
Abstract
Voltage-gated Na+ (NaV) channels regulate homeostasis in bacteria and control membrane electrical excitability in mammals. Compared to their mammalian counterparts, bacterial NaV channels possess a simpler, fourfold symmetric structure and have facilitated studies of the structural basis of channel gating. However, the pharmacology of bacterial NaV remains largely unexplored. Here we systematically screened 39 NaV modulators on a bacterial channel (NaChBac) and characterized a selection of compounds on NaChBac and a mammalian channel (human NaV1.7). We found that while many compounds interact with both channels, they exhibit distinct functional effects. For example, the local anesthetics ambroxol and lidocaine block both NaV1.7 and NaChBac but affect activation and inactivation of the two channels to different extents. The voltage-sensing domain targeting toxin BDS-I increases NaV1.7 but decreases NaChBac peak currents. The pore binding toxins aconitine and veratridine block peak currents of NaV1.7 and shift activation (aconitine) and inactivation (veratridine) respectively. In NaChBac, they block the peak current by binding to the pore residue F224. Nonetheless, aconitine has no effect on activation or inactivation, while veratridine only modulates activation of NaChBac. The conservation and divergence in the pharmacology of bacterial and mammalian NaV channels provide insights into the molecular basis of channel gating and will facilitate organism-specific drug discovery.
Similar content being viewed by others

Introduction
Electrical signaling is a highly conserved biological mechanism throughout the evolution of plants and animals1,2. From prokaryotic organelles to vertebrates, ion channels are key regulators of cellular homeostasis, electrolyte balance and signaling1. Single-celled eukaryotes first evolved voltage-gated calcium channels (CaV), which are believed to have evolved subsequently into voltage-gated Na+ (Nav) channels predating the origin of nervous systems in animals3. In prokaryotes, NaV channels might have evolved independently to regulate cellular homeostasis, though their exact functions are not well understood.
In humans, the malfunction of NaV channels, either through genetic mutations or off-target drug interactions, results in severe pathologies including cardiac arrhythmia and epilepsy4. Given their critical physiological function, pathological relevance and utility as therapeutic targets, it is essential to understand the interaction of NaV channels with pharmacological agents. This task has been facilitated by the recent determination of prokaryotic5,6,7 and eukaryotic8,9,10,11,12,13 NaV channel structures. However, these structures do not provide a clear picture of the dynamic conformational changes that underlie state and voltage-dependent effects of pharmacological agents. Therefore, functional studies must be integrated with structural insights to elucidate the molecular mechanisms of compound-channel interaction14,15.
Eukaryotic NaV channels are large, complex membrane-bound proteins composed of four homologous domains that are connected via long intracellular linkers16. In contrast, prokaryotic NaV channels are formed by co-assembly of four identical subunits6,7. Bacterial NaV channels are useful as model systems for their more complex eukaryotic counterparts because their crystal structures in various states have been solved, providing unprecedented insights into ion selectivity, gating, as well as drug interaction mechanisms5,6,7,17,18,19. For example, the crystal structure of NaVAb (from Arcobacter butzleri) with lidocaine and flecainide reveals that the potency of resting state block by these local anesthetics (LAs) is determined by the size of fenestrations that connect the lipids in the cell membrane with the inner pore of the channel, the so-called hydrophobic pathway for drug entry20. This notion is corroborated by pharmacological characterization, as lidocaine and benzocaine block NaChBac channel when applied extracellularly, suggesting the conservation of this hydrophobic pathway in related channels. Despite such progress, the pharmacology of bacterial voltage-gated Na+ (BacNaV) channels remains largely underexplored.
In this study, we systematically investigate the interaction of NaChBac (from Bacillus halodurans) with 39 Na+ channel modulators from various drug classes. Comparing the pharmacology of NaChBac with human NaV1.7 channels reveals that while some drug interaction mechanisms are conserved, others exhibit striking divergence.
Results
Effects of local anesthetic (LA) site-binding small molecules on the NaChBac channel
A group of 16 small molecule compounds known to block mammalian Nav channels, including antiarrhythmics, anticonvulsants, muscle relaxants, and local anesthetics were selected for evaluation. To carry out the initial screening, compounds were tested at a concentration that was 50% higher than the known IC50 values for the least-sensitive mammalian NaV channel isoform. When tested against NaChBac channels, 11 out of 16 compounds caused robust block, whereas five compounds, carbamazepine, oxcarbazepine, co102862, QX-222 and QX-314 were relatively ineffective, resulting in only 15–31% inhibition (Fig. 1A, Supplement Fig. 1A, Supplement Table I). Among the inactive compounds, QX-222 and QX-314 are permanently charged. Our results are consistent with a previous report that QX-314 has no effect on the NaChBac channel when applied extracellularly21. In contrast to QX-222 and QX-314, the anticonvulsants carbamazepine, oxcarbazepine, and co102862 are highly hydrophobic and are almost completely neutral at physiological pH. Thus, a majority of LA site-binding small molecules block NaChBac channels, with the exception of the positively charged or highly hydrophobic compounds.

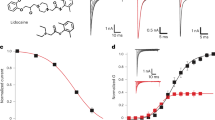
The local anesthetics lidocaine and ambroxol differently modulate NaV1.7 and NaChBac channel gating. (A) Chemical structures of four selected small molecule pore blockers, ambroxol, lidocaine, carbamazepine, and QX-314. Representative NaChBac current traces are shown before and after the application of compounds. Testing concentrations for the compounds are shown in Supplement Table I. (B) Lidocaine’s effect on the NaV1.7 (top) and NaChBac (bottom) channels. Current traces (left), current–voltage (I–V) (middle), conductance–voltage (G–V), and steady-state inactivation (SSI) relationships are shown for both NaV1.7 and NaChBac measured before and after application of 200 µM lidocaine. Lidocaine blocks both NaChBac and NaV1.7 channels. (C) Ambroxol’s effect on NaV1.7 (top) and NaChBac (bottom) channels. Representative current traces (left), I-V (middle), G–V, and SSI relationships (right) are shown for NaV1.7 and NaChBac channels measured before and after 50 µM ambroxol. Ambroxol has similar effects as lidocaine on both NaV1.7 and NaChBac.
We further examined two of the ‘hit’ compounds (defined by > 40% block at test concentration), lidocaine and ambroxol on NaChBac. In response to 200 µM lidocaine, both NaV1.7 and NaChBac exhibited a similar level of tonic block, as shown in representative current traces and current–voltage (I–V) relationships (Fig. 1B). Lidocaine had a minimal effect on the voltage-dependence of activation (G–V relationship) of NaV1.7, but significantly shifted the steady-state inactivation (SSI) curve to more negative potentials (Fig. 1B, top right, ΔV1/2 = − 22.9 ± 2.0, p = 0.0003). These effects were described previously22,23,24 and indicate that lidocaine stabilizes NaV1.7 channels in an inactivated conformation. Lidocaine induced a smaller hyperpolarizing shift in the SSI curve of NaChBac channel currents (Fig. 1B, bottom right, ΔV1/2 = − 11.1 ± 3.2.0, p = 0.007). Lidocaine also increased the slope factor (k[n]) of the SSI curve of NaV1.7, an effect not observed for NaChBac (Table 1). This difference may be explained by differences in the mechanism of inactivation for the two channel types. NaChBac channel lacks the fast inactivation gate in NaV1.7 channel, hence its inactivation might be mediated by an alternative and slower mechanism, such as pore collapse.
Similar to lidocaine, 200 µM ambroxol showed robust inhibition of both NaV1.7 and NaChBac channels (Fig. 1C). Ambroxol induced a significant hyperpolarizing shift in the SSI curve of NaV1.7 (Fig. 1C, top right, ΔV1/2 = − 19.9 ± 2.9, p = 0.001), but the effect was not as pronounced in NaChBac (Fig. 1C, bottom right, ΔV1/2 = − 11.6 ± 3.5, p = 0.03). Overall, although lidocaine and ambroxol exhibit similar blockade of NaChBac and NaV1.7 channels, they modulate the inactivation process of these channels to different extents.
Effect of voltage sensing domain (VSD)-binding toxins on NaChBac channels
The LA binding site resides in the pore domain, with highly conserved primary amino acid sequences across mammalian and bacterial NaV channels21. Unlike the pore domain, the sequence of the voltage sensing domains (VSDs) exhibits much higher variability. As a result, peptide toxins that bind to the VSDs exhibit higher channel specificity when compared to LA compounds25. Since the effect of VSD-binding toxins on NaChBac has not been examined systematically, we tested 15 such toxins on the amplitude and gating of NaChBac channel currents at concentrations known to alter gating of mammalian NaV channels. 11 toxins did not show significant effect, whereas four peptide toxins exhibited modulation on the NaChBac channel, including GsAF-I, GrTx1, GsAF-II, and BDS-I (Supplement Fig. 1B). GsAF-I, GrTx1, and GsAF-II are categorized into site 4 toxins and bind to the VSD of domain II (DII-VSD), whereas BDS-I is a site 3 toxin and interacts with the VSD of domain IV (DIV-VSD) of mammalian NaV channels26.
We chose the site 4 toxin GsAF-I and site 3 toxin BDS-I for further analysis, because these two toxins were potent inhibitors of NaChBac channels. In contrast, ProTx-II (site 4) and ATX-II (site 3) had no effect on current amplitude or kinetics of NaChBac currents (Fig. 2A). GsAF-I blocks the peak NaChBac currents and induced small shifts in the voltage dependence of inactivation (SSI) (Fig. 2B, bottom and Table 1). The overall effect of GsAF-I on NaV1.7 is similar to NaChBac, except in NaV1.7, GsAF-I caused a greater inhibition of peak currents and a shallower SSI curve (k[n]control = − 6.3 ± 0.3, k[n]GsAF-I = − 10 ± 0.3) (Fig. 2B, top), suggesting that in the presence of GsAF-I, more channels start to inactivation at very negative potentials.

Site 3 and Site 4 VSD-binding toxins can exert unique effects on the NaChBac channel. (A) Simulated structure of two site 4 toxins, GsAF-I and ProTXII, and two site 3 toxins, BDS-I and ATXII (top). Representative current traces of NaChBac are shown before and after the toxin application (bottom). GsAF-I and BDS-I blocked Na+ conductance, while ProTXII and ATXII did not. Concentrations of the compounds are shown in Supplement Table I. (B) Effect of GsAF-I on NaV1.7 and NaChBac. GsAF-I (0.8 µM) inhibited NaV1.7 peak current by ~ 50%. GsAF-I (1.6 µM) inhibited NaChBac peak current by ~ 50%. Representative current traces (left), I–V (middle), G–V and SSI (right) relationships are shown before (black traces and symbols) and after GsAF-I application (red traces and symbols). (C) Representative current traces (left), I–V (middle), G–V and SSI (right) relationships are shown for NaV1.7 and NaChBac channels before (black traces and symbols), and after BDS-I (red traces and symbols). BDS-I blocked NaChBac, but augmented NaV1.7 current.
BDS-I is thought to bind to DIV-VSD of NaV1.7, the VSD that is essential for controlling fast inactivation27,28,29. BDS-I increased peak NaV1.7 channel current at test potentials < + 20 mV, slowed its rate of inactivation and induced a depolarizing shift in the SSI curve, but did not alter activation gating (Fig. 2C, top). In contrast, BDS-I decreased peak NaChBac channel currents at test potentials < + 20 mV and slowed the rate of current activation (Fig. 2C, bottom). In addition, BDS-I induced a similar positive shift in both the voltage-dependence of activation (Fig. 2C, bottom right, ΔV1/2 = 16.5 ± 3.8 mV, p = 0.001), and SSI (ΔV1/2 = 14.3 ± 2.4 mV, p = 0.006), perhaps reflecting the tight coupling between channel activation and inactivation. Thus, while GsAF-I and BDS-I both inhibit peak NaChBac currents, only BDS-I markedly alters the gating of these channels.
Lack of effects of isoform-specific compounds on NaChBac channels
Similar to the VSD-targeting toxins, isoform-specific compounds including G0766 (PF-771), G4936 (GX-936)30 and A80346731 have high specificities, as they do not bind to the common LA binding sites30,31. It’s not surprising that they showed no effects on the NaChBac channel (Supplement Fig. 1D and Supplement Table I), suggesting these compounds’ binding sites are not conserved in NaChBac.
Distinct modulations of NaV1.7 and NaChBac by pore-binding toxins
We tested several toxins that are known to bind to the pore domain of mammalian Na+ channels, including tetrodotoxin (TTX), u-conotoxins KIIIA, GIIIB, aconitine and veratridine, on the NaChBac channel. The site 1 toxins TTX and u-conotoxins KIIIA and GIIIB did not significantly alter currents of NaChBac, whereas the site 2 toxins aconitine and veratridine significantly blocked peak inward Na+ current of NaChBac (Fig. 3A and Supplement Fig. 1C). Aconitine and veratridine exhibit dual effects on the mammalian Na+ channels, modulating their gating to increase currents in addition to inhibiting peak Na+ conductance32,33. We assessed the effects of aconitine and veratridine on NaV1.7 and NaChBac to probe for conserved interaction mechanisms. The effect of aconitine (7 µM) on NaV1.7 current was highly voltage dependent. This can be appreciated in the current traces shown in Fig. 3B (top left), where the current elicited by a depolarizing pulse from − 100 to − 50 mV was increased, whereas the current induced by a pulse from − 100 to − 20 mV was decreased by aconitine (Fig. 3B, top left). The I–V plots of Fig. 3B (middle panel) illustrates the effects of aconitine on current magnitude across a wide range of test potentials, with enhancement observed for voltages below − 25 mV, robust inhibition at more depolarized potentials and a negative shift in the peak of the I-V relationship. Aconitine also shifted the G-V and SSI relationships for NaV1.7 channel currents (Fig. 3B, top). Intriguingly, inhibition of peak NaChBac channel currents was voltage-independent and was not accompanied by a shift in the I–V, G–V or SSI relationships (Fig. 3B, bottom). Aconitine is also a more potent inhibitor of NaChBac (IC50 = 1.3 µM) than of NaV1.7 (IC50 = 7.4 µM) channels (Supplemental Fig. 2. In summary, although aconitine inhibits both NaV1.7 and NaChBac, it only alters the gating of the NaV1.7 channels.

Pore binding toxins aconitine and veratridine differentially modulate NaV1.7 and NaChBac. (A) Structures of four toxins that are known to bind to the pore domain of eukaryotic NaV channels. Representative current traces are shown before and after toxin application. Aconitine and veratridine, which are site-2 toxins, showed robust block of the NaChBac channel. In contrast, site 1 toxins, tetrodotoxin and µ-conotoxin KIIIA exert minimal block on NaChBac. Concentrations of the compounds are shown in Supplement Table I. (B) Aconitine (7 µM and 2 µM) were used to achieve ~ 50% block of the NaV1.7 and NaChBac channels, respectively. To illustrate the voltage-dependent response to aconitine, two sets of representative current traces are shown for both NaV1.7 and NaChBac. The top set is elicited by depolarizing pulse from − 120 to − 50 mV. The bottom set shows the response to a larger depolarizing pulse from − 120 to − 20 mV. The I–V, G–V, and SSI relationships are presented for NaV1.7 (top) and NaChBac (bottom) before and after aconitine application. (C) Effect of 7 µM veratridine on NaChBac and NaV1.7. Representative current traces before and after veratridine are shown. Inset for NaV1.7 current highlights the effect of veratridine on sustained Na+ current. I–V, G–V, and SSI relationships are also presented to quantify peak inward currents and voltage dependence of steady-state gating.
Veratridine inhibited both NaV1.7 and NaChBac channels at the test concentration of 7 µM (Fig. 3C, left and middle panels), but differentially affects their gating. In contrast to aconitine, veratridine primarily interfered with the inactivation gating of NaV1.7, causing a leftward shift of the SSI relationship with almost no effect on the G–V relationship (Fig. 3C, top right). Although veratridine enhanced the voltage dependence of inactivation, it prevented channels from fully inactivating, manifested as an increase in the sustained inward Na+ current measured at the end of a test pulse (Fig. 3C, top left). Conversely, while veratridine accelerated NaChBac inactivation (Fig. 3C, bottom left), it did not alter the voltage dependence of SSI (Fig. 3C, bottom right). Furthermore, veratridine induced a depolarizing shift in the G–V relationship of NaChBac. In summary, although veratridine blocks peak Na+ current of both channel types, it differentially modulates their gating, specifically altering SSI of NaV1.7 and inhibiting activation of NaChBac.
F224 in the NaChBac channel is critical for its interaction with aconitine and veratridine
To gain a better understanding of the interaction mechanisms between NaChBac and aconitine or veratridine, we used site-directed mutagenesis to probe for the potential location of their binding sites. Based on the sequence alignment of inner pore residues of NaV1.4, NaV1.7, NaVMs, NaVAb and NaChBac, we focused on several key residues that were previously shown to be important for site-2 toxin binding in human NaV1.4 channel34,35 (Fig. 3A). We mutated the conserved residues in NaChBac. We tested four NaChBac mutant channels, each containing one of the following point mutations: F221A, F224A, N225K, or F227A. Among them, F221A and N225K mutant channels did not functionally express (Supplement Fig. 3). F224A exhibited a lower peak current, a much faster rate of inactivation (Fig. 4B, Supplement Fig. 3) and an altered G–V relationship (rightward shifted with reduced slope; Fig. 4D) compared to WT NaChBac. F227A exhibited a hyperpolarizing shift in the G–V curve compared to WT channels (Fig. 4D). Aconitine (7 µM) caused about 80% inhibition of WT channel currents but had no effect on F224A channel currents and a reduced effect on F227A channels (Fig. 4B, C). The voltage dependence of activation (G–V curves) was not significantly altered by aconitine for any of the three channels (Fig. 4D). Veratridine (7 µM) inhibited peak currents of WT channels by 55.6 ± 2.0%, F227A channels by 65.1 ± 1.7%, but caused minimal or no block of F224A channels (Fig. 4B, C). Veratridine did not alter the G–V relationship of F224A channels, but caused a larger depolarizing shift in the G–V curve of F227A channels (ΔV1/2 = 20.1 ± 2.3 mV, WT ΔV1/2 = 7.7 ± 1.8 mV, p = 0.004) (Fig. 4E), further suggesting an enhanced effect of veratridine in the presence of the F227A mutation. Although both aconitine and veratridine interact strongly with F224, they may possess different spatial orientations within the pore, resulting in distinct interactions with the neighboring residue F227. We also tested how the two NaChBac mutations affected LA binding by assessing lidocaine blockade (Fig. 4B, C). We observed a decreased but still robust block by lidocaine of F224A channels, and no change in block of F227A channels.

Identification of key interacting residues for aconitine and veratridine in NaChBac. (A) Sequence alignment of the S6 transmembrane segments of S6 of NaV1.4, NaV1.7 DIV, NaVMs, NaVAb, and NaChBac. The residues that were previously shown to be important for site-2 toxin batrachotoxin (BTX) in NaV1.435 are labeled in green. The location of F224 and F227 residues of NaChBac are noted. (B) Representative current traces for WT NaChBac (left), F224A NaChBac (middle) and F227A NaChBac (right). Current traces recorded before and after the application of aconitine, veratridine, and lidocaine are shown. (C) Summary of percentage of peak current block induced by aconitine, veratridine, and lidocaine for WT, F224A and F227A NaChBac channels. Each data point represents recordings from one cell. Bars indicate mean ± S.E.M. for all cells. (D) G–V relationships for WT, F224A and F227A NaChBac channels before and after application of aconitine. (E) G–V relationships for WT, F224A and F227A NaChBac channels before and after application of veratridine.
Methods and materials
Cell lines
pcDNA3.1 (−) Hygro vector containing NaChBac or mutant cDNA was transfected into HEK 293 cells with Lipofectamine 2000 (Thermo Fisher, Waltham, MA). The transfected cells were cultured with selection antibiotics for a week, then single colonies were isolated and used for expansion. 24 colonies were tested with SyncroPatch 768PE for NaChBac expression. The colony with the highest expression was further expanded and used for characterization in this study. CHO cells stably expressing human NaV1.7 channels were constructed as described previously36. Cell lines were cultured in high glucose DMEM (HEK 293) or Ham’s F12 for (CHO), supplemented with 10% FBS, 2 mM L-glutamine and antibiotics in 5% CO2 at 37 °C.
Electrophysiology recordings and compound applications
SyncroPatch 768PE (Nanion, Munich, Germany) was used to perform all automated patch clamp experiments. Chips with medium resistance (5-8MΩ) were used. The intracellular recording solution contained (in mM): 50 CsCl, 60 CsF, 10 NaCl, 20 EGTA and 10 HEPES (pH 7.2, osmolarity 285 mOsm), and extracellular recording solution contained (in mM): 80 NaCl, 60 NMDG, 4 KCl, 2 CaCl2, 1 MgCl2, 5 Glucose and 10 HEPES (pH 7.4, osmolarity 300 mOsm). Whole cell recordings were performed as previously described12,36. Series resistance compensation was set to 80%. Cell catching, sealing, whole-cell configuration formation, control solution application, recording, compound application and recording were performed sequentially. The holding membrane potential (Vm) for all experiments was set at − 120 mV unless otherwise noted. For current–voltage (IV) relationship recordings, NaV1.7 and NaChBac channels were elicited by depolarizing voltage steps from − 80 mV to + 60 mV (5 mV increments) for 50 ms and 500 ms, respectively. Steady-state inactivation (SSI) protocol were performed by preconditioning Vm from − 120 to + 20 mV (5 mV increments) for 500 ms (NaV1.7) or 5 s (NaChBac), then followed with stimulation pulse at − 10 mV for 20 ms (NaV1.7) and 200 ms (NaChBac). For compound tonic block testing, a pulse from − 120 to − 20 mV was applied every 2 s, 5 min before and 20 min after applying the compound. All compounds were purchased from Alomone Labs (Israel), except compounds G0766 and G4936 were produced in house at Genentech. Small molecule compounds and water-soluble toxins were reconstituted in DMSO and extracellular recording solution, respectively. For compounds that were reconstituted in DMSO, control solution containing the same amount of DMSO was used. For small peptide toxins, 0.1% BSA was added to recording solutions to prevent non-specific binding during perfusion.
Data analysis and statistics
Data were analyzed using Clampfit (v10; Molecular Devices), MATLAB (R2018b; MATLAB), and Excel (Microsoft). G–V and SSI curves were fitted to a Boltzmann function: y = 1/(1 + exp [(V − V1/2)/k]). Statistics for comparison of data recorded between before and after compound administration were performed using a paired Student t-test (Microsoft Excel). One-way ANOVA was used to compare drug block for different NaChBac mutant channels. Data are presented as mean ± SEM, from 5–20 cells.
Discussion
Bacterial and mammalian sodium channels are separated by hundreds of millions of years of evolution. High resolution structures of bacterial NaV channels have provided significant insights into the molecular basis of Na+ channel gating and ion selectivity. However, the molecular basis for the interactions between bacterial NaV channels with pharmacological agents has remained understudied. Here, we systematically tested 39 eukaryotic NaV channel-modulators and discovered intriguing similarities and differences in their interactions with NaV1.7 and NaChBac channels. Our study indicates that the divergence in pharmacology is determined by structural discrepancies, as well as by differences in gating dynamics between NaChBac and NaV1.7 channels.
Among a panel of 16 LA compounds known to bind to the pore of mammalian NaV channels, 11 compounds exhibited robust block of NaChBac, whereas 5 compounds exhibit minimal effects on NaChBac when applied extracellularly, including three highly hydrophobic compounds (carbamazepine, oxcarbazepine and Co102862). It was proposed that these compounds access their binding site by diffusion from the membrane lipid bilayer into the inner pore37. This proposed hydrophobic diffusion pathway has since been identified as lateral fenestrations in the NaVAb channel structure6,20. In supporting the relevance of this pathway to drug block, single residue mutations that decrease the size of fenestrations showed graded effects on resting-state block by the LAs flecainide, lidocaine, and benzocaine20. Compared to their robust effect on NaV1.7, the relatively bulky tricyclic compounds, such as Co102862 had minimal effect on NaChBac. It is possible that Co102862 preferentially binds to the inactivated state of NaV channel (not present in NaChBac)38. Alternatively, the NaChBac fenestrations are too small to allow access of this compound to its inner pore.
Given that VSD-targeting toxins exhibit high specificities on mammalian NaV channels, it is not unexpected that only 4 out of 15 toxins were able to modulate the NaChBac channel. From further assessment of GsAF-I and BDS-I, we observed that both toxins shifted the voltage dependence of inactivation of NaV1.7, but in opposite directions. In mammalian NaV channels, the conformation of VSD of Domain IV (DIV-VSD) tightly regulates channel inactivation27,39,40. Inhibition of DIV-VSD activation (rightward shift) usually hinders inactivation, while leftward shift in voltage-dependence of DIV-VSD activation often promotes inactivation14,41. Therefore, it is likely that both GsAF-I and BDS-I interact with the DIV-VSD in the NaV1.7 channel, but in a different manner. In contrast to four distinct VSDs of NaV1.7 that contribute uniquely to channel gating, NaChBac has four symmetrical VSDs that equally contributes to gating. In NaChBac, although both GsAF-I and BDS-I inhibit peak Na+ currents, they have drastically different effects on channel gating: BDS-I caused inhibited channel activation (rightward shift of G-V curve), while GsAF-I had no significant effect. This data suggest that GsAF-I and BDS-I may have different affinities for VSDs at distinct conformational states, a mechanism that was demonstrated in ProTx-II modulation of NaV1.712. Intriguingly, we observed that compounds that promote or stabilize the inactivated state in NaV1.7, including LAs, GsAF-I and aconitine, induced a small leftward shift or no effect on the G-V relationship of NaChBac. Conversely, compounds that inhibit inactivation in NaV1.7, such as BDS-I and veratridine, also induced significant rightward shifts in the G–V curves of NaChBac. These findings suggest a conservation between the DIV-VSD of the mammalian NaV channels and the VSD of NaChBac in the toxin binding mechanism. Further, as NaChBac has symmetrical and simple VSD to pore coupling, by observing VSD-toxins’ effect on NaChBac G–V, we can speculate on their interactions with the mammalian NaV channels’ DIV-VSDs.
Aconitine and veratridine are site-2 neurotoxins, defined by their activator activity resulting from binding to the intracellular pore of mammalian NaV channels33,34. We demonstrated that both aconitine and veratridine bind to the F224 residue on NaChBac. Aconitine blocks peak current without affecting voltage-dependence of channel activation, while veratridine blocks peak current, speeds up inactivation and shifts activation to the depolarized direction. Interestingly, it was reported recently that BTX also binds to the F224 residue on NaChBac42. However, it causes hyperpolarization of activation and prevents deactivation of the channel42. Therefore, although the putative binding sites for aconitine, veratridine and BTX may overlap to some extent, their interaction with the inner pore results in distinct gating effects on NaChBac. Small molecules, such as lidocaine and PI1, bind to the Threonine residue (T220 in NaChBac) adjacent to the lipid-facing fenestration, as resolved in the NaVab and NaVMs structures20,43. Comparing to the small molecule binding sites, our findings suggest that aconitine and veratridine seem to occupy a lower position in the cavity, underlying their distinct modulation of NaChBac gating properties compared to small molecules. Notably, despite the four-fold symmetry of the NaChBac channel, due to the steric hindrance, pore-modulating compounds are likely to only bind to one subunit20,43, resulting in an asymmetric drug conformation in the pore, which further explains why site-2 toxins exert differential gating effects although sharing a common binding site.
Our current study also provides insights into organism-specific drug discovery. Bacterial ion channels are fundamental to the survival and function of bacteria, in terms of maintaining ion and pH homeostasis, controlling cell mobility, and cellular communications44,45,46. As a result, bacterial ion channels have emerged as new target for developing new antibiotics to combat the problem of multi-drug resistance (MDR)47. Here we identified a broad selection of compounds that modulate NaChBac function. Particularly, compounds such as aconitine exhibit higher potency on NaChBac compared to NaV1.7. These compounds and their related mechanisms can potentially be further explored for developing new antibiotics targeting the bacterial NaV channels.
References
Hille, B. Ion Channel Excitable Membranes 1–37 (Sunderland, Massachusetts, 2001).
Payandeh, J. & Minor, D. L. Bacterial voltage-gated sodium channels (BacNaVs) from the soil, sea, and salt lakes enlighten molecular mechanisms of electrical signaling and pharmacology in the brain and heart. J. Mol. Biol. https://doi.org/10.1016/j.jmb.2014.08.010 (2015).
Liebeskind, B. J., Hillis, D. M. & Zakon, H. H. Evolution of sodium channels predates the origin of nervous systems in animals. Proc. Natl. Acad. Sci. USA https://doi.org/10.1073/pnas.1106363108 (2011).
Echt, D. S. et al. Mortality and morbidity in patients receiving encainide, flecainide, or placebo. N. Engl. J. Med. 324, 781–788 (1991).
McCusker, E. C. et al. Structure of a bacterial voltage-gated sodium channel pore reveals mechanisms of opening and closing. Nat. Commun. https://doi.org/10.1038/ncomms2077 (2012).
Payandeh, J., Scheuer, T., Zheng, N. & Catterall, W. A. The crystal structure of a voltage-gated sodium channel. Nature 475, 353–358 (2011).
Zhang, X. et al. Crystal structure of an orthologue of the NaChBac voltage-gated sodium channel. Nature 486, 130–134 (2012).
Shen, H. et al. Structure of a eukaryotic voltage-gated sodium channel at near-atomic resolution. Science 355, 4369 (2017).
Pan, X. et al. Structure of the human voltage-gated sodium channel Nav1.4 in complex with β1. Science 362, eaau2486 (2018).
Shen, H., Liu, D., Wu, K., Lei, J. & Yan, N. Structures of human Nav17 channel in complex with auxiliary subunits and animal toxins. Science 363, 1303–1308 (2019).
Jiang, D. et al. Structure of the cardiac sodium channel. Cell https://doi.org/10.1016/j.cell.2019.11.041 (2020).
Xu, H. et al. Structural basis of Nav1.7 inhibition by a gating-modifier spider toxin. Cell 176, 702.e14-715.e14 (2019).
Clairfeuille, T. et al. Structural basis of a-scorpion toxin action on Na v channels. Science https://doi.org/10.1126/science.aav8573 (2019).
Zhu, W., Varga, Z. & Silva, J. R. Molecular motions that shape the cardiac action potential: insights from voltage clamp fluorometry. Prog. Biophys. Mol. Biol. https://doi.org/10.1016/j.pbiomolbio.2015.12.003 (2016).
Zhu, W. et al. Predicting patient response to the antiarrhythmic mexiletine based on genetic variation: personalized medicine for long QT syndrome. Circ. Res. https://doi.org/10.1161/CIRCRESAHA.118.314050 (2019).
Gellens, M. E. et al. Primary structure and functional expression of the human cardiac tetrodotoxin-insensitive voltage-dependent sodium channel. Proc. Natl. Acad. Sci. USA 89, 554–558 (1992).
Payandeh, J., Gamal El-Din, T. M., Scheuer, T., Zheng, N. & Catterall, W. A. Crystal structure of a voltage-gated sodium channel in two potentially inactivated states. Nature https://doi.org/10.1038/nature11077 (2012).
Chakrapani, S., Sompornpisut, P., Intharathep, P., Roux, B. & Perozo, E. The activated state of a sodium channel voltage sensor in a membrane environment. Proc. Natl. Acad. Sci. USA 107, 5435–5440 (2010).
Sula, A. et al. The complete structure of an activated open sodium channel. Nat. Commun. https://doi.org/10.1038/ncomms14205 (2017).
Gamal El-Din, T. M., Lenaeus, M. J., Zheng, N. & Catterall, W. A. Fenestrations control resting-state block of a voltage-gated sodium channel. Proc. Natl. Acad. Sci. USA 115, 13111–13116 (2018).
Lee, S., Goodchild, S. J. & Ahern, C. A. Local anesthetic inhibition of a bacterial sodium channel. J. Gen. Physiol. 139, 507–516 (2012).
Ragsdale, D. S., McPhee, J. C., Scheuer, T. & Catterall, W. A. Molecular determinants of state-dependent block of Na+ channels by local anesthetics. Science 265, 1724–1728 (1994).
Burashnikov, A., Di Diego, J. M., Zygmunt, A. C., Belardinelli, L. & Antzelevitch, C. Atrium-selective sodium channel block as a strategy for suppression of atrial fibrillation: differences in sodium channel inactivation between atria and ventricles and the role of ranolazine. Circulation 116, 1449–1457 (2007).
Vedantham, V. & Cannon, S. C. The position of the fast-inactivation gate during lidocaine block of voltage-gated Na+ channels. J. Gen. Physiol. 113, 7–16 (1999).
Gilchrist, J., Olivera, B. M. & Bosmans, F. Animal toxins influence voltage-gated sodium channel function. Handb. Exp. Pharmacol. https://doi.org/10.1007/978-3-642-41588-3_10 (2014).
Liu, P., Jo, S. & Bean, B. P. Modulation of neuronal sodium channels by the sea anemone peptide BDS-I. J. Neurophysiol. https://doi.org/10.1152/jn.00785.2011 (2012).
Capes, D. L., Goldschen-Ohm, M. P., Arcisio-Miranda, M., Bezanilla, F. & Chanda, B. Domain IV voltage-sensor movement is both sufficient and rate limiting for fast inactivation in sodium channels. J. Gen. Physiol. 142, 101–112 (2013).
Mangold, K. E. et al. Mechanisms and models of cardiac sodium channel inactivation. Channels 11, 517–533 (2017).
Hsu, E. J. et al. Regulation of Na(+) channel inactivation by the DIII and DIV voltage-sensing domains. J. Gen. Physiol. 149, 389–403 (2017).
Chernov-Rogan, T. et al. Mechanism-specific assay design facilitates the discovery of Nav17-selective inhibitors. Proc. Natl. Acad. Sci. USA https://doi.org/10.1073/pnas.1713701115 (2018).
Browne, L. E., Blaney, F. E., Yusaf, S. P., Clare, J. J. & Wray, D. Structural determinants of drugs acting on the Nav 1.8 channel. J. Biol. Chem. https://doi.org/10.1074/jbc.M807569200 (2009).
Rao, S. & Sikdar, S. K. Modification of α subunit of RIIA sodium channels by aconitine. Pflugers Arch. Eur. J. Physiol. 439, 349–355 (2000).
Catterall, W. A. et al. Voltage-gated ion channels and gating modifier toxins. Toxicon 49, 124–141 (2007).
Stevens, M., Peigneur, S. & Tytgat, J. Neurotoxins and their binding areas on voltage-gated sodium channels. Front. Pharmacol. https://doi.org/10.3389/fphar.2011.00071 (2011).
Wang, S. Y. & Wang, G. K. Batrachotoxin-resistant Na+ channels derived from point mutations in transmembrane segment D4–S6. Biophys. J. https://doi.org/10.1016/S0006-3495(99)77465-5 (1999).
Li, T. et al. High-throughput electrophysiological assays for voltage gated ion channels using SyncroPatch 768PE. PLoS ONE 12, 1–18 (2017).
Hille, B. Local anesthetics: hydrophilic and hydrophobic pathways for the drug-receptor reaction. J. Gen. Physiol. 69, 497–515 (1977).
Ilyin, V. I. et al. V102862 (Co 102862): a potent, broad-spectrum state-dependent blocker of mammalian voltage-gated sodium channels. Br. J. Pharmacol. https://doi.org/10.1038/sj.bjp.0706058 (2005).
Varga, Z. et al. Direct measurement of cardiac Na + channel conformations reveals molecular pathologies of inherited mutations. Circ. Arrhythmia Electrophysiol. https://doi.org/10.1161/CIRCEP.115.003155 (2015).
Zhu, W. et al. Mechanisms of noncovalent β subunit regulation of Na V channel gating. J. Gen. Physiol. 149, 813–831 (2017).
Peters, C. H., Yu, A., Zhu, W., Silva, J. R. & Ruben, P. C. Depolarization of the conductance-voltage relationship in the NaV1.5 mutant, E1784K, is due to altered fast inactivation. PLoS ONE https://doi.org/10.1371/journal.pone.0184605 (2017).
Finol-Urdaneta, R. K. et al. Batrachotoxin acts as a stent to hold open homotetrameric prokaryotic voltage-gated sodium channels. J. Gen. Physiol. 151, 186–199 (2019).
Bagnéris, C. et al. Prokaryotic NavMs channel as a structural and functional model for eukaryotic sodium channel antagonism. Proc. Natl. Acad. Sci. USA 111, 8428–8433 (2014).
Koishi, R. et al. A superfamily of voltage-gated sodium channels in bacteria. J. Biol. Chem. https://doi.org/10.1074/jbc.M313100200 (2004).
Prindle, A. et al. Ion channels enable electrical communication in bacterial communities. Nature https://doi.org/10.1038/nature15709 (2015).
Krulwich, T. A., Ito, M. & Guffanti, A. A. The Na+-dependence of alkaliphily in Bacillus. Biochim. Biophys. Acta https://doi.org/10.1016/S0005-2728(00)00285-1 (2001).
Iscla, I. et al. A new antibiotic with potent activity targets MscL. J. Antibiot. https://doi.org/10.1038/ja.2015.4 (2015).
Author information
Authors and Affiliations
Contributions
W.Z. contributed to experimental design, data collection, analysis, manuscript writing and figure preparation. J.C. contributed to experimental design and manuscript writing. T.L. and J.S. reviewed and edited the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhu, W., Li, T., Silva, J.R. et al. Conservation and divergence in NaChBac and NaV1.7 pharmacology reveals novel drug interaction mechanisms. Sci Rep 10, 10730 (2020). https://doi.org/10.1038/s41598-020-67761-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-67761-5
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
