 min after the administration of NH4Cl exhibited a marked increase in the phosphorescence lifetime of Ir-1 (Fig. 6A,B). The average phosphorescence lifetimes (τp) and pO2 of different ROIs in the pericentral region (ROI1) and periportal regions (ROI2 and ROI3) showed that the lifetimes decreased over time, and the O2 levels recovered to the original levels in approximately 1 h.

(A,B) Variation of PLIM images of hepatic lobules after administration of NH4Cl following Ir-1 injection into the tail vein of a mouse. Scale bar: 200 µm. (C,D) Variation of average phosphorescence lifetime and pO2 of ROIs in the pericentral region (ROI1) and periportal regions (ROI2 and ROI3) shown in B. Error bar: S.D.
To clarify the difference in in vivo probe performance between Ir-1 and Ir-2, we performed O2 imaging experiments on liver tissues using another Ir(III) complex, (btp)2Ir(acac-2OH) (Ir-3), which has a similar structure to Ir-1 (Fig. S8A). The \(\tau_{{\text{p}}}^{0}\) and kq of Ir-3 in AML 12 cells were determined to be 4.80 µs and 4.82 × 103 mmHg−1 s−1. The PLIM image (Fig. S8B) in the hepatic lobule of an Ir-3-administered mouse exhibited a clear lifetime gradient and gave an average pO2 of 16 ± 3.2 mmHg in CV, 21 ± 3.4 mmHg in IR, and 24 ± 3.8 mmHg in PV (Fig. S8C). The hepatic O2 levels obtained with Ir-3 were close to those obtained with Ir-1, although Ir-3 gave somewhat smaller pO2 values.
In contrast to the results in the liver tissue, Ir-1 and Ir-2 gave similar O2 tensions in the kidney (Fig. S9). Intravenously administered probes rapidly migrated from the vasculature into the proximal tubule cells and were hardly excreted in the urinary space during the observation period. The average O2 tension of tubular cells in each image was derived using calibration lines obtained for human kidney 2 (HK-2) cells to be 45 ± 9.8 mmHg for Ir-1 and 36 ± 4.3 mmHg for Ir-2 (Fig. S10).
Discussion
We first verified that Ir-1 and Ir-2 exhibit desired photophysical properties as O2 probes in cells and cell spheroids. Although the τp0 values (5–10 μs) of Ir-1 and Ir-2 are much smaller than those of Pt(II)- and Pd(II)-porphyrins, their oxygen sensitivity is sufficiently high due to their large kq values. In addition, the moderately long lifetimes of the Ir(III) complexes have the advantage of increasing the counting efficiency in PLIM measurements and thus shortening the image acquisition time.
One key issue in applying small-molecule intracellular oxygen probes for in vivo O2 measurements is the method of lifetime calibration. We quantified the tissue pO2 based on the kq and τp0 determined using a cell line that is the same as or close to the cell type in the target organ in which the probe is accumulated. The O2 quenching rate constants in HT-29 and AML 12 cells were reduced by an order of magnitude compared with those in solution (see Table 1). Since lipophilic Ir-1 and Ir-2 are likely to accumulate in organelle membranes after they pass through the plasma membranes of living cells, the much smaller kq values in cells can be attributed, at least in part, to the decrease in the diffusion rates of O2 and Ir(III) complexes in organelle membranes. Further reductions in kq values can be caused by the binding of excited probe molecules to proteins in the organelle membrane in which they are incorporated. The τp0 values of Ir-1 and Ir-2 in cells were decreased by 10–15% compared with those in MeCN. One possible reason lies in the concentration quenching in organelle membranes. The τp0 of Ir-1 partitioned into DMPC liposomes tended to shorten with increasing probe concentration in solution (Table S1), suggesting that concentration quenching may have occurred. Cross-sensitivity to endogenous species other than O2 cannot be ruled out, but its contribution appears to be relatively small because the τp0 values of Ir-1 and Ir-2 in cells are close to those in MeCN. It was also confirmed that phosphorescence lifetime is almost independent of pH and the presence of glutathione under physiological conditions (Tables S2 and S3).
One of the major advantages of Ir-1 and Ir-2 is their extremely high cellular uptake efficiencies. The O2 imaging experiments using HT-29 cell spheroids (Fig. 3) demonstrated that Ir-1 and Ir-2 are efficiently internalized into the spheroids with a diameter of ~ 200 μm. The penetrative abilities of some molecular O2 probes into cell spheroids reported so far are compared in Table S4. These probes include glucose conjugates of Pt(II)-meso-tetrakis-(pentafluorophenyl)porphyrin20, Pt(II)-5, 10, 15, 20-tetrakis-(4-carboxyphenyl)porphyrin36, a Pt(II) complex bearing a cyclometalating 3-di(2-pyridyl)benzene-based moiety35, and click-assembled oxygen-sensing nanoconjugates with Pd(II) tetracarboxytetrabenzoporphyrin as a phosphorescent core24. Comparing each probe for its concentration in culture medium and the incubation time required for observation of clear luminescence images, it can be seen that Ir-1 and Ir-2 have excellent cell permeability. Furthermore, PLIM measurements with these probes made it possible to track oxygen levels and distributions in spheroids. These properties are very useful for investigating the oxygen status in tissues.
The oxygen gradient in the hepatic lobule has been demonstrated by applying the hypoxia marker 2-nitroimidazole to liver tissue sections53. High-resolution visualization of hepatic oxygen distribution in vivo has been performed by Paxian et al.44 by intravital emission microscopy using [Ru(phen)3]2+ (Tris(1,10-phenanthroline)ruthenium(II)) as an O2 probe. They observed a continuous increase in emission intensity of [Ru(phen)3]2+ from periportal to pericentral regions in rat liver, implying an O2 gradient within the liver tissue. However, the emission intensity depends on the probe distribution in the tissue, so that intensity-based measurements cannot assess the precise pO2 and O2 gradient. As for hepatic tissue oxygen levels, Kietzmann and Jungermann54,55 have reported the pO2 to be 60–65 mmHg in the periportal blood and 30–35 mmHg in the perivenous blood, and they suggested the pO2 level to be approximately 15 mmHg lower in periportal and perivenous cells. Tsukada and Suematsu56 have investigated the average pO2 in hepatic microcirculation by phosphorescence lifetime measurements of BSA (bovine serum albumin)-bound Pd-TCPP (Pd(II)-meso-tetra(4-carboxyphenyl)porphine) in hepatic tissues of mice. They obtained an average pO2 of 59.8 mmHg in portal vessels and 38.9 mmHg in central venules. Our PLIM measurements with Ir-1 showed that the average pO2 of hepatocytes near PV and CV were 39 ± 4.2 mmHg and 24 ± 6.1 mmHg, respectively. Although the hepatic oxygen levels obtained in our study are somewhat lower than those reported for intravascular pO2, these are considered to be reasonable values given the oxygen levels to which hepatocytes are exposed. To the best of our knowledge, the PLIM images shown in Fig. 5 are the first to visualize the oxygen concentration gradient in hepatic lobules in vivo using an intracellular O2 probe.
Imaging the spatiotemporal pO2 changes within the liver microarchitecture provides useful information about the oxygen response and dynamics of the liver at the cellular level during physiological stimulation. Our PLIM measurements with Ir-1 showed that intravenous administration of NH4Cl resulted in a rapid decrease in hepatic tissue O2 levels, and its recovery rate depended on the position in the lobule (Fig. 6). Hepatocytes are known to display considerable functional differences depending on their position along the porto-central axis of the liver lobule, and ammonia detoxification in the liver occurs by two pathways: consumption by urea synthesis in the periportal area and removal by glutamine synthesis in perivenous area. Since these processes require O2 consumption, ammonia stimulation may produce hypoxic regions in the hepatic lobules depending on the O2 consumption rate of hepatocytes.
In HT-29 cell spheroids, Ir-1 and Ir-2 showed almost the same O2 levels and distribution, whereas in hepatic tissues of living mice, Ir-2 gave much lower oxygen levels: 7 ± 2.7 mmHg for the periportal region and 3 ± 1.8 mmHg for the perivenous region (Table 2). In kidneys, on the other hand, both probes accumulated in tubular cells and showed similar oxygen levels: 45 ± 9.8 mmHg for Ir-1 and 36 ± 4.3 mmHg for Ir-2 (Fig. S9). These results suggest that the administration of Ir-2 to mice causes hypoxia in hepatic tissues due to some specific metabolic processes in the liver. Since spectral measurements (Fig. S7) revealed that the luminescent properties of Ir-1 and Ir-2 were maintained in liver tissue, it is likely that Ir-2 is excreted in the bile through solubilization processes that do not affect the luminescent properties such as glucuronidation at the OH group. The luminescence spectrum of the extract from bile 6 h after probe administration showed that Ir-1 was excreted through the bile duct along with bile (Fig. S11). Considering that Ir-3, which has the same acac moiety as Ir-1, gave similar liver tissue O2 levels as Ir-1, the lipophilic Tris-ligand structure of Ir-2 may be related to its metabolism in the liver.
Conclusions
Small-molecule O2 probes, Ir-1 and Ir-2, were efficiently taken up into cells when added to the culture media of monolayer cells and cell spheroids, and this enabled reversible pO2 measurements in the cells. PLIM measurements of mouse livers following intravenous administration of Ir-1 or Ir-2 allowed high-resolution O2 imaging of hepatic tissues, exhibiting an O2 gradient from the pericentral to periportal regions in hepatic lobules. The phosphorescence lifetime of Ir-1 gave reasonable hepatic O2 levels after calibrating the lifetime using cultured AML 12 cells. Furthermore, Ir-1 allowed visualization of the pO2 changes in hepatic tissues stimulated by ammonia. However, Ir-2 gave a much lower pO2 compared with Ir-1, which may be due to the toxicity of Ir-2, which promoted detoxification in the liver. These results reveal that Ir(III) complexes allow imaging of spatiotemporal changes in oxygen levels within the tissue microarchitecture in vivo, but some complexes may influence oxygen consumption in the liver when used for oxygen imaging of hepatic tissues.
Methods
Materials
The Ir(III) complexes used in this study were synthesized and characterized according to the methods described in the Supplementary Information.
Acetonitrile (MeCN; Kanto Chemical, spectroscopic grade) and NH4Cl (Kanto Chemical, special grade) were used as received. Antimycin A from Streptomyces sp. (Ant A) and carbonyl cyanide 4-(trifluoromethoxy) phenylhydrozone (FCCP, 98%) were purchased from Sigma-Aldrich. Cell Counting Kit-8 (CCK-8) was purchased from Dojindo Laboratories.
1H-NMR spectra were recorded with a JEOL JNM-ECS400 (400 MHz) spectrometer in DMSO-d6. 1H-NMR chemical shifts were referenced to tetramethylsilane. The apparent resonance multiplicity was described as s (singlet), d (doublet), dd (double doublet), t (triplet), and m (multiplet). ESI–MS spectra were recorded on an Applied Biosystems API 2000 mass spectrometer.
Photophysical properties in solution
Absorption spectra were recorded on a UV/Vis spectrophotometer (Ubest-V550, JASCO). Emission spectra were measured with a system consisting of a monochromatized Xe arc lamp, a sample holder, and a photonic multichannel analyzer (C10027-01, Hamamatsu Photonics), and were fully corrected for spectral sensitivity between 200–950 nm. Phosphorescence lifetimes in solution were measured with a lifetime measurement system (Quantaurus-Tau C11367G, Hamamatsu Photonics) based on the time-correlated single photon counting (TCSPC) method. Phosphorescence quantum yield was measured with an absolute photoluminescence quantum yield measurement system (C9920-02, Hamamatsu Photonics) that consisted of a Xe arc lamp, a monochromator, an integrating sphere, a multichannel detector, and a personal computer57.
Cell and cell spheroid cultures
Human colorectal adenocarcinoma (HT-29) cells and HeLa cells were purchased from the American Type Culture Collection (ATCC). Alpha mouse liver 12 cells (AML12 cells) were kindly provided by Prof. T. Inagaki of the Laboratory of Epigenetics and Metabolism, IMCR, Gunma University. Human kidney 2 cells (HK-2 cells) were kindly provided by Prof. M. Nangaku of the Graduate School of Medicine, the University of Tokyo. HT-29 cells were incubated in McCoy’s 5A medium (Gibco) containing 10% fetal bovine serum (FBS), penicillin (50 units/mL), and streptomycin (50 µg/mL). AML12 cells and HK-2 cells were incubated at 37 °C in a mixture of Dulbecco’s Modified Eagle’s Medium (DMEM) and Ham’s F-12 Nutrient Mixture containing 10% FBS, penicillin (50 units/mL), and streptomycin (50 µg/mL). All cells were grown in a humidified 5% CO2 enriched atmosphere at 37 °C. In PLIM imaging, all cells were cultured on glass-based dishes, and the medium was changed to McCoy’s 5A or DMEM/F-12 without phenol red and FBS prior to the measurements.
HT-29 cell spheroids were prepared using a low attachment 96-well NanoCulture Plate (NCP; NCP-LH96, ORGANOGENICS) with a micro-honeycomb pattern on the film that comprises the bottom of the plate. McCoy’s 5A medium (100 μL) containing 10% FBS, penicillin (50 units/mL), and streptomycin (50 µg/mL) was added to each well in the NCP, and the NCP was centrifuged at 10 × 100 rpm for 5 min to remove the bubbles in the media in the wells. Then HT-29 cells (1.0 × 105 cells/mL) suspended in 100 μL McCoy’s 5A medium were added to each well in the NCP. The cells were incubated in the NCP at 37 °C, 5% CO2 for 4 days to form the HT-29 cell spheroids.
PLIM imaging of cultured cells
PLIM images were measured using an inverted fluorescence microscope (IX73, Olympus) equipped with a picosecond diode laser (BDL-SMC, Becker & Hickl; wavelength: 488 nm, pulse width: 40–90 ps, repetition rate: 50 MHz) and a confocal scanning system (DCS-120, Becker & Hickl). A 4 × objective (UPlanSApo; NA 0.16, Olympus), 10 × objective (UPlanSApo; NA 0.40, Olympus), 20 × objective (UCPlanFLN; NA 0.70, Olympus), 40 × oil objective (UPlanFLN; NA 1.30, Olympus), or 100 × oil objective (UPlanSApo; NA 1.40, Olympus) were used for PLIM imaging. The emission, as guided through a 1 mm pinhole, was collected through a longpass filter and detected with a hybrid detector module (HPM-100–40, Becker & Hickl). Time-resolved emission measurements were performed with a time-correlated single photon counting (TCSPC) system (Simple-Tau-150-DX, Becker & Hickl). The emission signals were acquired at a resolution of 128 × 128 pixels, and the decay curve in each pixel was fitted to single- or double-exponential decay function. In the case of double-exponential decay, the amplitude average lifetime was used for Stern–Volmer analyses. PLIM images were analyzed with SPCImage data analysis software (Becker and Hickl). In PLIM imaging of cell spheroids, HT-29 sell spheroids were incubated in the McCoy’s 5A medium containing a final concentration of 500 nM Ir-1 or Ir-2 for 24 h at 37 °C, 5% CO2 prior to the measurements.
In vivo PLIM measurements
All protocols for animal experiments were approved by the Ethical Committee on Animal Experiments of Gunma University (18-018), and all animal experiments were conducted in accordance with the institutional guidelines. In vivo PLIM measurements were carried out using six- to eight-week-old Balb/c male mice (CLEA Japan). After general anesthesia, probe solution (200 µL) containing 250 nmol Ir(III) complex in 10% DMSO/saline was injected into the tail vein, and the liver or kidney was exposed for ~ 10 min before PLIM experiments.
Calibration of the phosphorescence lifetime
The oxygen partial pressure in cells was obtained from the phosphorescence lifetime based on a calibration using cultured cells. HT-29, AML12, or HK-2 cells were seeded on glass-based dishes and stained with Ir-1 or Ir-2 (500 nM, 2 h). PLIM images were acquired for the stained cells at 21%, 15%, 10%, 5%, and 0% O2 in an incubator at 37 °C. Here, in the 5–21% O2 experiments, cells were incubated with Ant A (10 µM, > 0.5 h) to block cellular respiration, and in the 0% O2 experiments, fresh medium containing Na2SO3 (500 mM) was used to remove existing O2 in the medium. The average phosphorescence lifetimes of the PLIM images taken under different pO2 were plotted according to the Stern–Volmer equation, τp0/τp = 1 + kqτp0pO2, to determine the bimolecular quenching rate constant, kq. The pO2 of cells, spheroids, and tissues was obtained from the phosphorescence lifetime (τp) using the kq and τp0.
Subcellular localization of probes
HeLa cells were cultured in DMEM (Gibco) with 10% FBS, penicillin (50 units/mL), and streptomycin (50 μg/mL), and were grown under a 5% CO2 atmosphere at 37 °C. The cells were seeded into glass bottom imaging dishes (Greiner) and allowed to adhere for 24 h, incubated with Ir-1 (500 nM) or Ir-2 (1 μM) for 2 h, washed 3 times with DMEM, and then the medium was replaced with DMEM (FluoroBrite, Gibco) without FBS. Luminescence microscopic images were obtained with an inverted microscope (IX71, Olympus) equipped with an electron multiplying CCD camera (Evolve 512, PHOTOMETRICS) driven by MetaMorph software.
Evaluation of cytotoxicity using CCK-8 assay
HT-29 cells (2.5 × 104 cells/well) were seeded into a 96-well flat bottom plates (Greiner) for 48 h. The cells were incubated with various concentrations of each probe for 24 h at 37 °C under a 5% CO2 atmosphere. The medium was removed, and the cells were washed gently with McCoy’s 5A medium without phenol red. Cell Counting Kit-8 reagent (CCK-8, Dojindo) was added to each well, and incubation was continued for 1 h58. The absorbance at 450 nm of each well referenced at 650 nm was recorded using a microplate reader (Infinite 200 PRO, Tecan). Cell viability (% of control) was evaluated as (Asample − Ablank)/(Acontrol − Ablank) × 100, where Asample is the absorbance of cells exposed to the probe, Acontrol is the absorbance of cells without probe, and Ablank is the absorbance of wells without cells.
Mitochondrial membrane potential
HT-29 cells (3.0 × 104 cells/well) were seeded into a 96-well flat bottom plates (Greiner) for 48 h. The cells were stained with Ir-1 or Ir-2 (1 μM, 2 h). The medium was removed, and the cells were washed gently with McCoy’s 5A medium without phenol red. 5,5′,6,6′-tetrachloro-1,1′-3,3′-tetraethyl-benzimidazolylcarbocyanine iodide (JC-1, Thermo Fisher Scientific) (5 μM) was added to each well, and incubation was continued for 30 min. The medium was removed, and the cells were washed gently with McCoy’s 5A medium without phenol red. The emission intensities of JC-1 were measured using a microplate reader (Infinite 200 PRO, Tecan) equipped with a gas control module (GCM, Tecan). The excitation wavelength was 488 nm, and the monitor wavelength was 595 nm for Red emission (aggregate) and 540 nm for green emission (monomer).
References
Mimura, I. & Nangaku, M. The suffocating kidney: tubulointerstitial hypoxia in end-stage renal disease. Nat. Rev. Nephrol. 6, 667–678 (2010).
Arden, G. B. & Sivaprasad, S. Hypoxia and oxidative stress in the causation of diabetic retinopathy. Curr. Diabetes Rev. 7, 291–304 (2011).
Vaupel, P. & Mayer, A. Hypoxia in cancer: significance and impact on clinical outcome. Cancer Metastasis Rev. 26, 225–239 (2007).
Rivas, L. et al. Micro-needle implantable electrochemical oxygen sensor: ex-vivo and in-vivo studies. Biosens. Bioelectron. 153, 112028 (2020).
Bolger, F. B. et al. Characterisation of carbon past electrodes for real-time amperometric monitoring of brain tissue oxygen. J. Neurosci. Methods 195, 135–142 (2011).
O’Connor, J. P. B., Robinson, S. P. & Waterton, J. C. Imaging tumour hypoxia with oxygen-enhanced MRI and BOLD MRI. Br. J. Radiol. 92, 20180642 (2019).
Feldman, S. C. et al. The blood oxygen level-dependent functional MR imaging signal can be used to identify brain tumors and distinguish them from normal tissue. AJNR Am. J. Neuroradiol. 30, 389–395 (2009).
Löck, S. et al. Residual tumour hypoxia in head-and-neck cancer patients undergoing primary radiochemotherapy, final results of a prospective trial on repeat FMISO-PET imaging. Radiother. Oncol. 124, 533–540 (2017).
Khan, M., Williams, B. B., Hou, H., Li, H. & Swartz, H. M. Repetitive tissue pO2 measurements by electron paramagnetic resonance oximetry: current status and future potential for experimental and clinical studies. Antioxid. Redox Signal 9, 1169–1182 (2007).
Weaver, J., Burks, S. C., Lie, K. J., Kao, J. P. Y. & Rosen, G. M. In vivo EPR oximetry using an isotropically-substitutes nitroxide: Potential for quantitative measurement of tissue oxygen. J. Magn. Reson. 271, 68–74 (2016).
Okuda, K. et al. 2-nitroimidazole-tricarbocyanine conjugate as a near-infrared fluorescent probe for in vivo imaging of tumor hypoxia. Bioconjugate Chem. 23, 324–329 (2012).
Piao, W. et al. Development of azo-based fluorescent probes to detect different levels of hypoxia. Angew. Chem. Int. Ed. 52, 13028–13032 (2013).
Uddin, M. I. et al. In vivo imaging of retinal hypoxia in a model of oxygen-induced retinopathy. Sci. Rep. 6, 31011 (2016).
Papkovsky, D. B. & Dmitriev, R. I. Imaging of oxygen and hypoxia in cell and tissue samples. Cell. Mol. Life Sci. 75, 2963–2980 (2018).
Roussakis, E., Li, Z., Nichols, A. J. & Evans, C. L. Oxygen-sensing methods in biomedicine from the macroscale to the microscale. Angew. Chem. Int. Ed. 54, 8340–8362 (2015).
Papkovsky, D. B. & Dmitriev, R. I. Biological detection by optical sensing. Chem. Soc. Rev. 42, 8700–8732 (2013).
Li, J.-B. et al. Recent progress in small-molecule near-IR probes for bioimaging. Trends Chem. 1, 224–234 (2019).
Yoshihara, T., Hirakawa, Y., Hosaka, M., Nangaku, M. & Tobita, S. Oxygen imaging of living cells and tissues using luminescent molecular probes. J. Photochem. Photobiol. C Photochem. Rev. 30, 71–95 (2017).
Tobita, S. & Yoshihara, T. Intracellular and in vivo oxygen sensing using phosphorescent iridium(III) complexes. Curr. Opin. Chem. Biol. 33, 39–45 (2016).
Dmitriev, R. I. et al. Small molecule phosphorescent probes for O2 imaging in 3D tissue models. Biomater. Sci. 2, 853–866 (2014).
Dmitriev, R. I., Ropiak, H. M., Ponomarev, G. V., Yashunsky, D. V. & Papkovsky, D. B. Cell-penetrating conjugates of coproporphyrins with oligoarginine peptides: rational design and application for sensing intracellular O2. Bioconjugate Chem. 22, 2507–2518 (2011).
Esipova, T. V. et al. Oxyphor 2P: a high-performance probe for deep-tissue longitudinal oxygen imaging. Cell Metab. 29, 1–9 (2019).
Roussakis, E., Spencer, J. A., Lin, C. P. & Vinogradov, S. A. Two-photon antenna-core oxygen probe with enhanced performance. Anal. Chem. 86, 5937–5945 (2014).
Nichols, A. J., Roussakis, E., Klein, O. J. & Evans, C. L. Click-assembled, oxygen-sensing nanoconjugates for depth-resolved, near-infrared imaging in a 3D cancer model. Angew. Chem. Int. Ed. 53, 3671–3674 (2014).
Hara, D. et al. Tracking the oxygen status in the cell nucleus with a hoechst-tagged phosphorescent ruthenium complex. ChemBioChem 19, 956–962 (2018).
Yoshihara, T., Yamaguchi, Y., Hosaka, M., Takeuchi, T. & Tobita, S. Ratiometric molecular sensor for monitoring oxygen levels in living cells. Angew. Chem. Int. Ed. 51, 4148–4151 (2012).
Yasukagawa, M., Yamada, K., Tobita, S. & Yoshihara, T. Ratiometric oxygen probes with a cell-penetrating peptide for imaging oxygen levels in living cells. J. Photochem. Photobiol. A Chem. 383, 111983 (2019).
Liu, Y. et al. Quantitative mapping of liver hypoxia in living mice using time-resolved wide-field phosphorescence lifetime imaging. Adv. Sci. 1902929, 1–11 (2020).
Felder, A. E., Wanek, J., Teng, P. Y., Blair, N. P. & Shahidi, M. A method for volumetric retinal tissue oxygen tension imaging. Curr. Eye Res. 43, 122–127 (2018).
Akiyama, H. et al. Ir(III) complex-based oxygen imaging of living cells and ocular fundus with a gated ICCD camera. Photochem. Photobiol. Sci. 17, 846–853 (2018).
Esipova, T. V. et al. Two new “protected” oxyphors for biological oximetry: properties and application in tumor imaging. Anal. Chem. 83, 8756–8765 (2011).
Jahn, K., Buschmann, V. & Hille, C. Simultaneous fluorescence and phosphorescence lifetime imaging microscopy in living cells. Sci. Rep. 5, 14334 (2015).
Becker, W., Su, B. & Bergmann, A. Simultaneous fluorescence and phosphorescence lifetime imaging. Proc. SPIE 7903, 790320 (2011).
Zhdanov, A. V., Golubeva, A. V., Okkelman, I. A., Cryan, J. F. & Papkovsky, D. B. Imaging of oxygen gradients in giant umbrella cells: an ex vivo PLIM study. Am. J. Physiol. Cell Physiol. 309, C501–C509 (2015).
Raza, A. et al. Oxygen mapping of melanoma spheroids using small molecule platinum probe and phosphorescence lifetime imaging microscopy. Sci. Rep. 7, 10743 (2017).
Kurokawa, H. et al. High resolution imaging of intracellular oxygen concentration by phosphorescence lifetime. Sci. Rep. 5, 10657 (2015).
Dmitriev, R. I., Zhdanov, A. V., Nolan, Y. M. & Papkovsky, D. B. Imaging of neurosphere oxygenation with phosphorescent probes. Biomaterials 34, 9307–9317 (2013).
Zhdanov, A. V. et al. Quantitative analysis of mucosal oxygenation using ex vivo imaging of healthy and inflamed mammalian colon tissue. Cell. Mol. Life Sci. 74, 141–151 (2017).
Lecoq, J. et al. Simultaneous two-photon imaging of oxygen and blood flow in deep cerebral vessels. Nat. Med. 17, 893–898 (2011).
Parpaleix, A., Houssen, Y. G. & Charpak, S. Imaging local neuronal activity by monitoring PO2 transients in capillaries. Nat. Med. 19, 241–246 (2013).
Spencer, J. A. et al. Direct measurement of local oxygen concentration in the bone marrow of live animals. Nature 508, 269–273 (2014).
Christodoulou, C. et al. Live-animal imaging of native haematopoietic stem and progenitor cells. Nature 578, 278–283 (2020).
Şencan, I. et al. Two-photon phosphorescence lifetime microscopy of retinal capillary plexus oxygenation in mice. J. Biomed. Opt. 23, 126501 (2018).
Paxian, M., Keller, S. A., Cross, B., Huynh, T. T. & Clemens, M. G. High-resolution visualization of oxygen distribution in the liver in vivo. Am. J. Physiol. Gastrointest. Liver Physiol. 286, G37–G44 (2004).
Zhang, S. et al. Phosphorescent-light emitting iridium complexes serve as a hypoxia-sensing probe for tumor imaging in living animals. Cancer Res. 70, 4490–4498 (2010).
Yoshihara, T. et al. Intracellular and in vivo oxygen sensing using phosphorescent Ir(III) complexes with a modified acetylacetonato ligand. Anal. Chem. 87, 2710–2717 (2015).
Hirakawa, Y. et al. Intravital phosphorescence lifetime imaging of the renal cortex accurately measures renal hypoxia. Kidney Int. 93, 1483–1489 (2018).
Yoshihara, T., Hirakawa, Y., Nangaku, M. & Tobita, S. Hydrophilic Ir(III) complexes for in vitro and in vivo oxygen imaging. In RSC Detection Science: Quenched-Phosphorescence Detection of Molecular Oxygen: Applications in Life Sciences, Chap. 4 (eds Papkovsky, D. B. & Dmitriev, R. I.) 71–90 (Royal Society of Chemistry, London, 2018).
Hijmans, B. S., Grefhorst, A., Oosterveer, M. H. & Groen, A. K. Zonation of glucose and fatty acid metabolism in the liver: mechanism and metabolic consequences. Biochimie 96, 121–129 (2014).
Kietzmann, T. Liver zonation in health and disease: hypoxia and hypoxia-inducible transcription factors as concert masters. Int. J. Mol. Sci. 20, 2347 (2019).
Montalti, M., Credi, A., Prodi, L. & Gandolfi, M. T. Handbook of Photochemistry 3rd edn. (CRC-Taylor & Francis, Boca Raton, 2006).
Hasebe, N. et al. Phosphorescence quenching of neutral and cationic iridium(III) complexes by molecular oxygen and aromatic electron acceptors. J. Photochem. Photobiol. A Chem. 324, 134–144 (2016).
Arteel, G. E., Thurman, R. G., Yates, J. M. & Raleigh, J. A. Evidence that hypoxia markers detect oxygen gradients in liver: pimonidazole and retrograde perfusion of rat liver. Br. J. Cancer 72, 889–895 (1995).
Kietzman, T. Metabolic zonation of the liver: The oxygen gradient revisited. Redox Biol. 11, 622–630 (2017).
Jungermann, K. & Kietzmann, T. Oxygen: modulator of metabolic zonation and disease of the liver. Hepatology 31, 255–260 (2000).
Tsukada, K. & Suematsu, M. Visualization and analysis of blood flow and oxygen consumption in hepatic microcirculation: application to an acute hepatitis model. J. Vis. Exp. e3996, 1–5 (2012).
Suzuki, K. et al. Reevaluation of absolute luminescence quantum yields of standard solutions using a spectrometer with an integrating sphere and a back-thinned CCD detector. Phys. Chem. Chem. Phys. 11, 9850–9860 (2009).
Yoshihara, T. et al. Mitochondria-targeted oxygen probes based on cationic iridium complexes with a 5-amino-1,10-phenanthroline ligand. J. Photochem. Photobiol. A Chem. 299, 172–182 (2015).
Acknowledgements
This work was supported in part by JSPS KAKENHI (Grant Numbers: JP 18H03509 to S. T. and JP 19K22947 to T.Y.) and a Grant-in-Aid for Scientific Research on Innovative Areas (No. 26111012 to S.T.). The authors thank Prof. Takeshi Inagaki for providing AML 12 cells.
Author information
Authors and Affiliations
Contributions
T.Y., K.M. and S.T. designed the experiment. K.M., A.K. and S.S. carried out the experiments. S.T., N.G. and K.M. wrote the manuscript. All authors reviewed the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary Video.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Mizukami, K., Katano, A., Shiozaki, S. et al. In vivo O2 imaging in hepatic tissues by phosphorescence lifetime imaging microscopy using Ir(III) complexes as intracellular probes. Sci Rep 10, 21053 (2020). https://doi.org/10.1038/s41598-020-76878-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-76878-6
This article is cited by
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
