
Abstract
Transglutaminase 2 (TG2), also known as tissue transglutaminase, is a calcium-dependent enzyme that has a variety of intracellular and extracellular substrates. TG2 not only increases in osteoarthritis (OA) tissue but also affects the progression of OA. However, it is still unclear how TG2 affects cartilage degradation in OA at the molecular level. Surgically induced OA lead to an increase of TG2 in the articular cartilage and growth plate, and it was dependent on TGFβ1 in primary chondrocytes. The inhibition of TG2 enzymatic activity with intra-articular injection of ZDON, the peptide-based specific TG2 inhibitor, ameliorated the severity of surgically induced OA as well as the expression of MMP-3 and MMP-13. ZDON attenuated MMP-3 and MMP-13 expression in TGFβ- and calcium ionophore-treated chondrocytes in a Runx2-independent manner. TG2 inhibition with ZDON suppressed canonical Wnt signaling through a reduction of β-catenin, which was mediated by ubiquitination-dependent proteasomal degradation. In addition, TG2 activation by a calcium ionophore enhanced the phosphorylation of AMPK and FoxO3a and the nuclear translocation of FoxO3a, which was responsible for the increase in MMP-13. In conclusion, TG2 plays an important role in the pathogenesis of OA as a major catabolic mediator that affects the stability of β-catenin and FoxO3a-mediated MMP-13 production.
Similar content being viewed by others


Introduction
Osteoarthritis (OA), which is the most common form of arthritis, is characterized by damage to the extracellular matrix of cartilage, resulting in the loss of hyaline articular cartilage1. This change is accompanied by subchondral bone remodelling and osteophyte formation at the joint margin1. In this process, the proteases produced by chondrocytes, especially matrix metalloproteinase (MMP)-13 and A Disintegrin and Metalloproteinase with Thrombospondin motifs (ADAMTS)-5, have a major role in the matrix degradation, and proteolytic generation of matrix fragments has been suggested to drive chondrocyte hypertrophy2,3. However, the regulatory mechanisms of MMPs and AMDATS production, as well as the post-transcriptional modification that stabilizes or activates these proteases, have not been fully elucidated4.
Transglutaminase 2 (TG2) is a 78-kDa calcium-dependent enzyme of the protein-glutamine γ-glutamyl-transferase family5,6. In the presence of calcium, TG2 functions as a catalytic enzyme that modifies the protein-bound glutamine side chains, resulting in protein crosslinking through transamidation or deamidation6,7. When this protein binds to GTP intracellularly, it becomes closed conformationally and acts as a G-protein associated signaling molecule8. Through its enzymatic and signaling functions, TG2 has diverse biological roles, such as an enhancer of inflammation, apoptosis regulation, cellular differentiation and extracellular matrix interaction6. In chondrocyte biology, TG2 is known not only as a marker for hypertrophic chondrocyte but also as an inducer of chondrocyte maturation9,10. TG2 knockout mice showed less severe cartilage degradation and enhanced osteophyte formation compared to control wild-type mice in a surgically induced OA model11.
Recently, we reported that enhanced subchondral bone remodelling in early OA can increase the calcium level in articular cartilage, and this change was related to MMP-3 and MMP-13 production12. This increased calcium level in the articular cartilage of early OA can affect the enzyme activity of TG2, which may affect the production and stability of MMPs, and consequently accelerate cartilage degradation. However, the molecular mechanisms of the catabolic role of TG2 in the progression of OA are still unclear. In the present study, we investigated the effect of inhibition of TG2 enzyme activity on the severity of surgically induced OA. At the molecular level, TG2 stabilized β-catenin to increase Wnt signaling and enhanced nuclear translocation of FoxO3a, which was responsible for the production of MMP-3 and MMP-13.
Methods
Surgical induction of OA and tissue preparation
Surgical OA was induced by destabilization of the medial meniscus (DMM) in mice as previously described12. Briefly, after 12-week-old C57BL/6 male mice were anaesthetized, the skin over the left knee was opened, and the meniscotibial ligament of the medial meniscus was sectioned in total 15 mice among which 5 mice were sacrificed at 4 weeks without treatment, and 5 mice were treated with ZDON and 5 with control DMSO for 8 weeks. The sham operation just visualized the meniscotibial ligament but not resected in total 13 mice (n = 5 untreated and sacrificed at 4 weeks, n = 4 treated with ZDON, n = 4 treated with vehicle for 8 weeks). The treatment group mice were intra-articularly injected with 10 μl of ZDON (200 μM), a TG2 inhibitor (Calbiochem, #616467, Darmstadt, Germany) or DMSO alone as a control once a week for 7 weeks and sacrificed at 8 weeks after DMM surgery. After sacrifice, the knee joints were fixed in 4% paraformaldehyde (Merck, Darmstadt, Germany) for 24 h and decalcified in 10% EDTA for 3 weeks. Embedded blocks were sectioned at a thickness of 5 μm and stained with safranin-O. OA severity was scored by two blinded observers using the Osteoarthritis Research Society International (OARSI) grading system13.
All animal protocols were approved by the Institutional Animal Care and Use Committee of the Daegu Fatima Hospital (approved protocol number F-16–4). Mice were maintained in standard laboratory conditions and conformed to the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH publication, 8th Edition, 2011).
Western blot analysis
The Western blot analysis was performed as reported previously14. Total cell lysates were separated by SDS-PAGE and transferred to polyvinylidene difluoride (PVDF) membranes (Amersham Biosciences, Buckinghamshire, UK). The membranes were blocked with 5% bovine serum albumin (BSA) at room temperature (RT) for 1 h, and incubated with the primary antibodies listed next for 1 h with shaking. The primary antibodies against TG2, β-catenin, p-β-catenin, non-p-β-catenin, Ampka, p-Ampka, Foxo3a, p-Foxo3a, p65, p-p65, mTOR, p-mTOR, Axin2, Tcf1, Sirt1, and IκB were purchased from Cell Signaling Technology (Danvers, MA); antibodies against Sox9, Runx2, Mmp3, Mmp13, and Adamts5 were from Abcam (Cambridge, UK); the antibodies against LaminB and haemagglutinin (HA) were from Santa Cruz Biotechnology (Dallas, TX), and β-actin antibody was from Sigma Aldrich (St. Louis, MO). Membranes were washed, and incubated with a horseradish peroxidase (HRP)-conjugated secondary antibody for 1 h. The membrane signal was visualized using Supersignal Chemiluminescent Substrates (Thermo Fisher Scientific, Waltham, MA).
Immunofluorescence and immunohistochemical staining
The immunofluorescence and immunohistochemical staining were performed on the tissue samples of surgical knee OA as reported previously14. For immunofluorescence staining, the rehydrated sections were retrieved in sodium citrate buffer (10 mM sodium citrate, 0.05% Tween 20, pH 6.0). After the sections were blocked with 2% BSA in PBS for 1 h, they were incubated with the primary antibodies for TG2 (1:200 dilution), β-catenin (1:400), non-p-β-catenin (1:400), FoxO3a (1:200), and p-FoxO3a (1:200) or normal rabbit IgG (1:200) in 1% BSA in PBS at 4 ℃ overnight. For immunohistochemistry, the retrieved sections were immunostained for MMP3 (1:200) and MMP13 (1:400) in 1% BSA in PBS for 16 h at 4 ℃ and then with goat anti-rabbit IgG conjugated with HRP (DakoCytomation, Glostrup, Denmark) for 1 h at RT.
For immunocyto-fluorescence, primary chondrocytes were plated on 8-chamber slides (3 × 104 cells/well, Thermo Fisher Scientific) and treated with TGF-β1 (10 ng/ml), with or without calcium ionophore (2 mM) or ZDON (100 μM). The cells were fixed with 4% formalin for 5 min, permeabilized with 0.25% Triton X-100 in PBS for 3 min, blocked with 5% BSA for 1 h, and incubated with rabbit anti-non-p-β-catenin (1:400) and mouse anti-TG2 (1:200) antibodies at RT for 1 h.
Isolation and culture of primary chondrocytes
Primary chondrocytes were prepared from the femur and tibia of postcoital day 15.5 ICR mouse embryos (E15.5) as previously described15. Briefly, the bones were incubated in α-minimum essential medium (α-MEM) supplemented with 0.2% BSA, 0.25 mM ascorbic acid, 1 mM β-glycerophosphate, 0.25% l-glutamine, and 0.25% penicillin/streptomycin for 24 h at 37 °C. The bones were then incubated in 0.25% trypsin–EDTA for 15 min at 37 °C, followed by further incubation with 3 mg/ml type II collagenase (Sigma Aldrich) in Dulbecco’s modified essential medium (DMEM) for 2 h at 37 °C with gentle shaking. Cells were filtered through a 40 μm nylon mesh (BD Biosciences) and collected by centrifugation. The cells cultured in a 2:3 mixture of DMEM and Ham’s F12 (DMEM/F12) with 10% foetal bovine serum (FBS), and 0.25% l-glutamine. For starvation and treatment, DMEM/F12 with 1% FBS was used.
Enzyme activity assay for TG2
Primary chondrocytes were seeded in 6-well plates (5 × 105 cells per well) and treated with TGFβ1 (10 ng/ml) or TGFβ1 plus calcium ionophore (2 mM) with or without ZDON (100 μM) for 24 h. Cell lysates were prepared with a lysis buffer [1X Tris-buffered saline (TBS) pH 7.6, 1% Triton X-100, and 1X protease inhibitors (Calbiochem)]. TG2 activity assays were performed using a TG2 Colorimetric Assay Kit according to the manufacturer's instructions (Novus Biologicals, Co.).
Ubiquitination assay
ATDC5 cells were seeded in 6-well plates (2 × 105 cells/well) and transfected with expression constructs of β-catenin (Santa Cruz) and HA-Ubiquitin16. After 24 h, cells were incubated in the media containing TGFβ1 (10 ng/ml) or TGFβ1 plus calcium ionophore (2 mM) with or without ZDON (100 μM) for 20 h, followed by exposure to 10 μM MG132 for 5 h. Then, cell lysates were prepared with 20 mM HEPES, pH 7.9 buffer containing 300 mM KCl, 10% glycerol, 10% NP-40, 1 mM DTT and 1 × protease inhibitors. Lysates were precleared with Protein A Sepharose (GE Healthcare, Bucks, UK) for 30 min, precipitated with anti-β-catenin antibody for 18 h, incubated with Protein A Sepharose beads for 2 h at 4 ℃ with gentle shaking and subjected to Western blot analysis.
Statistical analyses
Mann–Whitney U tests or Kruskal–Wallis one-way analysis of variance (ANOVA) tests were used to determine differences between means. All analyses were conducted using SPSS version 14.0 software (SPSS, Chicago, IL). The results are presented as the mean ± S.D., and statistical significance was defined as p values of < 0.05.
Results
Surgically induced OA leads to an increase of TG2 in the articular cartilage and growth plate, which is dependent on TGFβ
We first assessed the expression of TG2 in the surgically induced OA model at 4 weeks after DMM surgery (n = 5). TG2 was selectively expressed in the articular cartilage and growth plate of DMM-operated mice but not in the joint tissue of sham-operated mice (Fig. 1a). The TG2 expression in the growth plate apart from articular cartilage suggests the presence of paracrine factors in the regulation of TG2 because DMM surgery primarily induces damage to articular cartilage by the increased compressive load17. To identify paracrine factors regulating TG2 expression, we treated primary chondrocytes with major growth factors known to be important in cartilage biology. TG2 was selectively induced by TGFβ1 in a dose-dependent manner in primary chondrocytes (Fig. 1b,c, see Supplementary figures for the whole blot image).

TG2 is induced in the chondrocytes of articular cartilage and growth plates in surgically induced OA tissue. (a) Representative images (n = 5 per group) of safranin-O (right panel) and TG2 immunostained cartilage (left panel) from DMM- or sham-operated mice at 4 week after operation. Areas in black rectangles in the articular cartilage and growth plate of safranin-O staining are displayed in immunostained images. White-rectangled areas in immunostained images were enlarged on the right side. Black and white bars represent 200 and 100 μm, respectively. (b) TG2 expression by growth factors in vitro. Primary chondrocytes from E15.5 mouse long bones were treated with various growth factors that play a key role in chondrocyte biology for 24 h (TGFβ 10 ng/ml, Wnt3a 10 ng/ml, Ihh 10 ng/ml, BMP2 100 ng/ml and FGF18 10 ng/ml). TG2, Sox9 and Runx2 protein levels were detected by Western blotting. n = 3. (c) Western blotting showing the TG2 increase by TGFβ in a dose-dependent manner. n = 3.
Inhibition of TG2 attenuates the severity of surgically induced OA and the expression of MMP-3 and MMP-13
To investigate the role of TG2 in OA, we surgically induced OA in 12-week-old male mice and administered weekly intra-articular injections of 10 μL of ZDON (200 μM), a cell-permeable, peptide-based inhibitor of TG2 activity. Eight weeks after DMM surgery, ZDON-treated mice showed less severe cartilage damage than control mice (Fig. 2a). In addition, the expression of MMP-13 and MMP-3 in articular cartilage was significantly reduced in ZDON-treated mice (Fig. 2b).

Inhibition of TG2 attenuated the severity of surgically induced OA. (a) Representative images of safranin-O stained knee joint cartilage from sham- and DMM-operated mice (n = 8 and 10, respectively). C57BL/6 male mice at 12 week of age underwent an operation, and were intra-articularly injected with 10 μl of ZDON (200 μM) or DMSO alone as a control once a week for 7 weeks. Cartilage damage was scored by OARSI grading. (b) Representative images of immunostaining for MMP-13 in the articular cartilage. MMP-13-positive cells above the tide mark were quantified with the NIS-Elements programme (Nikon). The proportion of MMP-13-positive cells among DAPI-positive cells is displayed with dot graphs. n = 8 for sham and 10 for DMM group.
Calcium-mediated activation of TG2 enzymatic function is important in MMP-3 and MMP-13 expression and canonical Wnt and AMPKa/FoxO3a signaling
To investigate the mechanism of TG2-mediated cartilage degradation, we conducted an in vitro study using primary chondrocytes from E15.5 mouse long bone. Based on the fact that TG2 is induced by TGFβ1 and is a calcium-dependent enzyme, primary chondrocytes were treated with TGFβ1 (10 ng/ml) or a calcium ionophore, A23187 (2 μM). Enzyme activity assays of TG2 showed that TGFβ1 increased the enzyme activity of TG2 compared to that of the control, but the activity was not suppressed by ZDON. Calcium ionophore treatment in the presence of TGFβ1 significantly increased TG2 activity compared to that of the TGFβ1 alone group, and the activity was suppressed by ZDON (Fig. 3a). Based on the enzyme activity results, we assessed the role of TG2 in the production of major proteases and Runx2 in primary chondrocytes. Although TGFβ1 increased the protein level of TG2, TG2 was significantly decreased after combination treatment with the calcium ionophore. While ZDON did not suppress the TGFβ1-mediated increase of MMP-3 and MMP-13, it significantly suppressed MMP production by the calcium ionophore and TGFβ1. The TGFβ1-mediated ADAMTS-5 and Runx2 increase was decreased by ZDON. However, ZDON failed to suppress the expression of ADAMTS-5 and Runx2 in the presence of the calcium ionophore and TGFβ1 (Fig. 3b, Supplementary figure 9).

Inhibition of TG2 attenuated the protein expression of MMP-3, and MMP-13, and it was associated with decreased Wnt and AMPKa/FoxO3a signaling. (a) TG2 enzyme activity was measured using TG2-specific Colorimetric Assay Kit (Novus Biologics) with cell extracts from primary chondrocytes treated with ZDON (100 μM) in the presence of TGFβ1 (10 ng/ml) or calcium ionophore (2 μM) for 24 h. n = 2. (b) Primary chondrocytes from E15.5 mouse long bones were cultured with TGFβ1 (10 ng/ml) to increase the level of TG2 for 24 h and then treated with a calcium ionophore (2 μM) to activate enzyme activity of TG2 with or without ZDON (100 μM), an inhibitor of TG2, for 24 h. Total cellular lysates were analysed by Western blotting with the indicated antibodies. The band intensities were quantified using the ImageJ programme, normalized relative to the quantity of their respective β-actin bands, and expressed as a fold-value of the non-treated control. n = 5. (c) TGFβ1-incubated primary chondrocytes were stimulated with 10 μM of a calcium ionophore with or without ZDON, and lysates were harvested at the indicated times and analysed by Western blotting. n = 3.
To elucidate the role of TG2 enzyme activity in chondrocyte signaling, we stimulated starved primary chondrocytes with the calcium ionophore (10 μM) in the presence of ZDON. The ZDON pretreatment significantly reduced total β-catenin level, and the same pattern was observed for the Ser33/37/Thr41-phosphorylated β-catenin. The inhibition of TG2 enzyme activity by ZDON significantly suppressed the Thr172 phosphorylation of AMPKa and Ser253 phosphorylation of FoxO3a. However, activation of p65, the major component of canonical NF-kB signaling, and mTOR was not affected by the calcium ionophore or ZDON (Fig. 3c).
Inhibition of enzyme activity of TG2 enhances ubiquitination of β-catenin and suppresses canonical Wnt signaling
The ZDON-mediated decrease in total β-catenin prompted us to investigate the change in canonical Wnt signaling. Although ZDON resulted in an increasing trend in both total β-catenin and non-phosphorylated β-catenin in the presence or absence of TGFβ1, the canonical Wnt signaling target proteins Axin2 and Tcf1 were not affected or were instead decreased by ZDON at the protein level. With enzymatic activation of TG2 by the calcium ionophore and TGFβ1, ZDON attenuated the total and non-phosphorylated β-catenin levels, which was accompanied by suppression of Axin2 and Tcf1 (Fig. 4a). We then investigated whether suppression of TG2 enzyme activity can affect ubiquitination of β-catenin in the ATDC5 chondrocyte cell line. Inhibition of TG2 enzyme activity by ZDON significantly increased ubiquitination of β-catenin in the presence of the calcium ionophore and TGFβ1 (Fig. 4b). Immunofluorescence staining for intracellular localization of non-phosphorylated β-catenin showed that the calcium ionophore increased the nuclear localization of non-phosphorylated β-catenin, but ZDON decreased overall β-catenin level as well as its nuclear localization. Interestingly, TG2 showed a perinuclear distribution pattern in the endoplasmic reticulum and Golgi. ZDON-mediated inhibition of TG2 enzyme activity disrupted this distribution pattern (Fig. 4c). The proportion of total and non-phosphorylated β-catenin-positive cells in the articular cartilage was also decreased by ZDON in vivo (Fig. 4d).

Inhibition of TG2 activity reduced total β-catenin level and canonical Wnt signaling in vitro and in vivo. (a) Primary chondrocytes cultured with TGFβ1 (10 ng/ml) were treated with a calcium ionophore (2 μM) with or without ZDON (100 μM) for 24 h. Western blot analyses were conducted for total and non-phosphorylated β-catenin and canonical Wnt signaling target genes of Axin2 and Tcf1. n = 3. (b) β-catenin ubiquitination assay. In the presence of TGFβ1 and a calcium ionophore, β-catenin and HA-tagged ubiquitin were transfected into ATDC5 cells with or without ZDON or MG132, a proteasome inhibitor. After β-catenin pulldown, samples were subjected to Western blotting with anti-HA and β-catenin. Inputs are indicated in the lower panels. (c) Representative image of immunofluorescence analysis for non-p-β-catenin and TG2 in primary chondrocytes. Primary chondrocytes were treated with TGF-β1 (10 ng/ml), with or without the calcium ionophore (2 μM) or ZDON (100 μM), and were co-stained with antibodies against non-p-β-catenin and TG2. n = 2. (d) Representative immunostaining images for total β-catenin and non-phosphorylated β-catenin in the articular cartilage of the DMM-operated knee joint. The percentages of total β-catenin and non-phosphorylated β-catenin-positive cells were quantified and are displayed as dot graphs. n = 5 per group.
TG2 involves nuclear translocation of FoxO3a but does not affect NF-kB signaling
We then examined whether the inhibition of TG2 enzyme activity can affect FoxO3a and NF-kB signaling. While ZDON increased FoxO3a in non-treated control chondrocytes, it did not affect the FoxO3a level in TGFβ1- or calcium ionophore-treated chondrocytes. Sirt1, which is known to regulate FoxO3a, was increased by ZDON in control and TGFβ1-treated conditions but was not affected in calcium ionophore-treated conditions. Both p65 and IkB, the major molecule of canonical NF-kB signaling, were not changed by the inhibition of TG2 (Fig. 5a). Although the total FoxO3 level was not affected by the inhibition of TG2 enzymatic function, a subcellular fractionation analysis revealed a significant decrease in nuclear translocation of FoxO3 by ZDON in calcium ionophore-treated conditions. Nuclear translocation of p65 and IkB, which was reported as a major substrate of TG2 in inflammatory conditions, showed no difference after ZDON-mediated inhibition of TG2 (Fig. 5b). We then used a FoxO3a agonist, Iturin A, to confirm the role of FoxO3a in MMP production. Iturin A (10 μM) increased TG2 and FoxO3a levels in primary chondrocytes, and it was not suppressed by ZDON. Iturin A increased the protein level of MMP-13 but not MMP-3. In addition, the Iturin A-mediated increase of MMP-13 was completely suppressed by ZDON (Fig. 5c). In vivo expression of FoxO3a showed that ZDON increased total FoxO3a and decreased phosphorylated FoxO3a in sham-operated mice. In DMM-operated mice, ZDON decreased both total and phosphorylated FoxO3a in articular cartilage (Fig. 5d).

Inhibition of TG2 reduced the nuclear translocation of FoxO3a but did not affect that of NFkB signaling. (a) Representative Western blot analyses for total protein levels of FoxO3a, Sirt1, p65 and IkB. Primary chondrocytes cultured with TGFβ (10 ng/ml) for 24 h were stimulated with a calcium ionophore (2 μM) with or without ZDON (100 μM) for 24 h. n = 3. (b) Western blots of cytoplasmic and nuclear protein fractions of TG2, FoxO3a, p65 and IkB. Biochemical fractionation of cytosolic and nuclear proteins from TGFβ1- and calcium ionophore-treated primary chondrocytes was performed to quantify nuclear translocation of the indicated proteins. n = 3. (c) The effects of Iturin A (10 μM), a FoxO3a agonist, on TG2, FoxO3a and MMPs were determined via Western blot analysis. Primary chondrocytes cultured with TGFβ1 for 24 h were treated with Iturin A with or without ZDON for 24 h. n = 3. (d) Representative immunostaining image for total FoxO3a and phosphorylated FoxO3a in the articular cartilage of a DMM-operated knee joint. The percentages of total and phosphorylated FoxO3a-positive cells were quantified and are displayed as a dot graph. n = 8 for sham and 10 for DMM group.
Discussion
In this study, we found that TG2 induced in articular cartilage by TGFβ aggravates the severity of surgically induced OA and increases the MMP-3 and MMP-13 production. TG2 enhanced not only canonical Wnt signaling via β-catenin stabilization but also AMPKa/FoxO3a signaling.
This study showed articular chondrocyte-specific expression of TG2 in the early stage of OA. In addition, TG2 was specifically induced by TGFβ in a dose-dependent manner (Fig. 1). The activation of osteoclasts in the early stage of OA induces active remodelling of subchondral bone12,18. This process can release matrix-derived coupling factors, such as Insulin-like growth factor 1 (IGF-1) and TGFβ, and the inhibition of TGFβ in subchondral bone can attenuate the progression of surgically induced OA19. However, the genetic deletion of a key component of TGFβ signaling, including TGFβ receptor type II or Smad3, aggravates the severity of OA20,21. These conflicting results can be explained by the difference in the degree of TGFβ signaling blockade between a partial inhibition with chemicals or blocking antibody and total inhibition with a genetic model. The discrepancy could also result from the differential role of TGFβ according to the differentiation status of chondrocytes, namely, resting, proliferating and hypertrophic chondrocytes22. TGFβ may naturally have both anabolic and catabolic functions, and its overall effect can vary depending on the differentiation status of chondrocytes. Our study provides a possible explanation for the catabolic function of TGFβ via TG2 in subchondral bone. Active subchondral bone remodelling during early OA can increase both TGFβ and calcium levels in the subchondral milieu12. TGFβ can increase the expression of TG2 in hypertrophic chondrocytes in the neighbouring subchondral bone9, and this calcium-rich environment in subchondral bone may enhance the calcium-dependent enzyme activity of TG2.
Wnt signaling has been shown to play a key role in OA pathogenesis through the induction of proteases, especially MMP-1323,24, and the regulation of chondrogenic and hypertrophic differentiation25,26,27. TG2 has been confirmed to enhance canonical Wnt signaling in tumour and vascular smooth muscle cells (VSMCs)28,29,30. A recent study showed that TG2 does not affect the transcriptional level of β-catenin, but it increases the phosphorylation of glycogen synthase kinase (GSK)-3β, which was inactivated by phosphorylation28. This inhibition of GSK-3β results in the accumulation of β-catenin and enhancement of Wnt signalling28. Furthermore, TG2 directly interacts with low density lipoprotein related-protein 5 (LRP5) and activates canonical β-catenin signaling in VSMCs29,30. Interestingly, our study demonstrated that TG2 enhances canonical Wnt signaling through the increase in total β-catenin level by inhibition of its ubiquitination in primary chondrocytes (Fig. 4). Although there is some controversy about the role of Wnt signaling in OA progression, enhanced Wnt signaling is generally known to aggravate the progression of OA24,31,32. To our knowledge, this is first evidence showing that TG2 enzyme activity is needed to maintain β-catenin stability and Wnt signaling in chondrocytes.
AMPK is activated under metabolic stress conditions, such as hypoxia and nutrient deprivation, to induce cell-protective autophagy and to stabilize cellular metabolism by increasing glucose uptake and fatty acid oxidation33,34. This functions to protect chondrocytes from cellular stress and consequently attenuate the progression of OA35,36. FoxO3a, a major downstream target molecule of AMPK, is also known to have a protective role in chondrocytes under metabolic stress conditions37,38,39. FoxO3a increases PGC1α expression to mediate mitochondrial biogenesis, induces the autophagic programme in an mTOR-dependent and independent manner, and enhances the production of endogenous antioxidants to maintain the cellular redox balance37,40,41. The inhibition of TG2 enzyme activity significantly suppressed the calcium ionophore-mediated phosphorylation of AMPK and FoxO3a, as well as the nuclear translocation of FoxO3a, in our study (Figs. 3 and 5). A calcium ionophore can sufficiently increase the intracellular calcium level to activate intracellular TG2, and these changes can be similar to cellular stress conditions, such as tissue damage or apoptosis, that result in loss of calcium homeostasis. In this stressful condition, AMPK and FoxO3a can be activated to protect chondrocytes, and the enzymatically activated TG2 may be involved in this process. The molecular mechanism of TG2-mediated phosphorylation of AMPK and FoxO3a is not fully elucidated and requires further investigation.
In addition to its effects on phosphorylation, TG2 increased MMP-13 expression through FoxO3a. The FoxO3a-mediated increase of MMP-13 was completely suppressed by inhibition of TG2 enzyme activity in this study. The ability of FoxO3a to enhance transcription of MMP-13 was highlighted in a recent study that showed FoxO3a directly bound to the MMP-13 promoter and induced MMP-13 promoter activity in VSMCs42. Through MMP-13 induction, FoxO3a promotes atherosclerosis and degeneration of the arterial intima and medial layer42. FoxO3a-dependent MMP-13 induction was also observed in a melanoma cell line in serum deprivation conditions43. The current pathophysiologic mechanisms that are considered important in OA eventually converged to the production of MMP-134. Our data suggest that FoxO3a mediates the catabolic effect of TG2 through the induction of MMP-13.
Several lines of evidence showed that canonical NF-kB signaling contributes to hypertrophic differentiation as well as MMP and nitric oxide production as upstream signaling of HIF-2α in OA chondrocytes44,45,46. TG2 is known to constitutively activate NF-kB signaling through the enhanced degradation of IkBα via non-proteasomal and proteasomal degradation47. However, our data showed that the activation or inhibition of TG2 failed to affect NF-kB activation or nuclear translocation in primary chondrocytes (Figs. 3 and 5), suggesting that the TG2-mediated activation of NF-kB signaling may vary depending on the cellular context.
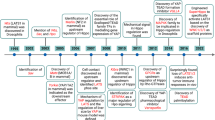
In conclusion, TG2 mainly induced by TGFβ can stabilize β-catenin to enhance canonical Wnt signaling and promote the nuclear translocation of FoxO3a, which can be responsible for MMP-13 production (Fig. 6). These findings shed light on the catabolic mechanism of TG2 in cartilage degeneration, and the inhibition of TG2 activity can be a good strategy in the development of disease-modifying OA drugs.

Schematic role of TG2 in OA derived from this study. TG2 mainly induced by TGFβ can be activated in calcium-rich milieu by active remodeling of subchondral bone. This activation of TG2 can stabilize β-catenin, which affects canonical Wnt signaling, and promote the phosphorylation of AMPK and FoxO3a, and nuclear translocation of FoxO3a. It can affect the production of MMPs and the progression of OA.
References
Glyn-Jones, S. et al. Osteoarthritis. Lancet386, 376–387 (2015).
Tchetina, E. V. et al. Chondrocyte hypertrophy can be induced by a cryptic sequence of type II collagen and is accompanied by the induction of MMP-13 and collagenase activity: implications for development and arthritis. Matrix Biol.26, 247–258 (2007).
Little, C. B. et al. Matrix metalloproteinase 13-deficient mice are resistant to osteoarthritic cartilage erosion but not chondrocyte hypertrophy or osteophyte development. Arthritis Rheum.60, 3723–3733 (2009).
Li, H., Wang, D., Yuan, Y. & Min, J. New insights on the MMP-13 regulatory network in the pathogenesis of early osteoarthritis. Arthritis Res. Ther.19, 248 (2017).
Adamczyk, M. Transglutaminase 2 in cartilage homoeostasis: novel links with inflammatory osteoarthritis. Amino Acids49, 625–633 (2017).
Lee, C. S. & Park, H. H. Structural aspects of transglutaminase 2: functional, structural, and regulatory diversity. Apoptosis22, 1057–1068 (2017).
Bianchi, N., Beninati, S. & Bergamini, C. M. Spotlight on the transglutaminase 2 gene: a focus on genomic and transcriptional aspects. Biochem. J.475, 1643–1667 (2018).
Lai, T. S., Lin, C. J. & Greenberg, C. S. Role of tissue transglutaminase-2 (TG2)-mediated aminylation in biological processes. Amino Acids49, 501–515 (2017).
Huebner, J. L., Johnson, K. A., Kraus, V. B. & Terkeltaub, R. A. Transglutaminase 2 is a marker of chondrocyte hypertrophy and osteoarthritis severity in the Hartley guinea pig model of knee OA. Osteoarthr. Cartil.17, 1056–1064 (2009).
Johnson, K. A. & Terkeltaub, R. A. External GTP-bound transglutaminase 2 is a molecular switch for chondrocyte hypertrophic differentiation and calcification. J. Biol. Chem.280, 15004–15012 (2005).
Orlandi, A. et al. Transglutaminase-2 differently regulates cartilage destruction and osteophyte formation in a surgical model of osteoarthritis. Amino Acids36, 755–763 (2009).
Jung, Y. K. et al. Calcium-phosphate complex increased during subchondral bone remodeling affects earlystage osteoarthritis. Sci. Rep.8, 487 (2018).
Glasson, S. S., Chambers, M. G., Van Den Berg, W. B. & Little, C. B. The OARSI histopathology initiative: recommendations for histological assessments of osteoarthritis in the mouse. Osteoarthr. Cartil.18(Suppl 3), S17-23 (2010).
Jung, Y. K. et al. Degrading products of chondroitin sulfate can induce hypertrophy-like changes and MMP-13/ADAMTS5 production in chondrocytes. Sci. Rep.9, 15846 (2019).
Kim, G. W. et al. CXC chemokine ligand 12a enhances chondrocyte proliferation and maturation during endochondral bone formation. Osteoarthr. Cartil.23, 966–974 (2015).
Park, N. R. et al. Core binding factor beta plays a critical role during chondrocyte differentiation. J. Cell Physiol.231, 162–171 (2016).
Kurosawa, H., Fukubayashi, T. & Nakajima, H. Load-bearing mode of the knee joint: physical behavior of the knee joint with or without menisci. Clin. Orthop. Relat. Res.1, 283–290 (1980).
Goldring, S. R. & Goldring, M. B. Changes in the osteochondral unit during osteoarthritis: structure, function and cartilage-bone crosstalk. Nat. Rev. Rheumatol.12, 632–644 (2016).
Zhen, G. et al. Inhibition of TGF-beta signaling in mesenchymal stem cells of subchondral bone attenuates osteoarthritis. Nat. Med.19, 704–712 (2013).
Shen, J. et al. Deletion of the transforming growth factor beta receptor type II gene in articular chondrocytes leads to a progressive osteoarthritis-like phenotype in mice. Arthritis Rheum.65, 3107–3119 (2013).
Chen, C. G., Thuillier, D., Chin, E. N. & Alliston, T. Chondrocyte-intrinsic Smad3 represses Runx2-inducible matrix metalloproteinase 13 expression to maintain articular cartilage and prevent osteoarthritis. Arthritis Rheum.64, 3278–3289 (2012).
van der Kraan, P. M., Blaney Davidson, E. N., Blom, A. & van den Berg, W. B. TGF-beta signaling in chondrocyte terminal differentiation and osteoarthritis: modulation and integration of signaling pathways through receptor-Smads. Osteoarthr. Cartil.17, 1539–1545 (2009).
Blom, A. B. et al. Involvement of the Wnt signaling pathway in experimental and human osteoarthritis: prominent role of Wnt-induced signaling protein 1. Arthritis Rheum.60, 501–512 (2009).
Zhu, M. et al. Activation of beta-catenin signaling in articular chondrocytes leads to osteoarthritis-like phenotype in adult beta-catenin conditional activation mice. J. Bone Miner. Res.24, 12–21 (2009).
Hartmann, C. & Tabin, C. J. Dual roles of Wnt signaling during chondrogenesis in the chicken limb. Development127, 3141–3159 (2000).
Yano, F. et al. The canonical Wnt signaling pathway promotes chondrocyte differentiation in a Sox9-dependent manner. Biochem. Biophys.. Res. Commun.333, 1300–1308 (2005).
Church, V., Nohno, T., Linker, C., Marcelle, C. & Francis-West, P. Wnt regulation of chondrocyte differentiation. J. Cell Sci.115, 4809–4818 (2002).
Condello, S., Cao, L. & Matei, D. Tissue transglutaminase regulates beta-catenin signaling through a c-Src-dependent mechanism. FASEB J.27, 3100–3112 (2013).
Faverman, L., Mikhaylova, L., Malmquist, J. & Nurminskaya, M. Extracellular transglutaminase 2 activates beta-catenin signaling in calcifying vascular smooth muscle cells. FEBS Lett.582, 1552–1557 (2008).
Beazley, K. E., Deasey, S., Lima, F. & Nurminskaya, M. V. Transglutaminase 2-mediated activation of beta-catenin signaling has a critical role in warfarin-induced vascular calcification. Arterioscler. Thromb. Vasc. Biol.32, 123–130 (2012).
Lietman, C. et al. Inhibition of Wnt/beta-catenin signaling ameliorates osteoarthritis in a murine model of experimental osteoarthritis. JCI Insight3, 3 (2018).
Zhu, M. et al. Inhibition of beta-catenin signaling in articular chondrocytes results in articular cartilage destruction. Arthritis Rheum.58, 2053–2064 (2008).
Steinberg, G. R. & Kemp, B. E. AMPK in health and disease. Physiol. Rev.89, 1025–1078 (2009).
Witczak, C. A., Sharoff, C. G. & Goodyear, L. J. AMP-activated protein kinase in skeletal muscle: from structure and localization to its role as a master regulator of cellular metabolism. Cell Mol. Life Sci.65, 3737–3755 (2008).
Terkeltaub, R., Yang, B., Lotz, M. & Liu-Bryan, R. Chondrocyte AMP-activated protein kinase activity suppresses matrix degradation responses to proinflammatory cytokines interleukin-1beta and tumor necrosis factor alpha. Arthritis Rheum.63, 1928–1937 (2011).
Zhou, S. et al. AMPK deficiency in chondrocytes accelerated the progression of instability-induced and ageing-associated osteoarthritis in adult mice. Sci. Rep.7, 43245 (2017).
Akasaki, Y. et al. FoxO transcription factors support oxidative stress resistance in human chondrocytes. Arthritis Rheumatol.66, 3349–3358 (2014).
Matsuzaki, T. et al. FoxO transcription factors modulate autophagy and proteoglycan 4 in cartilage homeostasis and osteoarthritis. Sci. Transl. Med.10, 428 (2018).
Davila, D. et al. Two-step activation of FOXO3 by AMPK generates a coherent feed-forward loop determining excitotoxic cell fate. Cell Death Differ.19, 1677–1688 (2012).
Olmos, Y. et al. Mutual dependence of Foxo3a and PGC-1alpha in the induction of oxidative stress genes. J. Biol. Chem.284, 14476–14484 (2009).
Kang, C. & Ji, L. Role of PGC-1alpha signaling in skeletal muscle health and disease. Ann. N. Y. Acad. Sci.1271, 110–117 (2012).
Yu, H. et al. FOXO3a (forkhead transcription factor o subfamily member 3a) links vascular smooth muscle cell apoptosis, matrix breakdown, atherosclerosis, and vascular remodeling through a novel pathway involving MMP13 (matrix metalloproteinase 13). Arterioscler. Thromb. Vasc. Biol.38, 555–565 (2018).
Storz, P., Doppler, H., Copland, J. A., Simpson, K. J. & Toker, A. FOXO3a promotes tumor cell invasion through the induction of matrix metalloproteinases. Mol. Cell Biol.29, 4906–4917 (2009).
Marcu, K. B., Otero, M., Olivotto, E., Borzi, R. M. & Goldring, M. B. NF-kappaB signaling: multiple angles to target OA. Curr. Drug Targets11, 599–613 (2010).
Mengshol, J. A., Vincenti, M. P., Coon, C. I., Barchowsky, A. & Brinckerhoff, C. E. Interleukin-1 induction of collagenase 3 (matrix metalloproteinase 13) gene expression in chondrocytes requires p38, c-Jun N-terminal kinase, and nuclear factor kappaB: differential regulation of collagenase 1 and collagenase 3. Arthritis Rheum.43, 801–811 (2000).
Saito, T. et al. Transcriptional regulation of endochondral ossification by HIF-2alpha during skeletal growth and osteoarthritis development. Nat. Med.16, 678–686 (2010).
Eckert, R. L. et al. Transglutaminase regulation of cell function. Physiol. Rev.94, 383–417 (2014).
Acknowledgements
This research was supported by a grant to S.H. of the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI), funded by the Ministry of Health & Welfare (Grant Number: HI15C1780), and a Grant to M.H. of the National Research Foundation (NRF), funded by the Ministry of Education of Korea government (Grant Number: 2018R1D1A1B07042715).
Author information
Authors and Affiliations
Contributions
Conceived and designed the experiments: M.H., Y.J., G.K., S.H. Performed the experiments: M.H., Y.J., G.K. Analysed the data: M.H., Y.J., G.K., S.H. Wrote the paper: M.H., S.H. Critical revision: M.H., Y.J., G.K., S.H. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Han, MS., Jung, YK., Kim, GW. et al. Transglutaminase-2 regulates Wnt and FoxO3a signaling to determine the severity of osteoarthritis. Sci Rep 10, 13228 (2020). https://doi.org/10.1038/s41598-020-70115-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-70115-w
This article is cited by
-
TGM2 accelerates migration and differentiation of BMSCs by activating Wnt/β-catenin signaling
Journal of Orthopaedic Surgery and Research (2023)
-
GPR56 signaling pathway network and its dynamics in the mesenchymal transition of glioblastoma
Journal of Cell Communication and Signaling (2023)
-
Type 2 transglutaminase in the nucleus: the new epigenetic face of a cytoplasmic enzyme
Cellular and Molecular Life Sciences (2023)
-
Selenium-sensitive histone deacetylase 2 is required for forkhead box O3A and regulates extracellular matrix metabolism in cartilage
Journal of Bone and Mineral Metabolism (2022)
-
Enzymatic Machinery of Ubiquitin and Ubiquitin-Like Modification Systems in Chondrocyte Homeostasis and Osteoarthritis
Current Rheumatology Reports (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
