
Abstract
Cabbage (Brassica oleracea var. capitata) is an important vegetable crop widely grown throughout the world, providing plentiful nutrients and health-promoting substances. To facilitate further genetics and genomic studies and crop improvement, we present here a high-quality reference genome for cabbage. We report a de novo genome assembly of the cabbage double-haploid line D134. A combined strategy of single-molecule real-time (SMRT) sequencing, 10× Genomics and chromosome conformation capture (Hi-C) produced a high quality cabbage draft genome. The chromosome-level D134 assembly is 529.92 Mb in size, 135 Mb longer than the current 02-12 reference genome, with scaffold N50 length being raised as high as 38 times. We annotated 44,701 high-quality protein-coding genes, and provided full-length transcripts for 45.59% of the total predicted gene models. Moreover, we identified novel genomic features like underrated TEs, as well as gene families and gene family expansions and contractions during B. oleracea evolution. The D134 draft genome is a cabbage reference genome assembled by SMRT long-read sequencing combined with the 10× Genomics and Hi-C technologies for scaffolding. This high-quality cabbage reference genome provides a valuable tool for improvement of Brassica crops.
Similar content being viewed by others


Introduction
Brassica oleracea is an important vegetable species widely grown throughout the world. The species comprises several subspecies showing extensive morphological and phytochemical diversity. As a diploid species, B. oleracea underwent a whole-genome triplication (WGT) event1, followed by two whole-genome duplication (WGD) event2,3 and a specific WGT4 of the Brassiceae lineage, thus becoming a model for studies on polyploid genome evolution. Currently, two B. oleracea genomes based on next-generation sequencing (NGS) technology are available: the TO1000 (kale-like; B. oleracea var. alboglabra) assembly and the 02-12 (cabbage; B. oleracea var. capitata) assembly5,6, but their errors and gaps make them difficult to use for many studies7,8,9,10. Recently, B. oleracea L. var. italica (broccoli) genome assembly was completed using long reads and optical maps11. Broccoli and cabbage belong to Brassica species, however their growth, morphology and molecular levels is extremely variable, showing the importance of generating several genome assemblies for different morphotypes of B. oleracea. Up to now, there is still no high quality, comprehensive assembled cabbage (B. oleracea var. capitata) genome, which hinders greatly basic genetics and genomics research, as well as crop improvement. Thus generating an accurate cabbage genome assembly is crucial.
To obtain a homozygous genome, the cabbage double-haploid (DH) line D134 was produced by microspore culture. We conducted whole-genome sequencing and de novo assembly for this line using single-molecule real-time (SMRT) cells on a PacBio Sequel platform combined with high-throughput chromosome conformation capture (Hi-C), next generation sequencing (NGS) and 10× Genomics technologies. This genome assembly is a draft genome for cabbage by Third-Generation Sequencing.
Materials and methods
Plant materials
A set of DH cabbage lines were previously obtained from a cross of the two cabbage inbred lines 96–100 and 01–20 (the hybrid was named as Zhonggan 18 for commercial use) using microspore culture, and this DH population has been extensively used in recent years to successfully mapped a series of important agronomic trait genes/QTLs for cabbage, including Fusarium wilt resistance, head weight, plant height, maturing period and seed number12,13,14,15,16. Moreover, one of these DH lines, D134, with excellent horticultural characteristics including early maturity, green leaf color and high resistance to Fusarium wilt, has been used constantly in cabbage breeding17,18. To better understand its genome background and lay a solid foundation for future cabbage breeding, D134 was chosen for genome sequencing using SMRT technique. High-quality genomic DNA was extracted from fresh leaf tissue using a modified cetyltrimethylammonium bromide (CTAB) protocol19, and stored at − 80 °C.
Genome size estimation and preliminary assembly
We conducted a genomic survey to estimate the genome size, GC content, homozygosity status and duplication content of D134 using a method based on K-mer distribution20. A short-insert-size (350 bp) library was constructed using a library construction kit (Illumina) and then sequenced on an Illumina HiSeq 2500 platform. The generated ~ 50 × high-quality reads were used to determine the distribution of K-mer values. We obtained a preliminary assembly by ALLPATHS-LG software21.
Genome sequencing and assembly
At least 50 kb genomic DNA was needed for 20-kb-insert-size library construction. The SMRTbell template was prepared following DNA fragmentation, DNA concentration detection, damage repair/end repair, adapter ligation and DNA purification. The DNA library was sequenced on a PacBio Sequel platform.
Read correction was performed using the PBcR wgs8.3rc1 assembly pipeline. Then, the error-corrected reads were aligned following the “overlap-layout-consensus” paradigm and assembled into contigs by using FALCON with the following parameters: seed_coverage = 60, length_cutoff_pr = 4,000, max_diff = 100, max_cov = 100. Finally, contig assembly correction was performed by mapping all the PacBio data against the generated contigs. Quiver algorithm (with default parameters) was used to polish the contig assembly, and Pilon (with default parameters) was used to perform error correction of contigs using the short paired-end reads generated from an Illumina HiSeq platform (with default parameters).
10× Genomics sequencing and scaffolding
We used a GemCode Instrument from 10× Genomics to prepare DNA samples and for automated barcoding. Approximately 1 ng of input DNA was used for the gel-bead-in-emulsion (GEM) reaction. Each input DNA fragment was encapsulated into a GEM and labeled with a unique 10× barcode. GEMs underwent isothermal incubation to generate 10×-barcoded amplicons. Differently barcoded amplicons were mixed and sheared into 500 bp to construct an NGS-ready library. The constructed library was finally sequenced on an Illumina HiSeq X Ten system. The linked reads of the 10× Genomics data were aligned to the contigs generated from PacBio data using bowtie2 with parameters (-D 1 -R 1 -N 0 -L 28 -i S,0,2.50 -n-ceil L,0,0.02 -rdg 5,10 -rfg 5,10 -no-unal), and fragScaff (v2-1)22 was used to build scaffolds using the barcoded sequencing reads with parameters (-fs1 '-m 3,000 -q 30 -E 30,000 -o 60,000′ -fs2 '-C 5′ -fs3 '-j 1 -u 3′).
Hi-C library construction and sequencing
Fresh leaves were cut into 2-cm pieces and fixed with 2% formaldehyde. Hi-C library construction was performed as previously described23. In brief, genomic DNA was extracted, and the fixed chromatin was digested by DpnII, followed by a fill-in reaction with biotinylated nucleotides and proximity ligation. The DNA was purified and then sheared into ~ 350-bp fragments using a Covaris S220 device. The DNA fragments were subjected to blunt-end repair, A-tailing, Illumina paired-end adapter ligation and PCR amplification. The Hi-C library was sequenced on an Illumina NovaSeq PE150 instrument.
Pseudomolecule construction
The Hi-C reads were mapped onto the draft assembly with BWA-MEM24 with default parameters. The alignment result was then filtered with mapping quality threshold 30, and duplicate and unmapped reads were removed with SAMTOOLS24. The scaffolds were breaked by SALSA with Hi-C clean data. Then the Hi-C clean data was aligned to the finally breaked contigs using BWA software with default parameters. Only the read pairs with both reads in the pair aligned to contigs and mapping quality higher than 30 are considered for scaffolding. According to the linkage information and restriction enzyme site, the string graph formulation was used to construct the scaffold graph with Lachesis (CLUSTER_N = 9, CLUSTER_MIN_RE_SITES = 2,300, CLUSTER_MAX_LINK_DENSITY = 9, CLUSTER_NONINFORMATIVE_RATIO = 0, ORDER_MIN_N_RES_IN_TRUNK = 20, ORDER_MIN_N_RES_IN_SHREDS = 15) and adjusted by juicer box25. The quality of the Hi-C assembly was assessed with Mummer software26 by collinearity analysis between the present assembly and the genome sequence of closely related species.
Repeat annotation
Repetitive elements in the D134 genome were predicted using a combination of homology prediction and de novo prediction27. For homology prediction, we used RepeatMasker and its in-house scripts (RepeatProteinMask) with default parameters to identify homologous repetitive elements based on Repbase28. For de novo prediction, we used RepeatMasker (RepeatMasker at https://repeatmasker.org) to identify repetitive elements based on a predicted database preliminarily generated by RepeatModeler (https://www.repeatmasker.org/RepeatModeler.html), RepeatScout (https://www.repeatmasker.org/) and LTR_finder (https://tlife.fudan.edu.cn/ltr_finder/) software and integrated using UCLUST software with default parameters29. And then all repeat sequences with lengths > 100 bp and gap ‘N’ less than 5% constituted the raw transposable element (TE) library. A custom library (a combination of Repbase and de novo TE library which was processed by uclust to yield a non-redundant library) was supplied to RepeatMasker for DNA-level repeat identification.
Gene annotation
We used a combination of several methods for gene prediction, including de novo prediction, homology based prediction and RNA-Seq assisted prediction. For gene predication based on Ab initio, by AUGUSTUS (https://bioinf.uni-greifswald. de/augustus/) and GlimmerHMM (https://ccb.jhu.edu/software/glimmerhmm/) were used in the automated gene prediction pipeline. For homology annotation, genes from sequenced genomes of Arabidopsis thaliana (TAIR10), B. oleracea (published), B. rapa, Cucumis sativus, Carica papaya, Solanum lycopersicum and Oryza sativa were downloaded (Supplementary Table S1) and matched using BLAST (https://blast.ncbi.nlm.nih.gov/Blast.cgi) (v2.2.26; E-value ≤ 1e−5) and then the matching proteins were aligned to the homologous genome sequences for accurate spliced alignments with GeneWise (https://www.ebi.ac.uk/~birney/wise2/) software which was used to predict gene structure contained in each protein region. For RNA-seq prediction, RNA-seq reads were aligned to the assembly using Hisat (v2.0.4)/TopHat (v2.0.11) with default parameters to identify exons region and splice positions and used as input for Stringtie (v1.3.3)/Cufflinks (v2.2.1) with default parameters for genome-based transcript assembly30. These predicted results were integrated into a non-redundant reference gene set with EVidenceModeler (EVM, https://evidencemodeler.sourceforge.net/) using PASA (Program to Assemble Spliced Alignment) terminal exon support and including masked transposable elements as input into gene prediction.
We generated functional annotation for the cabbage genes by aligning their predicted protein-coding regions to sequences in publicly available protein databases with BLAST (version 2.2.28+) (with a threshold of E-value ≤ 1e−5). The following protein databases were used: SwissProt (https://www.uniprot.org/), TrEMBL (https://www.uniprot.org/), KEGG (https://www.genome.jp/kegg/) and InterPro (https://www.ebi.ac.uk/interpro/).
ncRNA annotation
ncRNAs include transfer RNAs (tRNAs), ribosomal RNAs (rRNAs), microRNAs (miRNAs) and small nuclear RNAs (snRNAs). The tRNA genes were predicted by tRNAscan-SE with default parameters31. rRNAs are highly conserved; therefore, we aligned the D134 genomes with Arabidopsis rRNAs to identify cabbage rRNAs using BLAST (version 2.2.28+). miRNAs and snRNAs were identified using INFERNAL (https://infernal.janelia.org/) to search the RNA families (Rfam) database with default parameters.
Gene family identification and expansion analysis
We downloaded genome and annotation data for A. thaliana (TAIR10), R. sativus (GenBank accession number GCF_000801105.1), B. rapa (https://brassicadb.org/brad, version 3.0), B. oleracea (https://brassicadb.org/brad, version 1.1; ftp://ftp.ensemblgenomes.org, release 38), C. rubella (Phytozome.v1.0), C. sativus (GenBank accession number GCF_000004075.1), S. lycopersicum (ensembl.plant.v32), Daucus carota (GenBank accession number GCF_001625215.1), Populus trichocarpa (ensembl.plant.v32), Vitis vinifera (Phytozome v9.0), C. papaya (Phytozome.ASGPBv0.4), Gossypium raimondii (GenBank accession number GCF_000327365.1), O. sativa (Nipponbare, IRGSP-1.0), Zea mays (ensembl.plant.v32), Hordeum vulgare (ensembl.plant.v32), and Brachypodium distachyon (ensembl.plant.v32). Gene family identification and expansion analysis were performed as previously described. We chose the longest transcript to represent each gene and removed gene models with open reading frames shorter than 150 bp. Gene family clustering was performed using OrthoMCL32 based on the set of 44,701 predicted genes of B. oleracea (D134) and the protein sets of the thirteen other dicots and five monocots mentioned above. Based on the OrthoMCL clustering results, gene family expansion analysis was performed using CAFE (https://sourceforge.net/projects/cafehahnlab/).
Phylogenetic analysis and prediction of evolutionary history
We constructed a phylogenetic tree based on a concatenated sequence alignment of 432 single-copy gene families from D134 and the 17 other plant species using RAxML software with the maximum likelihood method33. Multiple sequence alignments were constructed with MUSCLE34. Divergence times were estimated by PAML MCMCTree35. The Markov chain Monte Carlo (MCMC) process was run for 1,000,000 iterations with a sample frequency of 50 after a burn-in period of 500,000 iterations. The default settings were used for other parameters of MCMCTree. Tracer was used to check for convergence. The following constraints were used for time calibrations: (i) the A. thaliana and O. sativa divergence time (20.4–30.9 Mya); (ii) the A. thaliana and P. trichocarpa divergence time (107.0–109.0 Mya); (iii) the B. distachyon and P. trichocarpa divergence time (140.0–200.0 Mya); and (iv) the S. lycopersicum and A. thaliana divergence time (107.0–125.0 Mya). The time calibrations were obtained from TimeTree (https://www.timetree.org/).
The branch-site model in CODEML from the PAML package36 was used to study positive Darwinian selection on single-copy gene families in B. oleracea (D134), B. oleracea (02-12), B. oleracea (TO1000), B. rapa and R. sativus. The alternative model for episodic evolution (ω > 1) was tested against the null model (neutral evolution, ω ≈ 1) by comparing likelihood ratios to a chi-square distribution (df = 1)36.
Identification of SNPs and indels
Identification of SNPs and indels was performed as previously described37. SNPs and indels (the latter with a length < 100 bp) between the D134 and 02-12 genomes were identified with Mummer as follows: (i) The D134 pseudochromosome sequence was mapped to its corresponding 02-12 pseudochromosome with nucmer with the parameters ‘-mumreference -g 1,000 -c 90 -l 40’. (ii) The delta filter was used to filter mapping noise and determine the one-to-one alignment blocks with the parameters ‘-r -q’. Alignments with aligned positions in one genome that were located more than 10 Mb away in another genome were further filtered. The aligned blocks between these two genomes were identified, and blank regions on the chromosomes that were potential low-similarity regions or multiple-aligned regions were filtered. (iii) Then, show-snps was used to obtain SNPs and small indels (< 100 bp). SNPs and indels shared between the D134 and TO1000 genomes were processed with the same method. The genome distributions of SNPs and indels between the D134 and 02-12/TO1000 genomes were also determined.
Results and discussion
Genome sequencing and assembly
To obtain a homozygous genome, the cabbage double-haploid (DH) line D134 was produced by microspore culture. We conducted whole-genome sequencing and de novo assembly for this line using SMRT cells on a PacBio Sequel platform combined with Hi-C, next generation sequencing (NGS) and 10X Genomics technologies. The SMRT cells yielded 8.34 million (64.72 Gb in total with an N50 of 13.3 kb and a mean length of 6.7 kb) PacBio single-molecule long reads (Supplementary Table S2–S6), corresponding to 98 × coverage of the 659.83-Mb cabbage genome as estimated by K-mer distribution analysis. The long reads were corrected using the PBcR wgs8.3rc1 assembly pipeline38 and preassembled following the “overlap-layout-consensus” paradigm, generating 870 contigs with an N50 size of 3.68 Mb (Table 1). These contigs were corrected and improved by 103.53 Gb of 10× Genomics short-read data. The initial assembly was 575.74 Mb (preliminary assembly1, 87.3% of the estimated genome size) and composed of 757 scaffolds with an N50 of 8.13 Mb (Tables 1, 2; Supplementary Table S2–S6). Assembly assessment with the core eukaryotic genes mapping approach (CEGMA)39 identified 97.58% complete genes and 99.19% complete and partial genes; assembly assessment with Benchmarking Universal Single-Copy Orthologs (BUSCO)40 identified 96.1% complete and single-copy genes among 1,440 genes, thus indicating the high quality of the D134 assembly.
The D134 assembly was further refined using Hi-C (in vivo fixation of chromosomes) libraries (~ 81.70 Gb of read pairs), resulting in an improved scaffold N50 of 56.58 Mb, with the longest scaffold being 71.59 Mb. We anchored and oriented 181 scaffolds onto nine pseudochromosomes (46.3–71.6 Mb in length), allowing the generation of a final chromosome-level assembly of 529.92 Mb (final assembly without unanchored contigs), covering 92.10% of the 575.39-Mb Hi-C improved assembly (preliminary assembly 2) (Tables 1, 2). The pseudomolecules are hereafter referred to as chromosomes and numbered according to the previously published assembly5,6.
Genome annotation
A comprehensive strategy combining de novo-based prediction, homology-based prediction and RNA-seq-based prediction was applied to annotate protein-coding genes in the D134 genome. The D134 genome comprises 44,701 protein-coding genes, with an average coding-sequence length of 1,057 and an average of 4.8 exons per gene (Supplementary Fig. S1, Supplementary Table S7). The majority of the predicted genes (44,097) were supported by the presence of transcripts from RNA-seq and known functional domains/known homologous proteins (Supplementary Fig. S1, S2; Supplementary Table S7, S8). In addition, we identified full-length transcripts for 45.59% (20,103) of the gene models with Pacific Bioscience SMRT long-read isoform sequencing of five different tissues: the root, stem, leaf, flower and silique. A total of 43,842 (98.10%) of cabbage gene models were functionally annotated based on protein databases including the SwissProt, TrEMBL, Kyoto Encyclopedia of Genes and Genomes (KEGG) and InterPro databases (Supplementary Fig. S3; Supplementary Table S9, S10).
Transposable elements (TEs) reportedly play important roles in shaping genome evolution and gene regulatory networks in many species41. We identified 56.47% of the D134 genome as repeat regions and 55.25% as TEs, including retrotransposons (39.51%), DNA transposons (12.38%), and unknown elements (2.66%). Previous studies indicated that long-read assemblies would improve the completeness of catalogue and genomic context of TEs11. We re-annotated the TEs in the long-read assembly broccoli HDEM, whose percentage was estimated to be 52.8%, making little difference to the D134 TE content. However, the long-read assemblies contained a much higher proportion of TEs than did the previously published short-read genomes of B. oleracea, namely, TO1000 (37.20%) and 02-12 (38.80%), and the genome of its close relative B. rapa (22.91%) (Supplementary Table S11). These differences are mainly attributed to the estimated relatively higher proportion of DNA transposons (12.38%) and especially long terminal repeat (LTR) retrotransposons (34.41%) in the D134 genome.
Evolution of the cabbage genome
We identified 66,401 gene families from 18 plant species using OrthoMCL (Fig. 1A). Among these gene families, 14,152 were shared by three Brassica species (D134, TO1000, and B. rapa) and Arabidopsis thaliana, and 1,920 were unique to D134 (Fig. 1B). Using genes extracted from a total of 432 single-copy families, we constructed a high-confidence phylogenetic tree and estimated the divergence times of 18 plant species. As shown in the phylogenetic tree, species of Cruciferae (B. oleracea (D134, 02-12, and TO1000), B. rapa, Raphanus sativus, Capsella rubella and A. thaliana) were clustered into a specific clade (Fig. 2; Supplementary Fig. S4). The divergence of C. rubella and A. thaliana from the other four Cruciferae occurred 14.0–24.0 million years ago (Mya), the divergence of R. sativus from the other four Brassica species occurred 21.0–30.9 Mya, and B. rapa diverged from B. oleracea approximately 6.2 Mya (Fig. 2; Supplementary Fig. S4). Moreover, we undertook a computational analysis of gene family sizes to study gene family expansion and contraction during the evolution of B. oleracea and related species (Fig. 2). Two hundred and fifty-seven gene families were expanded in the lineage leading to the Cruciferae, whereas 86 families decreased in size (Fig. 2). One hundred and thirty gene families were expanded in D134, compared to 160 in 02-12 and 489 in TO1000 (Fig. 2). Moreover, 363 gene families decreased in size in D134, compared to 309 in 02-12 and 246 in TO1000 (Fig. 2). The expanded gene families in D134 were enriched for protein tyrosine kinase activity, protein metabolic processes, pentose and glucuronate interconversions, and mismatch repair, while the contracted gene families were enriched for transmembrane transporter activity, polycyclic aromatic hydrocarbon degradation, and limonene and pinene degradation (Supplementary Table S12–S15). Compared with 02-12, TO1000, B. rapa and R. sativus, 157 positively selected genes were identified in D134. These genes were enriched for nuclease activity, noncoding RNA (ncRNA) processing, phenylalanine metabolism, and tyrosine metabolism (Supplementary Table S16, S17).

Distribution of genes in cabbage D134 and other representative plant species. (A) Orthologous genes found in different plant species. Ath, A. thaliana; Bdi, B. distachyon; Bol0, B. oleracea (D134); Bol1, B. oleracea (02-12); Bol2, B. oleracea (TO1000); Bra, B. rapa; Cru, C. rubella; Cpa, C. papaya; Csa, C. sativus; Dca, D. carota; Gra, G. raimondii; Hvu, H. vulgare; Osa, O. sativa; Ptr, P. trichocarpa; Rsa, R. sativus; Sly, S. lycopersicum; Vvi, V. vinifera; Zma, Z. mays. (B) Venn diagram showing unique and shared gene families among A. thaliana, B. rapa and B. oleracea (D134 and TO1000).

Phylogenetic tree showing divergence times and the evolution of gene family sizes. The phylogenetic tree shows the topology and divergence times for 18 plant species. MRCA, most recent common ancestor. The number in parentheses is the number of gene families in the MRCA as estimated by CAFÉ.
Global genome comparison of D134, 02-12 and TO1000
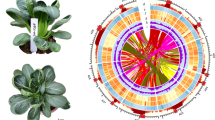
The 02-12 and TO1000 genome sequences were primarily assembled by whole-genome shotgun sequencing strategies using NGS technologies5,6, suggesting that the reference sequences do not cover the entire genome. These two sequenced genomes include numerous gaps, unanchored scaffolds, and even some misordered scaffolds7. In this study, the D134 assembly was 135 Mb and 74 Mb longer than the 02-12 and TO1000 reference genomes, respectively (Tables 1, 2). The gaps of D134 assembly are far less than 02-12, TO1000 and HDEM assembly, indicating that the D134 genome was vastly improved with SMRT, Hi-C and 10× Genomics technologies. When the pseudochromosomes of D134 were aligned to the pseudochromosomes of 02-12 and TO1000, approximately 45.78% of the 02-12 genome sequence and 65.19% of the TO1000 genome sequence was matched in one-to-one syntenic blocks with 68.39% and 55.39% of the D134 genome sequences, respectively (Fig. 3, Supplementary Fig. S5; Supplementary Table S18). Moreover, we identified 2,057,052 single nucleotide polymorphisms (SNPs) and 434,689 insertion/deletion polymorphisms (indels) in the syntenic sequences aligned between the D134 and 02-12 genomes and 3,963,977 SNPs and 581,173 indels in those between the D134 and TO1000 genomes (Supplementary Table S19 and S20). The distributions of SNPs and indels were positively correlated (Fig. 3, Supplementary Fig. S5). Most SNPs and indels were distributed in the intergenic regions (Supplementary Table S19 and S20).

Genomic landscape of D134 and 02-12. Chromosomes, gene density, TE density, SNP density, indel density and best-hit gene pairs are in order from outside to inside in the Circos images.
Conclusions
In conclusion, we report a genome assembly of the cabbage double-haploid line D134. A combined strategy of single-molecule sequencing, 10× Genomics and chromosome conformation capture produced a high quality cabbage draft genome. A total of 181 scaffolds accounting for 92.10% of the 575.39-Mb assembly (preliminary assembly 2) were anchored onto nine pseudochromosomes. The D134 assembly is 135 Mb longer than the current cabbage (02-12) reference genome, with scaffold N50 length being raised as high as 38 times. We annotated high-quality protein-coding genes in D134, and provided full-length transcripts for 45.59% of the total predicted gene models (44,701). The D134 displayed dramatic variations and plentiful transposable elements compared with 02-12 and TO1000 reference genomes. Moreover, we identified new gene families and gene family expansions and contractions during B. oleracea evolution. This study provides a high-quality cabbage reference genome and facilitates basic research on and improvement of Brassica crops.
Data availability
The accession number of genome sequencing data is CNP0000469 in China National GeneBank DataBase (CNGBdb).
References
Jaillon, O. et al. The grapevine genome sequence suggests ancestral hexaploidization in major angiosperm phyla. Nature 449, 463–467 (2007).
Bowers, J. E., Chapman, B. A., Rong, J. & Paterson, A. H. Unravelling angiosperm genome evolution by phylogenetic analysis of chromosomal duplication events. Nature 422, 433–438 (2003).
Jiao, Y. et al. Ancestral polyploidy in seed plants and angiosperms. Nature 473, 97–100 (2011).
Wang, X. et al. The genome of the mesopolyploid crop species Brassica rapa. Nat. Genet. 43, 1035–1039 (2011).
Parkin, I. A. et al. Transcriptome and methylome profiling reveals relics of genome dominance in the mesopolyploid Brassica oleracea. Genome Biol. 15, R77 (2014).
Liu, S. et al. The Brassica oleracea genome reveals the asymmetrical evolution of polyploid genomes. Nat. Commun. 5, 3930 (2014).
Lee, J. et al. Genotyping-by-sequencing map permits identification of clubroot resistance QTLs and revision of the reference genome assembly in cabbage (Brassica oleracea L.). DNA Res. 23, 29–41 (2015).
Liu, X. et al. Genetics and fine mapping of a yellow-green leaf gene (ygl-1) in cabbage (Brassica oleracea var. capitata L.). Mol. Breed. 36, 82 (2016).
Liu, X. et al. Genetics and fine mapping of a purple leaf gene, BoPr, in ornamental kale (Brassica oleracea L. var. acephala). BMC Genom. 18, 230 (2017).
Zhang, B. et al. Disruption of a CAROTENOID CLEAVAGE DIOXYGENASE 4 gene converts flower colour from white to yellow in Brassica species. New Phytol. 206, 1513–1526 (2015).
Belser, C. et al. Chromosome-scale assemblies of plant genomes using nanopore long reads and optical maps. Nat. Plants 4, 879 (2018).
Lv, H. et al. Mapping and analysis of a novel candidate Fusarium wilt resistance gene FOC1 in Brassica oleracea. BMC Genom. 15(1), 1094 (2014).
Lv, H. et al. Linkage map construction using InDel and SSR markers and QTL analysis of heading traits in Brassica oleracea var. capitata L.. Mol. Breed. 34, 87–98 (2014).
Lv, H. et al. Whole-genome mapping reveals novel QTL clusters associated with main agronomic traits of cabbage (Brassica oleracea var. capitata L.). Front. Plant Sci. 7, 989 (2016).
Lv, H. et al. Genome-wide InDel/SSR scanning reveals significant loci associated with excellent agronomic traits of a cabbage (Brassica oleracea) elite parental line ‘01–20’. Sci. Rep. 7, 41696 (2017).
Li, X. et al. Identification of a major QTL for seed number per silique in cabbage (Brassica oleracea L. var. capitata) using genotyping by sequencing. Euphytica 215, 133 (2019).
Lv, H. et al. Breeding of cabbage (Brassica oleracea L. var. capitata) with Fusarium wilt resistance based on microspore culture and biomarker selection. Euphytica 200, 465–473 (2014).
Liu, X. et al. Rapid introgression of the Fusarium wilt resistance gene into an elite cabbage line through the combined application of a microspore culture, genome background analysis, and disease resistance-specific marker assisted selection. Front. Plant Sci. 8, 354 (2017).
Zhang, G. et al. The Apostasia genome and the evolution of orchids. Nature 549, 379 (2017).
Kim, E. B. et al. Genome sequencing reveals insights into physiology and longevity of the naked mole rat. Nature 479, 223–227 (2011).
Butler, J. et al. ALLPATHS: de novo assembly of whole-genome shotgun microreads. Genome Res. 18, 810–820 (2008).
Adey, A. et al. In vitro, long-range sequence information for de novo genome assembly via transposase contiguity. Genome Res. 24, 2041–2049 (2014).
Zhu, W. et al. Altered chromatin compaction and histone methylation drive non-additive gene expression in an interspecifc Arabidopsis hybrid. Genome Biol. 18, 157 (2017).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
R Core Team. R: a language and environment for statistical computing (R Foundation for Statistical Computing, 2015).
Kurtz, S. et al. Versatile and open sofware for comparing large genomes. Genome Biol. 5, R12 (2004).
Zhang, G. et al. The Dendrobium catenatum Lindl. genome sequence provides insights into polysaccharide synthase, floral development and adaptive evolution. Sci. Rep. 6, 19029 (2016).
Bao, W., Kojima, K. K. & Kohany, O. Repbase Update, a database of repetitive elements in eukaryotic genomes. Mob. DNA 6, 11 (2015).
Edgar, R. C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26, 2460–2461 (2010).
Trapnell, C. et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 7, 562–578 (2012).
Lowe, T. M. & Eddy, S. R. tRNAscan-SE: A program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 25, 955–964 (1997).
Li, L., Stoeckert, C. J. Jr. & Roos, D. S. OrthoMCL: Identifcation of ortholog groups for eukaryotic genomes. Genome Res. 13, 2178–2189 (2003).
Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313 (2014).
Robert, C. E. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797 (2004).
Yang, Z. PAML 4: Phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 24, 1586–1591 (2007).
Zhang, J., Nielsen, R. & Yang, Z. Evaluation of an improved branch-site likelihood method for detecting positive selection at the molecular level. Mol. Biol. Evol. 22, 2472–2479 (2005).
Sun, S. et al. Extensive intraspecific gene order and gene structural variations between Mo17 and other maize genomes. Nat. Genet. 50, 1289–1295 (2018).
Berlin, K. et al. Assembling large genomes with single-molecule sequencing and locality-sensitive hashing. Nat. Biotechnol. 33, 623–630 (2015).
Parra, G., Bradnam, K. & Korf, I. CEGMA: A pipeline to accurately annotate core genes in eukaryotic genomes. Bioinformatics 23, 1061–1067 (2007).
Simão, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V. & Zdobnov, E. M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212 (2015).
Jiao, Y. et al. Improved maize reference genome with single-molecule technologies. Nature 546, 524–527 (2017).
Acknowledgements
This work was supported by grants from the National Key Research and Development Program of China (2017YFD0101804; 2016YFD0101702), the National Natural Science Foundation of China (31701927), the Central Public-interest Scientific Institution Basal Research Fund (Y2020PT01), the Science and Technology Innovation Program of the Chinese Academy of Agricultural Sciences (CAAS-ASTIP-IVFCAAS), and the earmarked fund for the Modern Agro-Industry Technology Research System, China (CARS-23). The work reported here was performed in the Key Laboratory of Biology and Genetic Improvement of Horticultural Crops, Ministry of Agriculture, Beijing 100081, China.
Author information
Authors and Affiliations
Contributions
L.H. and F.Z. produced the cabbage D134 line. F.Z., Y.L. and Z.Y. conceived and designed the experiments; L.H., W.Y. and J.J. performed genome comparison. L.H. and H.F. performed genome assembly and genome annotation. L.H., W.Y., H.F., and J.J. wrote the manuscript. Z.M., L.Z. and Y.L. coordinated and designed the study.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interest.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Lv, H., Wang, Y., Han, F. et al. A high-quality reference genome for cabbage obtained with SMRT reveals novel genomic features and evolutionary characteristics. Sci Rep 10, 12394 (2020). https://doi.org/10.1038/s41598-020-69389-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-69389-x
This article is cited by
-
Resynthesizing Brassica napus with race specific resistance genes and race non-specific QTLs to multiple races of Plasmodiophora brassicae
Scientific Reports (2024)
-
Large-scale gene expression alterations introduced by structural variation drive morphotype diversification in Brassica oleracea
Nature Genetics (2024)
-
Identification of QTLs for resistance to 10 pathotypes of Plasmodiophora brassicae in Brassica oleracea cultivar ECD11 through genotyping-by-sequencing
Theoretical and Applied Genetics (2023)
-
Comparison of ONT and CCS sequencing technologies on the polyploid genome of a medicinal plant showed that high error rate of ONT reads are not suitable for self-correction
Chinese Medicine (2022)
-
Genome sequencing sheds light on the contribution of structural variants to Brassica oleracea diversification
BMC Biology (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
