
Abstract
MicroRNAs (miRNAs) are implicated in every stage of carcinogenesis and play an essential role as genetic biomarkers of cancer. We aimed to evaluate microRNA-34a gene (MIR34A) expression in colorectal cancer (CRC) tissues compared with non-cancer one and to preliminarily explore the association of one related variant to CRC risk. A total of 116 paraffin-embedded colon specimens were enrolled. MiR-34a was quantified by qPCR, and rs2666433 (A/G) genotyping was performed by TaqMan Real-Time PCR. Also, the somatic mutation burden was assessed. MIR34A expression in the CRC specimens was significantly upregulated (median = 21.50, IQR: 7.0–209.2; P = 0.001) relative to the non-cancer tissues. Allele (A) was highly prevalent in CRC tissues represented 0.56 (P < 0.001). AA/AG genotype carriers were 5.7 and 2.8 more likely to develop cancer than GG carriers. Tumor-normal tissue paired analysis revealed genotype concordance in 33 out of 58 tissue samples. Approximately 43% of the specimens showed a tendency for G to A shift. Additionally, a higher frequency of somatic mutation (92%) was observed in adenocarcinoma (P = 0.006). MIR34A expression and gene variant did not show associations with the clinicopathological data. However, G > A somatic mutation carriers had more prolonged DFS and OS. Bioinformatics analysis revealed miR-34a could target 30 genes that are implied in all steps of CRC tumorigenesis. In conclusion, this study confirms MIR34A upregulation in CRC tissues, and its rs2666433 (A/G) variant showed association with CRC and a high somatic mutation rate in cancer tissues. MiR-34a could provide a novel targeted therapy after validation in large-scale studies.
Similar content being viewed by others


Introduction
Colorectal cancer (CRC) represents the third most frequently diagnosed cancer, and the fourth prime cause of cancer-related mortality worldwide1,2. Although the etiology of colorectal cancer is multifactorial, genetic and epigenetic alterations of proto-oncogenes and tumor suppressor genes remain the fundamental mechanism of tumorigenesis3. Increasing interest in non-coding genomic sequences has revealed the recent implication of several classes of non-coding RNAs (ncRNAs), including microRNAs (miRNAs) as a type of posttranscriptional regulators in carcinogenesis4. This group of ncRNAs could exhibit differential expression in many types of cancer, including CRC5, and their dysregulation could promote every stage of carcinogenesis, including cell proliferation, invasion, and metastasis, and confer resistance to apoptosis through interaction with several intracellular signaling networks6,7,8. The thermodynamics of miRNA-mRNA target interactions may be influenced by single nucleotide polymorphisms (SNPs) occurring in the mature sequence of miRNA, resulting in target gene dysregulation with consequent phenotype variations and/or cancer susceptibility9,10.
MiR-34a is one of the emerging microRNAs that are implicated in many cancers, including CRC11,12,13,14. It has been shown that the expression of miR-34a is reduced in primary CRC tissues15. Moreover, CpG methylation of the miR-34a gene (MIR34A) promoter is detected in some colon cancer cell lines16, and its expression could be induced upon p53 activation17. Also, miR-34a could lower cell cycle progression through p53-dependent induction of p21 to alter colon cancer cell proliferation through direct or indirect regulation of the E2F transcription factor family15. Contrary to evidence on the pro-apoptotic role of miR-34a, however, also exists in the literature. It has been demonstrated that miR-34a may cooperate with p21 and 14-3-3σ to override the apoptotic signals generated by p53 activation16. As a controversy of miR-34a role in CRC still exists and also as sequence variations in the miRNA-binding sites could affect either the expression level and/or the oncogenic or tumor-suppressing functions of cancer-associated miRNAs6,9,10, the current study aims to analyze MIR34A expression and rs2666433 (A/G) variant in preliminary samples of archived CRC tissue specimens in comparison to non-cancer tissues and correlate the results to the available clinicopathological data. This could help improve our understanding of the impact of such type of miRNAs in CRC and its potential role as a candidate for the future molecular-based individualized therapy of such lethal cancer.
Results
In silico analysis of miR-34a
Our bioinformatics analysis identified miR-34a-5p to be the most highly significant non-coding microRNA enriched in the colorectal cancer pathway (Fig. 1). It can complement and bind 30 target genes and fine-tuning their expression profile (Fig. 2).

Top microRNAs involved in the colorectal cancer pathway. (A) microRNAs involved in the colorectal cancer KEGG pathway (https://www.kegg.jp/kegg/kegg1.html)18,19. (B) Functional enrichment pathway analysis of miR-34a target genes. [Data source: Diana lab tools]. The top bar indicates the log (P-value) of the implication of the specified pathway in colon cancer. The direction towards red color indicates more significance as the log P < 0.05 equivalent to P < -1.30.

MicroRNA-34a targets the colorectal cancer pathway. Genes targeted by miRNA34a are colored in orange. In the CRC pathway (KEGG: hsa05210)18,19, miR-34a-5p significantly targets 30 genes (P = 0.0013); including B-Raf proto-oncogene, serine/threonine kinase (BRAF), Ras-Related C3 Botulinum Toxin Substrate 2 (RAC2), phosphoinositide-3-kinase regulatory subunit 2 (PIK3R2), phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit alpha (PIK3CA), RAF1, B-cell lymphoma-2 (BCL2), BCL2 associated agonist of cell death (BAD), Baculoviral IAP repeat containing 5 (BIRC5), transforming growth factor beta 1 (TGFB1), TGFB3, TGFB-receptor 2 (TGFBR2), TCF7L1 (Transcription Factor 7 Like 1), ARAF, Tumor Protein P53 (TP53), AKT serine/threonine kinase 2 (AKT2), Jun proto-oncogene, AP-1 transcription factor subunit (JUN), cyclin D1 (CCND1), mothers against DPP homolog 4 (SMAD4), catenin beta 1 (CTNNB1), MutS homolog 6 (MSH6), axis inhibition proteins 2 (AXIN2), MYC proto-oncogene, BHLH transcription factor (MYC), mitogen-activated protein kinase 1 (MAPK1), MAPK3, MAPK8, mitogen-activated protein kinase kinase 1 (MAP2K1), caspase-9 (CASP9), Ral guanine nucleotide dissociation stimulator (RALGDS), lymphoid enhancer binding factor 1 (LEF1), cytochrome C, somatic (CYCS).
Functional enrichment analysis of miR-34a
Both miR-34a-5p and miR-34a-3p were mostly significantly involved in two pathways: namely fatty acid biosynthesis (hsa00061) and fatty acid metabolism (hsa01212). miR-34a-5p was also identified to target specific cancer types; including colorectal cancer, thyroid cancer, non-small cell lung cancer, chronic myeloid leukemia, bladder cancer, pancreatic cancer, glioma, and melanoma, in addition to multiple cancer-related pathways as cell cycle, pathways in cancer, p53 signaling pathway, and proteoglycans in cancer.
In the CRC pathway (KEGG: hsa05210)18,19, miR-34a-5p significantly targets 30 genes (P = 0.0013); which are involved in all steps of colorectal development and progression (Fig. 2). These gene lists included apoptotic genes (BCL2, BAD, BIRC5, and CASP9), proliferative genes (CCND1, TGFB1, and TGFB3), tumor suppressor genes (TGFBR2, TP53, and SMAD4), DNA repair gene (MSH6), oncogene (CTNNB1), transcription factors (JUN, MYC, TCF7L1), and serine-threonine kinases (BRAF, RAF1, ARAF, AKT2, MAPK1, MAPK3, and MAPK8) (Fig. 2). Enrichment of miR-34a in hallmarks of cancer20 revealed to be involved in two main functions: namely resisting cell death (gray color, Fig. 3) and tumor invasion and metastasis (black color, Fig. 3).

MicroRNA-34a is involved in the cancer hallmarks. The thirty target genes in the colorectal cancer pathway are functionally enriched in diverse cancer hallmarks. Data source: Cancer Hallmarks Analytics Tool (https://chat.lionproject.net/)20. Each color indicates a specific cancer hallmark, as indicated in the left colored legend.
Genotyping of MIR34A variant
Genotype frequencies of rs2666433 agreed with HWE in patients (P = 0.34) and controls (P = 0.13). MAF (A allele) accounted for 0.31 in controls. According to the 1000 Genome Project, the same allele frequencies were 0.43 in Africans, 0.31 in East Asians, 0.23 in South Asians, 0.17 in Americans, and 0.09 in Europeans.
Impact of genotypes on cancer risk
On the comparison between malignant and adjacent colon tissues, A allele was highly prevalent in cancer tissues representing a frequency of 0.56, P < 0.001. Correspondingly, AA and AG genotypes were predominant in cancer specimens (34.5% and 43.1%) compared to counterpart non-cancerous tissues (13.8% and 34.5%), respectively. AA and AG were 5.7 and 2.8 more likely to develop cancer than GG (AA versus GG: OR 5.76, 95%CI: 2.02–16.43, AG versus GG: OR 2.88, 95% CI 1.20–6.93) (Table 1).
Somatic mutation burden analysis
Tumor-normal paired analysis revealed genotype concordance in 33 out of 58 tissue samples. However, the rest of the specimen (43.1%) showed a tendency for G to A shift; 8 (13.8%) controls with AG genotype were substituted to AA in paired adjacent cancer tissue, 13 non-malignant samples (22.4%) changed from GG to AG, and 4 samples (6.9%) with GG genotype showed double mutations to AA at both gene loci in malignant tissues derived from the same patients (Table 2).
Transcriptomic signature of miR-34a in CRC
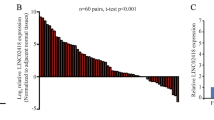
The mean quantitative threshold cycle (Cq) for RNU6B was 38.5 ± 3.23 in cancer specimens compared to 39.4 ± 5.38 in paired samples (P > 0.05). Hence, RNU6B was used in the downstream analysis as an endogenous control. In contrast, Cq of miR-34a-5p varied significantly between the study population; 35.1 ± 2.6 in cancer and 36.7 ± 3.8 in non-cancer (P < 0.001). Marked over-expression of miR-34a was observed in the cancer specimen with a median and quartile levels of 1436.8 (312.6–11,551.6) (Fig. 4A). Therefore, log-transformed values were used for further analyses. No differential expression level of miR-34a was found in patients with different rs2666433 genotypes (P = 0.82) (Fig. 4B).

The relative expression profile of the MIR34A gene in colon cancer specimens. Data are shown as medians and quartiles. Box plot values were log-transformed as data were non-parametric. The box defines upper and lower quartiles (25 and 75%, respectively), and the error bars indicate upper and lower adjacent limits. The fold change was normalized to RNU6B and calculated using the delta-delta quantitative cycle (Cq) method [= 2(-DDCq)] in comparison to non-cancer adjacent tissues. The red dotted line represents the control level. Mann–Whitney U and Kruskal–Wallis tests were applied. (A) Overall samples. (B) Stratified by rs2666433 genotype.
Association of MIR34A expression and variant with clinicopathological features
As depicted in Table 3 and Suppl. Fig. S1, no association was found between any of the patient characteristics and miR-34a expression or polymorphism. However, patients harboring G > A somatic mutation had a more prolonged DFS (P = 0.003) and OS (P < 0.001) than non-carriers. Also, unlike all other types of colon cancer, a higher frequency of somatic mutation (92%) was observed in adenocarcinoma (P = 0.006) (Table 4).
Discussion
Given the advancement in the "high-throughput genome-wide profiling" and "screening technologies", newly emerged miRNA signatures and several "miRNA–mRNA" crosstalk have been identified in CRC21. An example of those signatures is the MIR34A gene expression, which plays a critical role in all stages of colorectal carcinogenesis, starting from colon epithelium proliferation, dysplasia, early/late adenoma, and progression to malignant neoplasm (Fig. 2). The present study identified significant upregulation of miR-34a in CRC tissues relative to normal tissues. In contrast to other studies that reported p53- and other molecular players-mediated miR-34a down-regulation in CRC tissue/plasma samples15,22,23,24,25,26,27, our finding was in line with that of Aherne and colleagues, who found a significant increase of miR-34a tissue expression in early-stage CRC samples compared to non-malignant ones and in colorectal adenomas relative to polyp and normal tissues28. Interestingly, the latter findings corresponded to the same changes in miR-34a circulating levels in CRC patients, in an independent cohort explored by the same authors. Brunet et al. also, reported overexpression of miR-34a in CRC (stage III) tissue samples relative to normal ones, which support the oncogenic role of miR-34a in the CRC. The observed controversy in results̕ reproducibility in aforementioned studies could reflect variable miRNA expression signatures due to disparities in participant age and/or time of sample collection27, varied sex distribution in the specified study29, racial difference30, tumor sample heterogeneity, and different detection approaches28. Additionally, miR-34a has multiple targets even in the same type of cancer31 (Figs. 2 and 3), as well as being itself a target for other coding32,33 and non-coding RNAs34,35,36,37,38, creating multiple circRNAs/lncRNA-miRNA-mRNA crosstalk networks that either promote or inhibit carcinogenesis in a spatial-, temporal- and cell type-specific pattern. In this sense, more "gene–gene interaction" analyses will better uncover miR-34a and its regulatory genes implicated in the pathogenesis of CRC.
Several molecular pathways have been identified to mediate the miRNA-34a role in this context, including Notch-1 and Notch-2 pathway suppression, which implicated in self-renewal and colon stem cells differentiation39,40, tumor-initiating cells (cancer stem cells) regulation41,42, and Fos-related antigen-1 (FRA1) targeting23 which plays an essential role in mediating the crosstalk between the oncogenic RAS-ERK and TGFβ signaling networks implied in "epithelial-mesenchymal plasticity" during CRC progression43.
It has been reported that miRNA SNPs might also cause an aberrant function of the miRNA in regulating the putative target genes44. Previous researches have shown that MIR34A variants could modulate the susceptibility of individuals to multiple human cancers, including osteosarcoma45, colon cancer46, and breast cancer47. The present results revealed that MIR34A rs2666433 AA and AG genotype carriers were 5.7 and 2.8 more likely to develop cancer than GG carriers. A part of one recent publication related to the study of the MIR34A rs2666433 association with ischemic stroke in Chinese population48, the impact of this variant on CRC risk (or other types of cancer) has not been reported previously. Interestingly, we also found that nearly 43% of the cancer tissues showed a tendency for G to A shift, and a higher frequency of somatic mutation (92%) was observed in the adenocarcinoma subtype of CRC. Although the normal and cancer colon tissues were exposed to the same environmental insult, only the cancer tissues showed the transformation into malignancy, which confirms the contribution of the cell genetic and epigenetic makeup to this transformation. Recently, Sun et al., suggested that the rs2666433 variant may affect the binding of transcription factors to MIR34A promoter sequences49.
Furthermore, Wei et al. reported that ischemic stroke patients with rs2666433 (AA) genotype had a higher level of miR-34a than those with (GG + GA) genotypes48, suggesting that rs2666433 may influence miR-34a expression level in their population. However, we could not find a significant association between the specified microRNA variant and its tissue expression levels in the present samples. The authors confirm the specificity of miRNAs, which is related to the type of the disease (ischemic stroke vs. cancer), the type of cancer (the CRC in the present study), the type of samples (body fluids vs. tissues), and the study population (i.e. ethnicity) among others. The negative result could also be partly related to the limited sample size that warrants further large-scale studies to confirm this finding in CRC tissues. It's worth noting that although this limitation above, an essential element of the validity of our study is its agreement with HWE in both study groups, particularly the controls which exclude any genotyping errors or guided sample selection by the authors. Another raised limitation in this study could be related to evaluation of the study variant in FFPE normal colon tissue samples, which, however, is "a very common source for DNA extraction in the studies regarding microRNAs"47.
In conclusion, the present study revealed miR-34a upregulation in CRC tissues compared to paired non-cancer ones. Moreover, for the first time, the authors reported an association between MIR34A rs2666433 (A/G) variant and CRC risk in the study population with a high rate of the specified miRNA mutation in cancer tissues relative to controls. These results could support the previous evidence of miR-34a implication in CRC pathogenesis and its potential use as a biomarker with other molecular panels or as an individualized therapeutic target in the near future. For results validation, further large-scale studies, including several miRNAs combinations in ethnic different populations, are highly recommended.
Methods
Bioinformatics-based selection of microRNA and its variant
Screening microRNAs involved in the colorectal cancer pathway [KEGG: hsa05210]18,19, we found 621 miRNAs targeting this particular pathway, in which miR-34a-5p ranked the top among them (Fig. 1). Experimentally validated gene targets for miR-34a-5p and miR-34a-3p were obtained from DIANA (https://diana.imis.athena-innovation.gr/), miRDB (https://mirdb.org), miRTarBase v.20 (https://mirtarbase.mbc.nctu.edu.tw/), TargetScanHuman v6.2 (https://www.targetscan. org/), PicTar (https://pictar.mdc-berlin.de/), and miRNAMap v2.0. Network analysis of target genes was carried out via a network analyst, web server (https://www.networkanalyst.ca). Functional enrichment pathway analysis was performed using miRTar. Human tool (https://miRTar.mbc.nctu.edu.tw/) and DIANA-miRPath v3.0 (https://snf-515788.vm.okeanos.grnet.gr/index.php?r=mirpath/reverse) which integrate all miRNA targets at the coding sequences (CDS), 3′ untranslated region )3′UTR), or 5′UTR regions into significant Kyoto encyclopedia of genes and genomes (KEGG) pathways and gene ontology (GO). Next, miR-34a disease interactions were retrieved from the PhenomiR database (https://mips.helmholtz-muenchen.de/phenomir/), miR2Disease database (https://www.miR2Disease.org), miRCancer database (https://mircancer.ecu.edu/about.jsp), ExcellmiRDB (www.excellmirdb.brfjaisalmer.com/), and Human miRNA Disease Database (HMDD) v2.0 (https://cmbi.bjmu.edu.cn/hmdd).
The genetic variant of MIR34A (rs2666433; A>G) is located 2 kb upstream to the mature miRNA sequence and within the intron of the MIR34A host gene (MIR34AHG; n.333-1283T > C) (ensemble.org). It was registered with an overall minor allele frequency (MAF: A allele) in the 1000Genome Project of 0.260.
Sample collection
A total of 116 formalin-fixed, paraffin-embedded (FFPE) specimens were collected retrospectively, including 58 CRC samples and paired 58 non-cancer colon tissues. Specimens were obtained from patients who underwent colon resection for histologically confirmed carcinoma. Paired controls were adjacent tissues obtained from the surgical free margins of each specimen and recorded to be normal by microscopic examination before its parafinization. All retrieved cases were archived in the Department of Pathology, Mansoura University, between 2013 and 2017. Patient data were obtained from medical records. There was no history of neoadjuvant therapy before surgery. Direct contact of patients was performed to complete missing data and follow-up (the last contact was in July 2019). The available follow-up period ranged from 20 to 68 months. Samples with incomplete clinical data or follow-up period, history of receiving any treatment before surgery, and/or diagnosis with malignant disease primarily arising from other organs were excluded. The study was conducted according to the ethical and legal guidelines adopted by the Declaration of Helsinki. Ethical approval for this study was granted by the local Research Ethics Committee (No. MED-2018-3-9-F-7825). The informed consent from the patients was waived from the ethical committee as the authors worked on archived samples.
Histopathological examination
Specimens included adenocarcinoma (n = 39; 67.2%), mucinous carcinoma (n = 8; 13.8%), signet ring cell carcinoma (n = 6; 10.3%), and undifferentiated type (n = 5; 8.60%). Apart from the limited sample size in this pilot study, the low frequency of undifferentiated carcinoma subtype in our cases could be congruent with the relative low frequency of such type of CRC as evidenced previously50, and including only the confirmed immunohistochemical staining for cytokeratin (CK) cases which could additionally contribute to the low number of such cases. Sections were examined for histopathologic diagnosis and tumor, node, metastasis (TNM) staging by an expert pathologist51. Other sections (5 to 8 μm thick) for cancer and paired non-cancer tissues were collected in separate Eppendorf tubes for both miRNA expression and SNP identification analyses.
Gene expression profiling
Total RNAwas purified from the FFPE colon sections using a Qiagen miRNeasy FFPE Kit (Cat # 217504) following the manufacturer's instructions12. RNAconcentration and purity were assessed by a NanoDrop ND-1000 spectrophotometer (NanoDrop Tech., Inc. Wilmington, DE, USA), and the integrity was checked by gel electrophoresis. Specific complementary DNA(cDNA) was prepared using TaqMan MicroRNA Reverse Transcription (RT) kit (P/N 4366596; (Thermo Fisher Scientific, Applied Biosystems, Foster City, CA, USA) for miR-34a-5p (assay ID 000426) as described in our previous publication52. RNU6B exhibited a uniform and stable expression in colon tissues with no significant difference between cancer and non-cancer samples; thus was used as an endogenous control (assay ID 001093). T-Professional Basic, Biometra PCRSystem (Biometra, Germany) was used. Appropriate negative controls were applied in each run to exclude amplicon contamination. The PCRreactions were carried out in triplicate in StepOne Real-Time PCRSystem (Applied Biosystems) using specific TaqMan small RNA assay53. All the steps of the quantitative Real-Time reverse transcription-polymerase chain reaction (qRT-PCR) were run according to the Minimum Information for Publication of Quantitative Real-Time PCR Experiments (MIQE) guidelines54. The relative MIR34A expression levels were calculated using the LIVAK method (2-ΔΔCq), where Delta-Delta quantitative cycle (Cq) = (Cq MIR34A − Cq RNU6B)CRC − (Cq MIR34A − Cq RNU6B)NAT55.
Allelic discrimination analysis
QIAamp DNA FFPE tissue kit (QIAGEN, 56,404) was used for DNA extraction according to the guided protocol. DNA quality/purity was evaluated, as mentioned above. DNA samples from the 58 cancer specimens and 58 non-cancer tissues were genotyped for the MIR34A variant (rs2666433 (A/G), assay ID C___2800266_10) using Taqman Real-Time PCR method as detailed previously56. Appropriate negative controls were applied in each PCR run to avoid the false positive of amplicon contamination. Real-time PCR amplification was performed on StepOne Real-Time PCR System (Applied Biosystems) using the following conditions: an initial hold (95 °C for 10 min) followed by a 40-cycle two-step PCR (95 °C denaturation for 15 s and annealing/extension 60 °C for 1 min). Allelic discrimination was called by the SDS software version 1.3.1 (Applied Biosystems). Genotyping was performed by two persons independently blinded to case/control status. Ten percent of the randomly selected samples were re-genotyped in separate runs to exclude the possibility of false genotype calls, with 100% concordance of the results.
Statistical analysis
Data were managed using SPSS version 24.0, the R packages, and GraphPad Prism version 7.0. Genotype and allele frequencies were calculated within each group. Hardy–Weinberg equilibrium (HWE) was estimated online (https://www.oege.org/software/hwe-mr-calc.shtml) and tested by the goodness of fit. Overall comparison and subgroup analyses were performed. Adjusted odds ratios (OR) with a 95% confidence interval (CI) was calculated to identify the strength of the association between the SNP and cancer risk under various genetic association models48; allelic model (G versus A), homozygote comparison (GG versus AA), heterozygote comparison (AG versus AA), dominant model (GG + AG versus AA), and recessive model (GG versus AG + AA). The Wilcoxon matched-pair signed-rank test was carried out to compare the expression level between cancer samples and their corresponding adjacent non-cancer tissues. Chi-square (2) and Fisher's exact tests were used for qualitative parameters, while quantitative variables were shown as mean ± standard deviation (SD) or median (quartiles) according to data distribution. Spearman's correlation test was applied for correlation analysis. Overall survival time was counted (months) from the date of diagnosis to the date of death or last follow-up before study finalization. The Kaplan–Meier method and the Cox proportional hazard model were carried out to assess survival rates among groups. A two-tailed P-value < 0.05 was considered significant.
Ethical approval
This study had been approved by the local Research & Ethics Committee, NBU, Arar, Saudi Arabia. The informed consent from the patients was waived from the ethical committee as the authors worked on archived FFPE samples.
Data availability
All data generated or analyzed during this study are included in this published article (and its Supplementary Information files).
References
Keum, N. & Giovannucci, E. Global burden of colorectal cancer: emerging trends, risk factors, and prevention strategies. Nat. Rev. Gastroenterol. Hepatol. 16, 713–732. https://doi.org/10.1038/s41575-019-0189-8 (2019).
Dekker, E., Tanis, P. J., Vleugels, J. L. A., Kasi, P. M. & Wallace, M. B. Colorectal cancer. Lancet 394, 1467–1480. https://doi.org/10.1016/S0140-6736(19)32319-0 (2019).
Hong, S. N. Genetic and epigenetic alterations of colorectal cancer. Intest. Res. 16, 327–337. https://doi.org/10.5217/ir.2018.16.3.327 (2018).
Ranganathan, K. & Sivasankar, V. MicroRNAs - Biology and clinical applications. J. Oral Maxillofac. Pathol. 18, 229–234. https://doi.org/10.4103/0973-029X.140762 (2014).
Falzone, L. et al. Integrated analysis of colorectal cancer microRNA datasets: identification of microRNAs associated with tumor development. Aging (Albany NY) 10, 1000–1014. https://doi.org/10.18632/aging.101444 (2018).
Wu, W. K. et al. MicroRNA in colorectal cancer: from benchtop to bedside. Carcinogenesis 32, 247–253. https://doi.org/10.1093/carcin/bgq243 (2011).
Weng, W., Feng, J., Qin, H., Ma, Y. & Goel, A. An update on miRNAs as biological and clinical determinants in colorectal cancer: a bench-to-bedside approach. Future Oncol. 11, 1791–1808. https://doi.org/10.2217/fon.15.83 (2015).
Wang, J., Du, Y., Liu, X., Cho, W. C. & Yang, Y. MicroRNAs as regulator of signaling networks in metastatic colon cancer. Biomed. Res. Int. 2015, 823620. https://doi.org/10.1155/2015/823620 (2015).
Cammaerts, S., Strazisar, M., De Rijk, P. & Del Favero, J. Genetic variants in microRNA genes: impact on microRNA expression, function, and disease. Front Genet 6, 186. https://doi.org/10.3389/fgene.2015.00186 (2015).
Moszynska, A., Gebert, M., Collawn, J. F. & Bartoszewski, R. SNPs in microRNA target sites and their potential role in human disease. Open Biol. https://doi.org/10.1098/rsob.170019 (2017).
Shehata, R. H. et al. Deregulation of miR-34a and Its Chaperon Hsp70 in Hepatitis C virus-Induced Liver Cirrhosis and Hepatocellular Carcinoma Patients. Asian Pac J Cancer Prev 18, 2395–2401. https://doi.org/10.22034/APJCP.2017.18.9.2395 (2017).
Toraih, E. A. et al. MicroRNA-34a: a key regulator in the hallmarks of renal cell carcinoma. Oxid. Med. Cell Longev. 2017, 3269379. https://doi.org/10.1155/2017/3269379 (2017).
Guo, Y., Bao, Y. & Yang, W. Regulatory miRNAs in colorectal carcinogenesis and metastasis. Int. J. Mol. Sci. https://doi.org/10.3390/ijms18040890 (2017).
Toraih, E. A. et al. Dual biomarkers long non-coding RNA GAS5 and microRNA-34a co-expression signature in common solid tumors. PLoS ONE 13, e0198231. https://doi.org/10.1371/journal.pone.0198231 (2018).
Tazawa, H., Tsuchiya, N., Izumiya, M. & Nakagama, H. Tumor-suppressive miR-34a induces senescence-like growth arrest through modulation of the E2F pathway in human colon cancer cells. Proc. Natl. Acad. Sci. USA 104, 15472–15477. https://doi.org/10.1073/pnas.0707351104 (2007).
Lodygin, D. et al. Inactivation of miR-34a by aberrant CpG methylation in multiple types of cancer. Cell Cycle 7, 2591–2600. https://doi.org/10.4161/cc.7.16.6533 (2008).
Hermeking, H. p53 enters the microRNA world. Cancer Cell 12, 414–418. https://doi.org/10.1016/j.ccr.2007.10.028 (2007).
Kanehisa, M., Sato, Y., Kawashima, M., Furumichi, M. & Tanabe, M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 44, D457-462. https://doi.org/10.1093/nar/gkv1070 (2016).
Kanehisa, M. & Goto, S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30. https://doi.org/10.1093/nar/28.1.27 (2000).
Baker, S. et al. Cancer Hallmarks Analytics Tool (CHAT): a text mining approach to organize and evaluate scientific literature on cancer. Bioinformatics 33, 3973–3981. https://doi.org/10.1093/bioinformatics/btx454 (2017).
Chen, B. et al. Emerging microRNA biomarkers for colorectal cancer diagnosis and prognosis. Open Biol. 9, 180212. https://doi.org/10.1098/rsob.180212 (2019).
Chang, T. C. et al. Transactivation of miR-34a by p53 broadly influences gene expression and promotes apoptosis. Mol. Cell 26, 745–752. https://doi.org/10.1016/j.molcel.2007.05.010 (2007).
Wu, J. et al. MicroRNA-34a inhibits migration and invasion of colon cancer cells via targeting to Fra-1. Carcinogenesis 33, 519–528. https://doi.org/10.1093/carcin/bgr304 (2012).
Rokavec, M. et al. IL-6R/STAT3/miR-34a feedback loop promotes EMT-mediated colorectal cancer invasion and metastasis. J. Clin. Invest. 124, 1853–1867. https://doi.org/10.1172/JCI73531 (2014).
Gao, J. et al. miR-34a-5p suppresses colorectal cancer metastasis and predicts recurrence in patients with stage II/III colorectal cancer. Oncogene 34, 4142–4152. https://doi.org/10.1038/onc.2014.348 (2015).
Badr, E. A. E. et al. The clinical impact of miRNA34a and P53 gene expression in colon cancer. Biochem. Biophys. Rep. 16, 88–95. https://doi.org/10.1016/j.bbrep.2018.10.002 (2018).
Oner, M. G. et al. Combined inactivation of TP53 and MIR34A promotes colorectal cancer development and progression in mice via increasing levels of IL6R and PAI1. Gastroenterology 155, 1868–1882. https://doi.org/10.1053/j.gastro.2018.08.011 (2018).
Aherne, S. T. et al. Circulating miRNAs miR-34a and miR-150 associated with colorectal cancer progression. BMC Cancer 15, 329. https://doi.org/10.1186/s12885-015-1327-5 (2015).
Duttagupta, R., Jiang, R., Gollub, J., Getts, R. C. & Jones, K. W. Impact of cellular miRNAs on circulating miRNA biomarker signatures. PLoS ONE 6, e20769. https://doi.org/10.1371/journal.pone.0020769 (2011).
Cheng, H. et al. Circulating plasma MiR-141 is a novel biomarker for metastatic colon cancer and predicts poor prognosis. PLoS ONE 6, e17745. https://doi.org/10.1371/journal.pone.0017745 (2011).
Ebner, O. A. & Selbach, M. Quantitative proteomic analysis of gene regulation by miR-34a and miR-34c. PLoS ONE 9, e92166. https://doi.org/10.1371/journal.pone.0092166 (2014).
Xiao, X. et al. The miR-34a-LDHA axis regulates glucose metabolism and tumor growth in breast cancer. Sci. Rep. 6, 21735. https://doi.org/10.1038/srep21735 (2016).
Huang, X. et al. PDL1 And LDHA act as ceRNAs in triple negative breast cancer byregulating miR-34a. J. Exp. Clin. Cancer. Res. 36, 129. https://doi.org/10.1186/s13046-017-0593-2 (2017).
He, R. et al. circGFRA1 and GFRA1 act as ceRNAs in triple negative breast cancer by regulating miR-34a. J. Exp. Clin. Cancer Res. 36, 145. https://doi.org/10.1186/s13046-017-0614-1 (2017).
Xu, H. et al. NFIX circular RNA promotes glioma progression by regulating miR-34a-5p via notch signaling pathway. Front. Mol. Neurosci. 11, 225. https://doi.org/10.3389/fnmol.2018.00225 (2018).
Huang, X., Gao, Y., Qin, J. & Lu, S. The mechanism of long non-coding RNA MEG3 for hepatic ischemia-reperfusion: mediated by miR-34a/Nrf2 signaling pathway. J. Cell. Biochem. 119, 1163–1172. https://doi.org/10.1002/jcb.26286 (2018).
Tao, F., Tian, X., Lu, M. & Zhang, Z. A novel lncRNA, Lnc-OC1, promotes ovarian cancer cell proliferation and migration by sponging miR-34a and miR-34c. J. Genet. Genom. 45, 137–145. https://doi.org/10.1016/j.jgg.2018.03.001 (2018).
Li, Y. et al. Long non-coding RNA-SNHG7 acts as a target of miR-34a to increase GALNT7 level and regulate PI3K/Akt/mTOR pathway in colorectal cancer progression. J. Hematol. Oncol. 11, 89. https://doi.org/10.1186/s13045-018-0632-2 (2018).
Hermeking, H. The miR-34 family in cancer and apoptosis. Cell Death Differ. 17, 193–199. https://doi.org/10.1038/cdd.2009.56 (2010).
Taketo, M. M. Reflections on the spread of metastasis to cancer prevention. Cancer Prev. Res. (Phila) 4, 324–328. https://doi.org/10.1158/1940-6207.CAPR-11-0046 (2011).
Bu, P. et al. A microRNA miR-34a-regulated bimodal switch targets Notch in colon cancer stem cells. Cell Stem Cell 12, 602–615. https://doi.org/10.1016/j.stem.2013.03.002 (2013).
Bu, P. et al. A miR-34a-Numb feedforward loop triggered by inflammation regulates asymmetric stem cell division in intestine and colon cancer. Cell Stem Cell 18, 189–202. https://doi.org/10.1016/j.stem.2016.01.006 (2016).
Diesch, J. et al. Widespread FRA1-dependent control of mesenchymal transdifferentiation programs in colorectal cancer cells. PLoS ONE 9, e88950. https://doi.org/10.1371/journal.pone.0088950 (2014).
Adams, B. D., Kasinski, A. L. & Slack, F. J. Aberrant regulation and function of microRNAs in cancer. Curr. Biol. 24, R762-776. https://doi.org/10.1016/j.cub.2014.06.043 (2014).
Lv, H., Pei, J., Liu, H., Wang, H. & Liu, J. A polymorphism site in the premiR34a coding region reduces miR34a expression and promotes osteosarcoma cell proliferation and migration. Mol. Med. Rep. 10, 2912–2916. https://doi.org/10.3892/mmr.2014.2582 (2014).
Jiang, H. et al. rs35301225 polymorphism in miR-34a promotes development of human colon cancer by deregulation of 3’UTR in E2F1 in Chinese population. Cancer Cell Int. 17, 39. https://doi.org/10.1186/s12935-017-0402-1 (2017).
Kalapanida, D. et al. Evaluation of pre-mir-34a rs72631823 single nucleotide polymorphism in triple negative breast cancer: a case-control study. Oncotarget 9, 36906–36913. https://doi.org/10.18632/oncotarget.26385 (2018).
Wei, G. J. et al. A genetic variant of miR-34a contributes to susceptibility of ischemic stroke among Chinese population. Front. Physiol. 10, 432. https://doi.org/10.3389/fphys.2019.00432 (2019).
Sun, Y. et al. Sequence variation in microRNA-34a is associated with diabetes mellitus susceptibility in a southwest Chinese Han population. Int. J. Clin. Exp. Pathol. 11, 1638–1644 (2018).
Remo, A. et al. Morphology and molecular features of rare colorectal carcinoma histotypes. Cancers 11, 1036. https://doi.org/10.3390/cancers11071036 (2019).
Akkoca, A. N. et al. TNM and Modified Dukes staging along with the demographic characteristics of patients with colorectal carcinoma. Int. J. Clin. Exp. Med. 7, 2828–2835 (2014).
Toraih, E. A., Fawzy, M. S., Mohammed, E. A., Hussein, M. H. & El-Labban, M. M. MicroRNA-196a2 biomarker and targetome network analysis in solid tumors. Mol. Diagn. Ther. 20, 559–577. https://doi.org/10.1007/s40291-016-0223-2 (2016).
Toraih, E. A. et al. MicroRNA-target cross-talks: Key players in glioblastoma multiforme. Tumour Biol. 39, 1010428317726842. https://doi.org/10.1177/1010428317726842 (2017).
Bustin, S. A. et al. The MIQE guidelines: minimum information for publication of quantitative real-time PCR experiments. Clin. Chem. 55, 611–622. https://doi.org/10.1373/clinchem.2008.112797 (2009).
Livak, K. J. & Schmittgen, T. D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25, 402–408. https://doi.org/10.1006/meth.2001.1262 (2001).
Toraih, E. A. et al. Combined genotype analyses of precursor miRNA196a2 and 499a variants with hepatic and renal cancer susceptibility a preliminary study. Asian Pac. J. Cancer Prev. 17, 3369–3375 (2016).
Acknowledgements
This study was supported by research funding from the Deanship of Scientific Research (grant no. MED-2018-3-9-F-7825), Northern Border University (NBU), Arar, Saudi Arabia, and Taif University Researchers supporting project number (TURSP-2020/108), Taif, Saudi Arabia.
Author information
Authors and Affiliations
Contributions
M.S.F. conceived of the study, participated in the design of the study, carried out the molecular lab work, and drafted the manuscript, A.T.I. carried out the pathology lab work and helped draft the manuscript; S.A.A. participated in the design of the study, collected field data, and critically revised the manuscript; B.T.A. participated in the collection of field data and helped draft the manuscript; E.A.T. participated in the molecular lab work, carried out the statistical and in silico analyses and critically revised the manuscript. All authors gave final approval for publication and agreed to be held accountable for the work performed therein.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Fawzy, M.S., Ibrahiem, A.T., AlSel, B.T.A. et al. Analysis of microRNA-34a expression profile and rs2666433 variant in colorectal cancer: a pilot study. Sci Rep 10, 16940 (2020). https://doi.org/10.1038/s41598-020-73951-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-73951-y
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
