
Abstract
Betulaceae, the birch family, comprises six living genera and over 160 species, many of which are economically valuable. To deepen our knowledge of Betulaceae species, we have sequenced the genome of a hornbeam, Carpinus fangiana, which belongs to the most species-rich genus of the Betulaceae subfamily Coryloideae. Based on over 75 Gb (~200x) of high-quality next-generation sequencing data, we assembled a 386.19 Mb C. fangiana genome with contig N50 and scaffold N50 sizes of 35.32 kb and 1.91 Mb, respectively. Furthermore, 357.84 Mb of the genome was anchored to eight chromosomes using over 50 Gb (~130x) Hi-C sequencing data. Transcriptomes representing six tissues were sequenced to facilitate gene annotation, and over 5.50 Gb high-quality data were generated for each tissue. The structural annotation identified a total of 27,381 protein-coding genes in the assembled genome, of which 94.36% were functionally annotated. Additionally, 4,440 non-coding genes were predicted.
Measurement(s) | DNA • RNA • sequence_assembly • sequence feature annotation |
Technology Type(s) | DNA sequencing • RNA sequencing • genome assembly • sequence annotation |
Sample Characteristic - Organism | Carpinus fangiana |
Machine-accessible metadata file describing the reported data: https://doi.org/10.6084/m9.figshare.11382390
Similar content being viewed by others
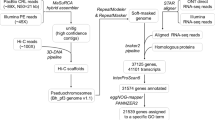
Background & Summary
Betulaceae, also known as the birch family, includes over 160 species of trees or shrubs1. It is divided into two subfamilies, Coryloideae and Betuloideae; Betuloideae comprises the genera Alnus and Betula, while Coryloideae comprises Corylus, Ostryopsis, Carpinus and Ostrya. These subfamilies and their genera are readily distinguished based on their different morphological characteristics, such as the samara of Coryloideae, the nuts of Betuloideae, and their different types of pollen2. In addition, cell biological investigations have revealed that Betulaceae species have very different chromosome numbers: the basic chromosome number is eight for Carpinus, Ostrya, Ostryopsis species, eleven for Corylus species, and fourteen for Alnus and Betula species3,4.
Several Betulaceae species, notably those belonging to the genera Betula, Alnus, and Carpinus, are important components of forests in temperate regions, mountains, and subtropical areas, as well as important sources of timber and materials for traditional Chinese medicine. Some species of Betula and Carpinus are used as ornamental trees and widely planted in large parks and gardens. Alnus species can form symbioses with nitrogen-fixing bacteria of the genus Frankia, helping to enhance soil fertility5. The fruits of Corylus, known as hazelnuts, are economically important. The birch family thus has remarkable ecological, economic, medicinal, and ornamental value. Additionally, Betulaceae is a relict family, and there are many reliable fossils of this family that have provided important paleobotanic insights6. However, only a few species of the family have been studied extensively in ways that could support their further development and utilization.
A few genomes of Betulaceae species have been published in recent years. The genomes of two Betuloideae members, Betula pendula (scaffold N50: 0.53 Mb)7 and Alnus glutinosa (scaffold N50: 0.10 Mb)8, were presented in 2017 and 2018, and the B. pendula genome was further anchored to fourteen chromosomes. The only published Coryloideae genomes are those of two ironwood trees from the genus Ostrya: O. rehderiana (scaffold N50: 2.31 Mb) and O. chinensis (scaffold N50: 0.81 Mb), which were reported in 20189. However, no genomes representing any of the other three genera in Coryloideae have been disclosed and there are no published chromosome-level genomes for this subfamily.
To enrich the available genomic resources for Betulaceae, we sequenced the whole genome of Carpinus fangiana (Fig. 1), a member of the most species-rich genus in Coryloideae10. A total of 77.85 Gb (~200x) next-generation data and 52.19 Gb (~130x) Hi-C data were used to assemble the genome. The assembly produced a genome having a total length of 386.19 Mb, with 357.84 Mb being anchored to eight chromosomes. To our knowledge, this is the first reported chromosome-level Coryloideae genome assembly. The contig N50 and scaffold N50 were 35.32 kb and 1.91 Mb, respectively. Structural annotation of the genome revealed a total of 27,381 protein-coding genes, of which 94.36% were functionally annotated. The genome was also predicted to contain 4,440 non-coding genes based on a comprehensive annotation. This chromosome-level genome of C. fangiana will greatly facilitate further biological studies on Betulaceae as well as the development and commercial exploitation of the genus.

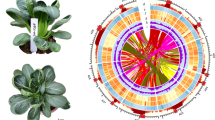
Photograph and location of the C. fangiana tree sampled for genome sequencing. (a) A photograph of a C. fangiana individual on Emei Mountain, Leshan, Sichuan, China. (b) Location of the C. fangiana sample used for genome sequencing.
Methods
Sampling, library construction and sequencing
Fresh leaves were collected from a wild C. fangiana tree in Ebian, Sichuan, China (N: 29° 1′44″; S: 102°59′30″; Fig. 1) and immediately dried over silica gel. Genomic DNA was then extracted from the dried leaves using the modified Cetyltrimethylammonium Ammonium Bromide (CTAB)11 method. Sequencing libraries with different insert sizes were constructed using a library construction kit (Illumina). Short paired-end libraries were constructed with insert sizes of 230, 500, and 800 bp, while the insert sizes used to construct mate pair libraries were 2, 5, 10, and 20 kb. The Illumina HiSeq 2000 platform was used to sequence 150 bp paired-end reads for all these libraries in accordance with the manufacturer’s instructions. These procedures generated a total of 115.12 Gb (~200x) raw data for C. fangiana genome assembly (Table 1).
A High-through chromosome conformation capture (Hi-C) library for the C. fangiana genome was also constructed. To this end, fresh leaves were fixed with formaldehyde to induce DNA cross-linking, after which the DNA was digested with HindIII. The resulting sticky ends were biotinylated and proximity-ligated to form chimeric junctions that were enriched for, and physically sheared into 300–700 bp fragments. These chimeric fragments were sequenced on the Illumina HiSeq platform, generating 52.54 Gb (~130x) of Hi-C data (Table 1).
We also harvested six tissues (bark, branch, bract, flower, fruit, leaf) for total RNA sequencing. These samples were flash frozen in liquid nitrogen, and total RNA was extracted using the modified CTAB method12. cDNA libraries were then constructed using the NEBNext Ultra RNA Library Prep Kit for Illumina (NEB). The Illumina HiSeq 2500 platform was used to sequence these libraries with a read length of 2 × 150 bp, generating over 5.50 Gb raw data for each tissue (Table 2).
Preprocessing and genome size estimation
Quality control checks on the raw genome data were preformed using FastQC (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/). Potential adapters in reads were removed using Scythe (http://github.com/vsbuffalo/scythe) and low-quality reads were discarded by Sickle (http://github.com/vsbuffalo/scythe). The program Lighter13 was then used to correct sequence errors in the remaining reads. For mate pair reads, we also used FastUniq14 to remove duplicates. In total, 77.85 Gb, ~200x high-quality next-generation sequencing data and 52.19 Gb, ~130x high-quality Hi-C data were generated for de novo assembly of the C. fangiana genome (Table 1).
Quality control of transcriptome data was performed using a custom Perl script. Reads were discarded if (1) the proportion of unidentified nucleotides in one read exceeded 5%, or (2) over 65% of the read’s bases had a phred quality below 8. After eliminating low-quality reads, the quantity of retained data for each tissue was above 5.50 Gb (Table 2). The RNA-seq reads were then assembled using Trinity15. CD-Hit16 was used to eliminate redundant transcript sequences, and candidate coding regions in the transcript sequences were identified by TransDecoder (https://transdecoder.github.io).
Before genome assembly, we estimated the C. fangiana genome’s size by performing a combined analysis using Jellyfish17 and GenomeScope18. Reads from the short-insert libraries were first processed by Jellyfish to assess their k-mer distribution, using a k value of 17. Then, GenomeScope was used to estimate the genome size based on the k-mer distribution (Fig. 2). The genome was thereby estimated to be around 396.74 Mb long.

K-mer distribution used to estimate the genome’s size. The distribution was determined based on the Jellyfish analysis using a k-mer size of 17.
Genome assembly
Preliminary de novo assembly of the C. fangiana genome was performed with Platanus19, which can effectively manage high-throughput data from heterozygous samples. Assembly using Platanus proceeded via three steps: (1) contig-assembly, in which de Bruijn graphs were constructed using the clean reads from short paired-end libraries and the sequences of contigs were then displayed in the graphs; (2) scaffolding, in which reads from all next-generation libraries (short paired-end and mate pair) were mapped to contigs, after which contigs considered to be linked were combined into scaffolds; (3) gap closing, in which reads that mapped onto scaffolds were collected to cover the gaps between them. GapCloser20 was used to further close the gaps based on reads from all the paired-end libraries, after which the automated HaploMerger2 pipeline21 was used to rebuild the above assembly and implement flexible and sensitive error detection. After discarding scaffolds smaller than 1 kb, a high-quality de novo assembled C. fangiana genome was obtained. The size of this genome (386.19 Mb) was 97.34% of the estimated value (396.74 Mb) and its GC content was 37.59%. The scaffold N50 and N90 values were 1.91 Mb and 0.43 Mb, while the contig N50 and N90 were 35.32 kb and 8.54 kb (Table 3).
The HiC-Pro22 program was used for quality assessment of the Hi-C data. Valid interaction pairs were mapped to and used for error correction of the contigs and scaffolds assembled based on the next-generation sequencing data. Next, the contigs and scaffolds were anchored to chromosomes using LACHESIS23. In total, 357.84 Mb of scaffolds were assembled into eight chromosomes (Table 4). Finally, we obtained a high-quality chromosome-level genome with a total size of 386.25 Mb. The contig N50 and scaffold N50 values of this chromosome-level assembly were 34.85 kb and 37.11 Mb, respectively (Table 3).
Heterozygosity assessment and repeat annotation
To assess the heterozygosity of the C. fangiana genome, we first mapped reads from the 500 bp library to the assembled genome using the BWA-MEM algorithm from the Burrows-Wheeler Aligner (BWA) package24. SAMtools25 was used to convert the mapping results to BAM format, sort them, and remove duplicates. The Picard package (http://broadinstitute.github.io/picard/) was used to replace read groups in the bam file. Two programs (RealignerTargetCreator and IndelRealigner) from the Genome Analysis ToolKit (GATK)26 package were used to avoid misalignments and account for the effects of indels. The SAMtools command ‘mpileup’ was used to generate a VCF format file, and the program bcftools from the SAMtools package was used to detect single nucleotide polymorphisms (SNPs). Finally, based on the SNPs, the heterozygosity was calculated to be 0.38% using a custom Perl script.
Repetitive sequences and transposable elements (TEs) in the C. fangiana genome were identified using a combined procedure incorporating de novo and homology-based approaches at the DNA and protein levels. Tandem repeats were annotated using Tandem Repeat Finder (TRF)27. A repeat library for the C. fangiana genome was generated using RepeatModeler (http://www.repeatmasker.org) to facilitate de novo annotation. RepeatMasker28 (http://www.repeatmasker.org) was used to identify and classify the TEs at the DNA level. We also used RepeatProteinMasker to perform a WU-BLASTX search against the TE protein database in order to identify and classify TEs at the protein level. Finally, long terminal repeats (LTR) were identified using LTR-FINDER29. In total, the C. fangiana genome was found to contain 158.69 Mb repetitive sequences, accounting for 41.08% of its length (Table 5). As shown in Table 5, the most common classifications assigned to these repetitive elements were Unknown (15.97% of the assembled genome) and LTRs (14.57% of the assembled genome).
Gene annotation
Structural annotation of gene models was performed by applying a combination of de novo, homology-based, and transcriptome-based methods to the repeat-masked genome. The de novo approach was implemented using Augustus30, Geneid31, GeneMark32, glimmerHMM33, and SNAP34. For homology-based prediction, TBLASTN35 was used to align predicted protein sequences from Arabidopsis thaliana, Vitis vinifera, Prunus persica, Ostrya chinensis, Ostrya rehderiana and Juglans regia to the C. fangiana genome with an E-value threshold of 1E-05. Then, GeneWise36 was used to obtain accurate spliced alignments by aligning homologous sequences to matched proteins. Transcriptome-based prediction was performed with the Program to Assemble Spliced Alignments (PASA)37, which was used to predict protein-coding regions based on the assembled transcripts of the six different C. fangiana tissues. The gene models obtained from the de novo, homology-based, and transcriptome-based annotations were combined to form a consensus gene set using EVidenceModeler (EVM)38. After strict filtering, a total of 27,381 non-redundant protein-coding genes were annotated in the C. fangiana genome (Table 6).
Functional annotation of the predicted protein genes was performed by using BLASTP with an E-value threshold of 1E-05 to search for homologous sequences in SwissProt (http://www.gpmaw.com/html/swiss-prot.html), TrEMBL (http://www.uniprot.org)39, and KEGG (http://www.genome.jp/kegg/) protein databases40. The program hmmscan of HMMER package (http://hmmer.org) was used to search the Pfam domains. InterProScan41 was used to annotate the protein motifs and domains, and the Blast2GO pipeline42 was used to obtain Gene Ontology (GO)43 IDs for each gene based on the NCBI NR database. In total, 25,836 protein-coding genes, corresponding to 94.36% of the total predicted gene models in the C. fangiana genome were successfully functionally annotated (Table 7).
We also annotated non-coding RNAs in the C. fangiana genome. tRNAscan-SE44 was used to detect putative transfer RNAs (tRNAs) with eukaryotic parameters, resulting in the identification of 632 tRNAs. To identify other non-coding RNAs, INFERNAL45 was used to perform searches against the Rfam46 database, resulting in the identification of 936 ribosomal RNAs (rRNAs), 197 microRNAs (miRNAs), 117 small nuclear RNAs (snRNAs), and 232 small nucleolar RNAs (snoRNAs) (Table 8).
Data Records
The sequencing data including the Illumina genome data (SRA accession: SRX6070999-SRX6071006), Hi-C data (SRA accession: SRX6071007), and Illumina transcriptome data (SRA accession: SRX6070994-SRX6070998, SRX6071008) were submitted to the NCBI Sequence Read Archive (SRA) database under BioProject accession number PRJNA54802747. The assembled genome was deposited at DDJB/ENA/GenBank under accession number VIBQ0000000048. Repeat annotations, gene model annotations and non-coding RNA annotations, the CDS sequences for the coding and non-coding genes, the protein sequences for the coding genes, as well as two custom Perl scripts were deposited at figshare49.
Technical Validation
Assessment of the genome assembly
We evaluated the completeness of the C. fangiana genome assembly in two ways. First, all the paired-end reads were mapped to the assembly genome with BWA. The aligned outputs were then analyzed using SAMtools. The mapping rate for each library was above 90% (Table 9). Furthermore, the coverage of the genome after gap elimination was 99.74%, with 95.05% having at least 100x coverage. Benchmarking Universal Single-Copy Orthologs (BUSCO)50 was also used to evaluate the completeness of the genome assembly. 95.30% of the “complete BUSCOs” were successfully identified in the assembly, and the proportion of “missing BUSCOs” was only 4.10% (Table 10). These results demonstrate the high reliability and completeness of the reported genome assembly.
Finally, we evaluated the assembly of the eight chromosomes. To this end, the anchored genome was split into ‘bins’ of 100 kb in length. The number of Hi-C read pairs covered by any two ‘bins’ was used to define the signal for the interaction between those ‘bins’, and these signal intensities were plotted in the form of a heat map. The signal intensities clearly divided the ‘bins’ into eight distinct groups, demonstrating the high quality of the chromosome assembly (Fig. 3).

Heat map of chromosomal interactions in the C. fangiana genome. Cfa01-Cfa08 represent the eight chromosomes in the C. fangiana genome. The horizontal and vertical coordinates represent the order of each ‘bin’ on the corresponding chromosome.
Improvement of gene annotation quality
To maximize the reliability of the gene annotation process, repeat regions in the assembled genome were masked before gene annotation. Mirroring the procedure used to filter gene annotation, EVM was initially used to merge the results obtained by de novo, homolog-based, and transcriptome-based predictions. Genes were then discarded if: (1) their CDS length was below 150 bp; (2) their putative coding regions could not be accurately translated into protein sequences; (3) they possessed early termination codons; or (4) they were only supported by de novo predictions. In addition, PASA was used to identify untranslated regions (UTRs).
Code availability
This work relied on many software tools. The versions, settings and parameters of these tools are given below.
(1) FastQC: version 0.11.5, default parameters; (2) Scythe: version 0.994 BETA, parameters: -q sanger --quiet; (3) Sickle: version 1.33, parameters: pe -t sanger -q 20 -l 50 -n --quiet; (4) Lighter: version 1.0.7, parameters: -K 21 360000000; (5) FastUniq: version 1.1, default parameters (6) Trinity: trinityrnaseq-2.6.4, parameters: --seqType fq --JM 260G; (6) CD-Hit: version 4.6, default parameters; (7) TransDecoder: version 5.2.0, default parameters; (8) Jellyfish: version 1.1.10, parameters: count command: -m 17 -s 4G -c 7, dump command: -c -t, histo command: default parameters; (9) GenomeScope: version 2.0, parameters: 17 (k-mer length) 150 (read length); (10) Platanus: version 1.2.1, default parameters for the all three steps, (11) GapCloser: version 1.12, parameter: -l 150; (12) HaploMerger2: version HaploMerger2_20151124, default parameters for the followed running processes: carrying out batchA to batchE with the recommended pipeline, among which batchA was repeated 3 times and batchD was repeated 2 times, respectively; (13) HiC-Pro: version 2.10.0, default parameters; (14) LACHESIS: released in 2017, parameters: CLUSTER_MIN_RE_SITES=36 CLUSTER_MAX_LINK_DENSITY=1 CLUSTER_NONINFORMATIVE_RATIO=8 ORDER_MIN_N_RES_IN_TRUN=22 ORDER_MIN_N_RES_IN_SHREDS 22; (15) BWA: version 0.7.12-r1039, default parameters; (16) SAMtools: version 1.5, parameters: view command: -bS, sort command: -O BAM, depth command: -Q 40, mpileup command: -DSug -C 50, default parameters for the rmdup, index and flagstat commands; (17) Picard: version 1.80, parameters: SORT_ORDER =coordinate RGPL =illumina RGPU =illumina; (18) GATK: version 3.3-0-g37228af, default parameters for the two programs RealignerTargetCreator and IndelRealigner; (19) bcftools: version 0.1.19-44428 cd, parameters: view –Ncg; (20) TRF: version 4.07b, parameters: Match=2 Mismatch=7 Delta=7 PM=80 PI=10 Minscore=50 MaxPeriod=500 -d –h; (21) RepeatModeler: version 1.0.4, parameters: -pa 30 -database Fan; (22) RepeatMasker: version open-4.0.5, parameters: -pa 30 -species all -nolow -norna -no_is -gff; (23) RepeatProteinMasker: version 2.1, parameters: -engine abblast -noLowSimple -pvalue 1e-04; (24) LTR-FINDER: version 1.05, default parameters; (25) Augustus: version 2.5.5, parameters: --species=arabidopsis; (26) Geneid: version 1.4, parameters: -3 -P; (27) GeneMark: version 3.47, parameters: -f gff3; (28) GlimmerHMM: version 3.0.4, default parameters; (29) SNAP: version 2006-07-28, default parameters; (29) GeneWise: version 2.4.1, parameters: -tfor/-trev -gff; (30) EVM: version 1.1.1, default parameters; (31) PASA: version 2.0.2, parameters: for Launch_PASA_pipeline.pl step: -C -R -r–ALIGNERS blat, gmap, default parameters for the below two steps: asa_asmbls_to_training_set.extract_reference_orfs.pl and pasa_asmbls_to_training_set.dbi; (32) BLASTP: version 2.2.30+, parameters: -evalue 1e-5 -outfmt 7; (33) Interproscan: version 5.25-64.0, parameters: -dp -f tsv; (34) tRNAscan-SE: tRNAscan-SE-2.0, default parameters; (35) BUSCO: version 2.0, parameters: -m genome -c 20.
References
Christenhusz, M. J. M. & Byng, J. W. The number of known plant species in the world and its annual increase. Phytotaxa. 261, 201–217 (2016).
Chen, Z. Phylogeny and phytogeography of the Betulaceae. Acta Phytotaxon. Sin. 32, 1–31 (1994).
Botta, R., Emanuel, E., Me, G., Sacerdote, S. & Vallania, R. Indagine cariologica in alcune specie del genere Corylus (In Italian). Rivista di Ortoflorfrutticoltura Italiana. 70, 323–329 (1986).
Snow, N. & Goldblatt, P. Chromosome number in Ticodendron (Fagales, Ticodendraceae). Ann. Mo. Bot. Gard. 79, 906–907 (1992).
Jin, W. Y., Cai, X. F., Na, M. K., Lee, J. J. & Bae, K. H. Triterpenoids and Diarylheptainoids from Alnus hirsuta inhibit HIF-1 in AGS Cells. Arch. Pharm. Res. 30, 412–418 (2007).
Crane, P. R. & Blackmore, S. Evolution, systematics, and fossil history of the Hamamelidae. (Published for the Systematics Association by the Clarendon Press, 1989).
Salojärvi, J. et al. Genome sequencing and population genomic analyses provide insights into the adaptive landscape of silver birch. Nat. Genet. 49, 904–912 (2017).
Griesmann, M. et al. Phylogenomics reveals multiple losses of nitrogen-fixing root nodule symbiosis. Science. 361, eaat1743 (2018).
Yang, Y. et al. Genomic effects of population collapse in a critically endangered ironwood tree Ostrya rehderiana. Nat. Commun. 9, 5449 (2018).
Lancaster, R. & Rix, M. 705. Carpinus fangiana. Curtis’s Botanical Magazine. 28, 103–110 (2011).
Doyle, J. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem Bull. 19, 11–15 (1987).
Chang, S. J., Puryear, J. & Cairney, J. A simple and efficient method for isolating RNA from pine trees. Plant Mol. Biol. Rep. 11, 113–116 (1993).
Song, L., Song, L., Florea, L. & Langmead, B. Lighter: fast and memory-efficient sequencing error correction without counting. Genome Biol. 15, 509 (2014).
Xu, H. et al. FastUniq: a fast de novo duplicates removal tool for paired short reads. PloS One. 7, e52249 (2012).
Grabherr, M. G. et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 29, 644–652 (2011).
Li, W. Z., Jaroszewski, L. & Godzik, A. Tolerating some redundancy significantly speeds up clustering of large protein databases. Bioinformatics. 18, 77–82 (2002).
Marçais, G. & Kingsford, C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics. 27, 764–770 (2011).
Vurture, G. W. et al. GenomeScope: fast reference-free genome profiling from short reads. Bioinformatics. 33, 2202–2204 (2017).
Kajitani, R. et al. Efficient de novo assembly of highly heterozygous genomes from whole-genome shotgun short reads. Genome Res. 24, 1384–1395 (2014).
Luo, R. et al. SOAPdenovo2: an empirically improved memory-efficient short-read de novo assembler. GigaScience. 1, 18 (2012).
Huang, S. et al. HaploMerger: reconstructing allelic relationships for polymorphic diploid genome assemblies. Genome Res. 22, 1581–1588 (2012).
Servant, N. et al. HiC-Pro: an optimized and flexible pipeline for Hi-C data processing. Genome Biol. 16, 1–11 (2015).
Burton, J. N. et al. Chromosome-scale scaffolding of de novo genome assemblies based on chromatin interactions. Nat. Biotechnol. 31, 1119–1125 (2013).
Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv preprint arXiv. 1303, 3997 (2013).
Li, H., Bob, H., Alec, W., Tim, F. & Jue, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 25, 2078–2079 (2009).
McKenna, A. et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303 (2010).
Benson, G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 27, 573–580 (1999).
Tarailo‐Graovac, M. & Chen, N. Using RepeatMasker to identify repetitive elements in genomic sequences. Curr. Protoc. Bioinformatics. 25, 4.10.1–4.10.14 (2009).
Xu, Z. & Wang, H. LTR_FINDER: an efficient tool for the prediction of full-length LTR retrotransposons. Nucleic Acids Res. 35, W265–W268 (2007).
Stanke, M., Diekhans, M., Baertsch, R. & Haussler, D. Using native and syntenically mapped cDNA alignments to improve de novo gene finding. Bioinformatics. 24, 637–644 (2008).
Parra, G., Blanco, E. & Guigó, R. Geneid in Drosophila. Genome Res. 10, 511–515 (2000).
Lukashin, A. V. & Borodovsky, M. GeneMark.hmm: new solutions for gene finding. Bioinformatics. 26, 1107–1115 (1998).
Majoros, W. H., Pertea, M. & Salzberg, S. L. TigrScan and GlimmerHMM: two open source ab initio eukaryotic gene-finders. Bioinformatics. 20, 2878–2879 (2004).
Korf, I. Gene finding in novel genomes. BMC Bioinformatics. 5, 59 (2004).
Gertz, E. M., Yu, Y. K., Agarwala, R., Schäffer., A. A. & Altschul, S. F. Composition-based statistics and translated nucleotide searches: improving the TBLASTN module of BLAST. BMC Biol. 4, 41 (2006).
Birney, E. et al. GeneWise and genomewise. Genome Res. 14, 988–995 (2004).
Haas, B. J. et al. Improving the Arabidopsis genome annotation using maximal transcript alignment assemblies. Nucleic Acids Res. 31, 5654–5666 (2003).
Haas, B. J. et al. Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome Biol. 9, R7 (2008).
Bairoch, A. & Apweiler, R. The SWISS-PROT protein sequence database and its supplement TrEMBL in 2000. Nucleic Acids Res. 28, 45–48 (2000).
Kanehisa, M. et al. BlastKOALA and GhostKOALA: KEGG tools for functional characterization of genome and metagenome sequences. J. Mol. Biol. 428, 726–731 (2016).
Mulder, N. & Apweiler, R. InterPro and InterProScan: tools for protein sequence classification and comparison. Methods Mol. Biol. 396, 59–70, (Springer, 2007).
Conesa, A., Sato, Y. & Morishima, K. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics. 21, 3674–3676 (2005).
Ashburner, M. et al. Gene ontology: tool for the unification of biology. Nat. Genet. 25, 25 (2000).
Lowe, T. M. & Eddy, S. R. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 25, 955–964 (1997).
Nawrocki, E. P., Kolbe, D. L. & Eddy, S. R. Infernal 1.0: inference of RNA alignments. Bioinformatics. 25, 1335–1337 (2009).
Griffiths-Jones, S. et al. Rfam: annotating non-coding RNAs in complete genomes. Nucleic Acids Res. 33, D121–D124 (2005).
NCBI Sequence Read Archive, https://identifiers.org/insdc.sra:SRP201422 (2019).
Yang, X. et al. Carpinus fangiana isolate Cfa_2016G, whole genome shotgun sequencing project. Genbank, https://identifiers.org/ncbi/insdc:VIBQ00000000.1 (2019).
Yang, X. Y., Wang, Z. F., Zhang, L., Hao, G. Q. & Yang, Y. Z. Data and material for the Carpinus fangiana genome. figshare, https://doi.org/10.6084/m9.figshare.8549204.v6 (2019).
Simão, F. A., Waterhouse, R. M., Loannidis, P., Kriventseva, E. V. & Zdobnov, E. M. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics. 31, 3210–3212 (2015).
Acknowledgements
This work was equally supported by the Strategic Priority Research Program of the Chinese Academy of Sciences (Grant No. XDB31000000) and National Key Research and Development Program of China (2017YFC0505203), and further by National Natural Science Foundation of China (31590821 and 31900201), the Fundamental Research Funds for the Central Universities (Grant No. lzujbky-2019-77) and the Lanzhou University’s “Double First-Class” Guided Project – Team Building Funding - Research Startup Fee for Jianquan Liu and Yongzhi Yang. We thank Dr. Mingcheng Wang and Cheng Zhang for their technical help.
Author information
Authors and Affiliations
Contributions
Yongzhi Yang designed and conceived this work; Lei Zhang and Guoqian Hao collected the materials and prepared DNA and RNA for sequencing; Zefu Wang and Xiaoyue Yang analyzed the data. Xiaoyue Yang wrote the manuscript with other authors’ help; Jianquan Liu and Yongzhi Yang revised the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
The Creative Commons Public Domain Dedication waiver http://creativecommons.org/publicdomain/zero/1.0/ applies to the metadata files associated with this article.
About this article

Cite this article
Yang, X., Wang, Z., Zhang, L. et al. A chromosome-level reference genome of the hornbeam, Carpinus fangiana. Sci Data 7, 24 (2020). https://doi.org/10.1038/s41597-020-0370-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-020-0370-5
This article is cited by
-
Genome structure-based Juglandaceae phylogenies contradict alignment-based phylogenies and substitution rates vary with DNA repair genes
Nature Communications (2023)
-
Leaf phenotypic variation in natural populations of Carpinus tschonoskii in China
Journal of Forestry Research (2023)
-
Genomic evidence for homoploid hybrid speciation between ancestors of two different genera
Nature Communications (2022)
