
Abstract
Zicaitai is a seasonal vegetable known for its high anthocyanin content in both stalks and leaves, yet its reference genome has not been published to date. Here, we generated the first chromosome-level genome assembly of Zicaitai using a combination of PacBio long-reads, Illumina short-reads, and Hi-C sequencing techniques. The final genome length is 474.12 Mb with a scaffold N50 length of 43.82 Mb, a BUSCO score of 99.30% and the LAI score of 10.14. Repetitive elements accounted for 60.89% (288.72 Mb) of the genome, and Hi-C data enabled the allocation of 430.87 Mb of genome sequences to ten pseudochromosomes. A total of 42,051 protein-coding genes were successfully predicted using multiple methods, of which 99.74% were functionally annotated. Notably, comparing the genome of Zicaitai with seven other species in the Cruciferae family revealed strong conservation in terms of gene numbers and structures. Overall, the high-quality genome assembly provides a critical resource for studying the genetic basis of important agronomic traits in Zicaitai.
Similar content being viewed by others
Background and Summary
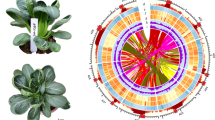
Brassica rapa var. purpuraria (NCBI: txid386281, Fig. 1) belongs to the Cruciferae family1,2 and is named “Zicaitai” for its purple stalks3. Zicaitai originated in the southern regions of China and then spread to the Yangtze River Basin, where it was subsequently widely domesticated. Its cultivation history can be traced back to ancient China, spanning more than a thousand years. To date, Zicaitai is a popular vegetable in China, and is also exported to other countries in Asia, Europe, and America.

Images of Zicaitai mature plant in laboratory research (a) and field experiment (b).
The stems and leaves of Zicaitai are the main edible portion and are rich in anthocyanin4,5,6. Anthocyanin is a water-soluble natural pigments, that possesses anti-cancer, anti-viral, and cardiovascular and cerebrovascular disease prevention properties7,8,9. Moreover, anthocyanin plays vital roles in attracting pollinators and seed dispersers, as well as protecting plants from abiotic and biotic stresses8,10,11,12,13. The anthocyanin biosynthesis pathway and the molecular mechanisms of anthocyanin accumulation are conserved and complicated in plants5,14. Briefly, it initiates with the synthesis of naringenin chalcone mediated by chalcone synthase (CHS), using 4-coumaroyl-CoA and malonyl-CoA as substrates. Subsequently, chalcone isomerase (CHI) converts naringenin chalcone to naringenin. Naringenin is then converted into dihydrokaempferol by flavanone 3-hydroxylase (F3H), which can be further hydroxylated into dihydroquercetin or dihydromyricetin by either flavonoid 3’-hydroxylase (F3’H) or flavonoid 3’,5’-hydroxylase (F3’5’H), respectively. Dihydroflavonol 4-reductase (DFR) converts the three dihydroflavonols into colorless leucoanthocyanidins, which are then transformed into colored anthocyanidins by anthocyanidin synthase (ANS). Finally, members of the glycosyltransferase enzyme family, such as flavonoid 3-O-glucosyltransferase (UFGT), attach sugar molecules to anthocyanidins, and the anthocyanidins may undergo further acylation by acyltransferases with aromatic acyl groups.
Nowadays, some candidate genes related to anthocyanin biosynthesis have been identified7,13,15,16,17,18,19. For example, Hayashi et al.9 have crossed a doubled haploid line of the turnip Brassica rapa cv. ‘Iyo-hikabu’, which is pigmented with anthocyanin, with a Chinese cabbage inbred line, ‘Y54’, which lacks anthocyanin pigmentation, and identified a novel locus (Anp) on chromosome A079 related with anthocyanin synthesis based on a bulked segregant analysis. Burdzinski and Wendell (2007) identified three markers linked to anthocyaninless, forming a linkage group spanning 46.9 cM, which were assigned to Brassica rapa linkage group R0920 based on 177 F2 offspring. Additionally, the pur gene, responsible for regulating purple leaf color, was successfully mapped to the end of chromosome A03 using an F2 population21. An insertion and deletion (InDel) marker developed from deletion/insertion in the promoter region of bHLH49 in the F2 population was found to significantly correlate with the stalk color trait2. EGL3, a positive regulator gene with potentially epistatic function, was localized to mediate the anthocyanin biosynthesis1. Two candidate genes controlling anthocyanin accumulation were identified in the F2 population derived from a cross between Zicaitai and caixin5, and they were homologous with AtEGL3, BrEGL3.1 and BrEGL3.2 genes. Although several studies have characterized anthocyanin in Brassica crops, there is limited information on the genes involved in anthocyanin biosynthesis in Zicaitai.
Understanding the genome structure and identifying candidate genes related to anthocyanin biosynthesis is crucial for Zicaitai. However, the lack of a high-quality reference genome for Zicaitai makes it challenging to identify candidate genes associated with important agronomic traits and breed excellent Zicaitai varieties. Hence, we have generated a chromosome-level genome assembly of Zicaitai using PacBio long-reads, Illumina short reads, and Hi-C sequencing data first in this study. The assembled genome has a total length of 474.12 Mb, with a scaffold N50 length of 43.82 Mb, and 90.88% of the genome sequence was successfully anchored onto ten pseudochromosomes. Through a combination of ab initio gene predictions, RNA-seq, and homologous protein evidence, a total of 42,051 protein-coding genes were identified, and 41,942 of them were functionally annotated. The genome sequence provides a valuable resource for exploring the molecular basis of agronomic traits in Zicaitai and will further facilitate its genetic improvements.
Methods
Sample collection
Young fresh leaves of Zicaitai were collected from one sample individual grown in a greenhouse in Guangzhou, Guangdong, China (N 23°06’, E 113°15’), and immediately frozen in liquid nitrogen for genomic DNA and RNA extraction.
DNA extraction and sequencing
Total high molecular weight (HMW) genomic DNA was extracted from Zicaitai young fresh leaves using the Tiangen Extraction Kit (Tiangen Biotech (Beijing) Co., Ltd.) for whole genome sequencing. The extraction process was according to the cetyltrimethylammonium bromide (CTAB) method, and concentration was ascertained by the Quant-iT PicoGreen® assay (Invitrogen, Waltham, MA, USA). The quality and quantity of the DNA samples were assessed using an ultraviolet spectrophotometer at 260 nm and 280 nm wave lengths. The DNA was fragmented with a Covaris M220 Focused-ultrasonicator Instrument. For genomic DNA sequencing, we employed three different approaches at Novogene Co., Ltd., Beijing, China. Firstly, DNA PCR-free libraries with insert sizes of 350 bp were constructed using the NEBNext Ultra DNA library Pre-Kit for Illumina short-reads sequencing. The resulting barcoded library were sequenced on the Illumina Hiseq 4000 platform to generate paired-end 150-bp reads. Subsequently, all the obtained reads were quality controlled by trimming adaptor sequences and low-quality reads using NGSQC v2.322 (-q 20, https://github.com/mjain-lab/NGSQCToolkit). Secondly, single-molecule real-time (SMRT) PacBio libraries were constructed using the PacBio 15-kb protocol and sequenced with a Pacbio Sequel IIe platform. Lastly, the Hi-C library was generated using the restriction endonuclease DpnII. The DpnII-digested chromatin was labeled with biotin-14-dATP, and in situ DNA ligation was performed. The DNA underwent extraction, purification, and shearing. After A-tailing, pull-down, and adapter ligation, the DNA library was sequenced on the Illumina Hiseq 4000 platform. The total data generated from long-read sequencing was 35.29 Gb, and the total data generated from short-read sequencing was 10.06 Gb (Table 1).
RNA extraction and sequencing
Simultaneously, fresh root, stem, leaf, flower, and pod of the same Zicaitai individual were collected for transcriptome sequencing. Total RNA was extracted using the TRIzol reagent (Thermo Fisher Scientific, MA, USA) according to the manufacturer’s protocol. RNA-seq library were sequenced on an Illumina Novaseq 6000 platform with paired-end 150 bp reads. The adapters and low-quality reads of the raw sequence reads were trimmed using NGSQC v2.322 (-q 20, https://github.com/mjain-lab/NGSQCToolkit). A total of ~15 Gb raw reads were yielded and used for the gene prediction.
Genome size estimation and assembly
For quality control, the adapter sequences and low-quality reads obtained from Illumina Hiseq 4000 platform were filtered using NGSQC v2.322 (-q 20, https://github.com/mjain-lab/NGSQCToolkit) and Trimmomatic v0.423 (adapter:2:30:10:2:True LEADING:3 TRAILING:3 MINLEN:50). The genome size was then estimated by using GenomeScope v2.024 and the 17-mer analysis with Jellyfish25 v2.2.7 (-C -m 17 -s 100 m) based on all the remaining reads. The final genome size was estimated to be 555.32 Mb.
For the genome assembly of Zicaitai, Hifiasm v0.16.126 was first used to assemble the initial assembly with PacBio CCS sequences. Secondly, NextPolish v1.3.127 was then applied three times to polish the draft genome assembly using Illumina short reads. Then, ALLHiC28, a Hi-C scaffolding pipeline, was used to align Hi-C reads to the draft assembly and subject them to quality control. Finally, 3D-DNA v18041929 was used to anchor primary contigs into chromosomes, and ambiguous fragments were removed manually with Juicebox v1.1330, a visualization software for Hi-C data. All of the above software runs with default parameters. The final genome assembly of Zicaitai was 474.12 Mb with a scaffold N50 of 43.82 Mb. The Hi-C analyses scaffolded ten pseudo-chromosomes (Fig. 2a), anchoring 90.88% (430.88 Mb) of the genome assembly. The average GC content of Zicaitai genome assembly was 38.54% (Table 2, Fig. 2b).

Hi-C chromatin interaction map and circos plot of the genome assembly. (a) Hi-C chromatin interaction map of the Zicaitai assembly. (b) The circos plot of genome characteristic of the Zicaitai. The rings from outside to inside indicate are: (a) GC content, (b) gene distribution, (c) TE density, and (d) ncRNA distribution in ten different chromosomes.
Genome completeness
The genome completeness was evaluated with BUSCO v5.4.731, searching against the embryophyta_odb10 database. The analysis found 99.30% (single-copied genes: 84.70%, duplicated genes: 14.60%), 0.20%, and 0.50% of the 42,051 projected genes in this genome as complete, fragmented, and missing sequences, respectively, indicating a highly complete genome assembly. Moreover, 242 genes were assembled in the CEGMA32 database (248 cor genes), suggesting a completeness score of 97.58%.
Genome annotation
The repetitive elements, protein-coding genes, and non-coding RNAs (ncRNAs) of the Zicaitai genome was annotated. The whole genome repeats were identified using a combined strategy based on homology alignment and de novo search. Tandem Repeat was extracted using TRF33 by ab initio prediction. The homolog prediction commonly used Repbase34 database employing RepeatMasker and RepeatProteinMask with default parameters to extract repeat regions. Ab initio prediction built the de novo repetitive elements database by LTR_FINDER35, RepeatScout36, and RepeatModeler237 (-LTRStruct), and then all repeat sequences with lengths more than 100 bp and gap ‘N’ less than 5% constituted the raw transposable element (TE) library. A total of 288.72 Mb repetitive elements were identified, constituting 60.89% of the total genome sequence. The most abundant repeating element was long terminal repeats (LTR, 47.96%), and unknown repeats (43.71%), followed by DNA transposons (6.08%) (Fig. 2b, Table 3, Fig. 3).

Repeat landscape plots illustrating TE accumulation history for Zicaitai genome, based on Kimura distance-based copy divergence analyses, with sequence divergence (CpG adjusted Kimura substitution level) illustrated on the x-axis, percentage of the genome represented by each TE type on the y-axis, and transposon type indicated by the colour chart on the right-hand side. CpG, region of DNA where a cytosine nucleotide is followed by a guanine nucleotide; LINE, long interspersed nuclear element; LTR, long terminal repeat; SINE, short interspersed nuclear element.
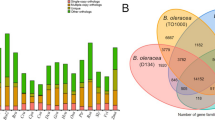
De novo gene prediction, homology-based prediction, and RNA-seq were applied for the annotation of protein-coding genes. The repeat-masked genome was analyzed using Augustus v2.438, GlimmerHMM v3.0.439, Geneid v1.4.540 and Genscan41 for de novo gene prediction. The protein sequences of Cruciferae species were downloaded from the NCBI Database as references for homology-based prediction. Transcriptome assemblies were also generated with Trinity v2.5.142 for the genome annotation. A consensus gene set was created by integrating the genes predicted by the aforementioned three methods using EVidenceModeler v1.1.143. Finally, a total of 42,051 protein-coding genes were predicted for the Zicaitai genome by combining the evidence from the transcriptome, ab initio, and homology-based predictions (Fig. 4a). The average length of the predicted protein-coding gene was 2,001 bp. The average lengths of the exon and intron were 228 bp and 235 bp, respectively. Each gene has an average of 4.82 exons. (Table 4). Gene functions were assigned according to the best match by aligning the protein sequences to the Swiss-Prot, GO, NR, InterPro, Pfam, and KEGG databases, respectively. A total of 41,942 (99.74%) genes were functionally annotated (Table 4, Fig. 4b).

Prediction and annotation of protein-coding genes in the Zicaitai genome.
For tRNA prediction, the program tRNAscan-SE44 was used, while for rRNA prediction, Blast45 program was used with relative species’ rRNA sequences as references. Other ncRNAs, including miRNAs and snRNAs, were identified by searching against the Rfam46 database using the infernal v1.147 software with default parameters. Finally, a total of 1,894 non-coding RNAs were predicted, including 4,885 transfer RNAs (tRNAs), 6,402 ribosomal RNAs (rRNAs), 511 micro-RNAs (miRNAs), and 2,774 small nuclear RNAs (snRNAs) (Table 5, Fig. 2b).
Data Records
The Illumina short reads, PacBio long-reads, Hi-C sequencing data, and RNA-seq data used for the genome assembly and annotation have been deposited in the NCBI Sequence Read Archive (SRA) database with the accession number SRP44163348. The chromosomal-level genome assembly sequence and annotation information have been deposited in the Figshare database49. The chromosomal-level genome assembly sequence was deposited in the GenBank database of NCBI with accession number JAUJLN00000000050.
Technical Validation
Evaluating the quality of the genome assembly
We evaluated the quality and completeness of the Zicaitai genome assembly using two approaches. First, we mapped short-reads to the genome to verify the accuracy, yielding mapping rates of 99.22%, which suggests the accuracy of the Zicaitai genome assembly. Second, BUSCO analysis found 99.30% of the 1,614 single-copy orthologues in the embryophyta_odb10 database to be complete (84.70% single-copied genes and 14.60% duplicated genes), with 0.2% fragmented and 0.5% missing (Table 4), indicating a remarkably complete assembly of the Zicaitai genome. Additionally, the whole-genome high long terminal repeat (LTR) assembly index (LAI) score is an important indicator of genome assembly quality and completeness. In this study, the LAI score for Zicaitai genome assembly was 10.14, indicating that the assembly quality of Zicaitai reached the reference genome level.
Gene annotation validation
To validate gene annotation, we studied the structure and number of genes in Zicaitai and seven other Cruciferae species based on protein annotation sequences retrieved from NCBI, including Brassica rapa (Brap), Brassica napus (Bnap), Brassica juncea (Bjun), Brassica nigra (Bnig), Brassica oleracea (Bole), Brassica carinata (Bcar), and Arabidopsis thaliana (Atha). A total of 42,051, 100,829, 96,553, 41,049, 57,386, 97,148, 43,923, and 27,221 protein-coding genes were identified in Zicaitai, Bjun, Bnap, Brap, Bnig, Bcar, Bole, and Atha, respectively (Table 6, Fig. 5). Except for tetraploid plants (Bnap, Bjun, and Bcar), the gene numbers of other diploid species were similar (about 40,000). The average lengths of transcripts, CDS, exons, and introns in Zicaitai and the other seven Cruciferae species were found to be almost identical. Additionally, the average number of exons per gene was also found to be equivalent across all species.

Genetic components of the Zicaitai genome and seven other Cruciferae species.
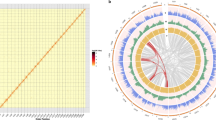
OrthoFinder v2.5.451 was used to infer sequence orthology. Phylogenetic trees were constructed using single copy gene family, and the differentiation time was estimated using the r8s program52. Based on the time tree, expansions and contractions of the gene family of Zicaitai and seven other Cruciferae species was estimated using CAFE v553 with a p value of 0.01. Finally, 236 and 222 gene families experienced expansions and contractions were found in Zicaitai, respectively (Fig. 6). Moreover, we have compared the genome sequences of Zicaitai and Brassica rape (Brara_Chiifu_V4.0), and the results indicated a significant degree of collinearity for the two genome sequences of Zicaitai and Brassica rape, with the exception of certain contigs located on chromosome 9 (Fig. 7).

Phylogenetic tree and gene families expansion and contraction of Zicaitai and seven other Cruciferae species. The scale at the bottom of the figure represents the divergence time.

Dot plot represents an alignment of two different genomes of Brassica rapa (x-axis) and Zicaitai (y-axis). Forward matches are shown in red, while reverse matches are shown in blue.
Code availability
This work did not utilize a custom script. Data processing was carried out using the protocols and manuals of the relevant bioinformatics software.
References
Zhang, X. et al. QTL-seq and sequence assembly rapidly mapped the gene BrMYBL2.1 for the purple trait in Brassica rapa. Sci Rep 10, 2328 (2020).
Li, G.-H. et al. A high-density genetic map developed by specific-locus amplified fragment (SLAF) sequencing and identification of a locus controlling anthocyanin pigmentation in stalk of Zicaitai (Brassica rapa L. ssp. chinensis var. purpurea). BMC Genomics 20, https://doi.org/10.1186/s12864-019-5693-2 (2019).
Tan, C. et al. Identification and characterization of the gene BraANS.A03 associated with purple leaf color in pak choi (Brassica rapa L. ssp. chinensis). Planta 258, 19, https://doi.org/10.1007/s00425-023-04171-7 (2023).
Liu, Y. et al. Comprehensive transcriptome-metabolome analysis and evaluation of the Dark_Pur gene from Brassica juncea that controls the differential regulation of anthocyanins in Brassica rapa. Genes (Basel) 13, https://doi.org/10.3390/genes13020283 (2022).
Guo et al. Anthocyanin profile characterization and quantitative trait locus mapping in zicaitai (Brassica rapa L. ssp chinensis var. purpurea). Molecular Breeding (2015).
Anna, P. Natural antioxidants and antioxidant capacity of Brassica vegetables: a review. (2005).
Zhang, N. & Jing, P. Anthocyanins in Brassicaceae: composition, stability, bioavailability, and potential health benefits. Crit Rev Food Sci Nutr 62, 2205–2220, https://doi.org/10.1080/10408398.2020.1852170 (2022).
Nistor, M. et al. Anthocyanins as Key Phytochemicals Acting for the Prevention of Metabolic Diseases: An Overview. Molecules 27, https://doi.org/10.3390/molecules27134254 (2022).
Hayashi, K. et al. Mapping of a novel locus regulating anthocyanin pigmentation in Brassica rapa. Breeding Science 60, 76–80, https://doi.org/10.1270/jsbbs.60.76 (2010).
Markham, A. & K.R. Flavonoids: Chemistry, Biochemistry and Applications. (Pesticide Science, 2006).
Liu, S. et al. SmbHLH60 and SmMYC2 antagonistically regulate phenolic acids and anthocyanins biosynthesis in Salvia miltiorrhiza. J Adv Res 42, 205–219, https://doi.org/10.1016/j.jare.2022.02.005 (2022).
Yan, H. et al. MYB-mediated regulation of anthocyanin biosynthesis. Int J Mol Sci 22, https://doi.org/10.3390/ijms22063103 (2021).
Liu, H., Liu, Z., Wu, Y., Zheng, L. & Zhang, G. Regulatory mechanisms of anthocyanin biosynthesis in apple and pear. Int J Mol Sci 22, https://doi.org/10.3390/ijms22168441 (2021).
Zhang, N. & Jing, P. Anthocyanins in Brassicaceae: composition, stability, bioavailability, and potential health benefits. Critical Reviews in Food Science and Nutrition, 1-15 (2020).
Sunil, L. & Shetty, N. P. Biosynthesis and regulation of anthocyanin pathway genes. Appl Microbiol Biotechnol 106, 1783–1798, https://doi.org/10.1007/s00253-022-11835-z (2022).
Mekapogu, M. et al. Anthocyanins in floral colors: biosynthesis and regulation in chrysanthemum flowers. Int J Mol Sci 21, https://doi.org/10.3390/ijms21186537 (2020).
Holton, T. A. & Cornish, E. C. Genetics and biochemistry of anthocyanin biosynthesis. The Plant Cell 7, 1071–1083 (1995).
Kim, J., Kim, D. H., Lee, J. Y. & Lim, S. H. The R3-Type MYB transcription factor BrMYBL2.1 negatively regulates anthocyanin biosynthesis in Chinese Cabbage (Brassica rapa L.) by repressing MYB-bHLH-WD40 complex activity. Int J Mol Sci 23, https://doi.org/10.3390/ijms23063382 (2022).
Ma, D. & Constabel, C. P. MYB repressors as regulators of phenylpropanoid metabolism in plants. Trends Plant Sci 24, 275–289, https://doi.org/10.1016/j.tplants.2018.12.003 (2019).
Burdzinski, C. & Wendell, D. L. Mapping the anthocyaninless (anl) locus in rapid-cycling Brassica rapa (RBr) to linkage group R9. BMC Genet 8, 64, https://doi.org/10.1186/1471-2156-8-64 (2007).
Wang, W. et al. Mapping the BrPur gene for purple leaf color on linkage group A03 of Brassica rapa. Euphytica 199, 293–302 (2014).
Dai, M. et al. NGSQC: cross-platform quality analysis pipeline for deep sequencing data. BMC Genomics 11(Suppl 4), S7, https://doi.org/10.1186/1471-2164-11-s4-s7 (2010).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120, https://doi.org/10.1093/bioinformatics/btu170 (2014).
Ranallo-Benavidez, T. R., Jaron, K. S. & Schatz, M. C. GenomeScope 2.0 and smudgeplot for reference-free profiling of polyploid genomes. Nat Commun 11, 1432, https://doi.org/10.1038/s41467-020-14998-3 (2020).
Marçais, G. & Kingsford, C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 27, 764–770, https://doi.org/10.1093/bioinformatics/btr011 (2011).
Cheng, H. et al. Haplotype-resolved assembly of diploid genomes without parental data. Nature Biotechnology 40, 1332–1335, https://doi.org/10.1038/s41587-022-01261-x (2022).
Hu, J., Fan, J., Sun, Z. & Liu, S. NextPolish: a fast and efficient genome polishing tool for long-read assembly. Bioinformatics 36, 2253–2255, https://doi.org/10.1093/bioinformatics/btz891 (2020).
Zhang, X., Zhang, S., Zhao, Q., Ming, R. & Tang, H. Assembly of allele-aware, chromosomal-scale autopolyploid genomes based on Hi-C data. Nat Plants 5, 833–845, https://doi.org/10.1038/s41477-019-0487-8 (2019).
Dudchenko, O. et al. De novo assembly of the Aedes aegypti genome using Hi-C yields chromosome-length scaffolds. Science 356, 92–95, https://doi.org/10.1126/science.aal3327 (2017).
Robinson, J. T. et al. Juicebox.js Provides a Cloud-Based Visualization System for Hi-C Data. Cell Syst 6, 256–258.e251, https://doi.org/10.1016/j.cels.2018.01.001 (2018).
Manni, M., Berkeley, M. R., Seppey, M., Simão, F. A. & Zdobnov, E. M. BUSCO update: novel and streamlined workflows along with broader and deeper phylogenetic coverage for scoring of Eukaryotic, Prokaryotic, and Viral Genomes. Mol Biol Evol 38, 4647–4654, https://doi.org/10.1093/molbev/msab199 (2021).
Parra, G., Bradnam, K. & Korf, I. CEGMA: a pipeline to accurately annotate core genes in eukaryotic genomes. Bioinformatics 23, 1061–1067, https://doi.org/10.1093/bioinformatics/btm071 (2007).
Benson, G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Research 27, 573–580, https://doi.org/10.1093/nar/27.2.573 (1999).
Bao, W., Kojima, K. K. & Kohany, O. Repbase update, a database of repetitive elements in eukaryotic genomes. Mob DNA 6, 11, https://doi.org/10.1186/s13100-015-0041-9 (2015).
Ou, S. & Jiang, N. LTR_FINDER_parallel: parallelization of LTR_FINDER enabling rapid identification of long terminal repeat retrotransposons. Mob DNA 10, 48, https://doi.org/10.1186/s13100-019-0193-0 (2019).
Price, A. L., Jones, N. C. & Pevzner, P. A. De novo identification of repeat families in large genomes. Bioinformatics 21, i351–i358, https://doi.org/10.1093/bioinformatics/bti1018 (2005).
Flynn, J. M. et al. RepeatModeler2 for automated genomic discovery of transposable element families. Proceedings of the National Academy of Sciences 117, 9451–9457, https://doi.org/10.1073/pnas.1921046117 (2020).
Keller, O., Kollmar, M., Stanke, M. & Waack, S. A novel hybrid gene prediction method employing protein multiple sequence alignments. Bioinformatics 27, 757–763, https://doi.org/10.1093/bioinformatics/btr010 (2011).
Majoros, W. H., Pertea, M. & Salzberg, S. L. TigrScan and GlimmerHMM: two open source ab initio eukaryotic gene-finders. Bioinformatics 20, 2878–2879, https://doi.org/10.1093/bioinformatics/bth315 (2004).
Parra, G., Blanco, E. & Guigó, R. GeneID in Drosophila. Genome Res 10, 511–515, https://doi.org/10.1101/gr.10.4.511 (2000).
Flicek, P. Gene prediction: compare and CONTRAST. Genome Biol 8, 233, https://doi.org/10.1186/gb-2007-8-12-233 (2007).
Haas, B. J. et al. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat Protoc 8, 1494–1512, https://doi.org/10.1038/nprot.2013.084 (2013).
Haas, B. J. et al. Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome Biol 9, R7, https://doi.org/10.1186/gb-2008-9-1-r7 (2008).
Lowe, T. M. & Eddy, S. R. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res 25, 955–964, https://doi.org/10.1093/nar/25.5.955 (1997).
Altschul, S. F., Gish, W., Miller, W., Myers, E. W. & Lipman, D. J. Basic local alignment search tool. J Mol Biol 215, 403–410, https://doi.org/10.1016/s0022-2836(05)80360-2 (1990).
Nawrocki, E. P. et al. Rfam 12.0: updates to the RNA families database. Nucleic Acids Res 43, D130–137, https://doi.org/10.1093/nar/gku1063 (2015).
Nawrocki, E. P. & Eddy, S. R. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics 29, 2933–2935, https://doi.org/10.1093/bioinformatics/btt509 (2013).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRP441633 (2023).
Zhang, Z. The genome sequence and annotation of Zicaitai, figshare, https://doi.org/10.6084/m9.figshare.23519952.v3 (2023).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc:JAUJLN000000000 (2023).
Emms, D. M. & Kelly, S. OrthoFinder: phylogenetic orthology inference for comparative genomics. Genome Biol 20, 238, https://doi.org/10.1186/s13059-019-1832-y (2019).
Sanderson, M. J. r8s: inferring absolute rates of molecular evolution and divergence times in the absence of a molecular clock. Bioinformatics 19, 301–302, https://doi.org/10.1093/bioinformatics/19.2.301 (2003).
Fábio, K. M., Dan, V., Ben, F., Matthew, W. H. CAFE 5 models variation in evolutionary rates among gene families. Bioinformatics, btaa1022, https://doi.org/10.1093/bioinformatics/btaa1022 (2020).
Acknowledgements
This work was funded by the Seed Industry Revitalization Project of Provincial Rural Revitalization Strategy Special Fund in 2022 [grant numbers: 2022-NJS-03-001, 2022-NPY-03-001]
Author information
Authors and Affiliations
Contributions
H.L.R. and J.Z. conceived the study. G.G.L. and J.W.Z. collected the samples. D.L.X., W.Y.X. and X.Y.Z. extracted the genomic DNA and conducted sequencing. Z.B.Z. performed bioinformatics analysis. H.L.R. and Z.B.Z. wrote the manuscript. H.Z. and Y.S.Z. improved and revised the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ren, H., Xu, D., Xiao, W. et al. Chromosome-level genome assembly and annotation of Zicaitai (Brassica rapa var. purpuraria). Sci Data 10, 759 (2023). https://doi.org/10.1038/s41597-023-02668-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-023-02668-0
