
Abstract
Auditory neuropathy is an important entity in childhood sensorineural hearing loss. Due to diverse etiologies and clinical features, the management is often challenging. This study used an integrative patient-history, audiologic, genetic, and imaging-based approach to investigate the etiologies and audiologic features of 101 children with auditory neuropathy. Etiologically, 48 (47.5%), 16 (15.8%), 11 (10.9%), and 26 (25.7%) children were categorized as having acquired, genetic, cochlear nerve deficiency-related, and indefinite auditory neuropathy, respectively. The most common causes of acquired and genetic auditory neuropathy were prematurity and OTOF mutations, respectively. Patients with acquired auditory neuropathy presented hearing loss earlier (odds ratio, 10.2; 95% confidence interval, 2.2–47.4), whereas patients with genetic auditory neuropathy had higher presence rate of distortion product otoacoustic emissions (odds ratio, 10.7; 95% confidence interval, 1.3–85.4). In patients with different etiologies or pathological sites, moderate to strong correlations (Pearson’s r = 0.51–0.83) were observed between behavioral thresholds and auditory steady-state response thresholds. In conclusion, comprehensive assessments can provide etiological clues in ~75% of the children with auditory neuropathy. Different etiologies are associated with different audiologic features, and auditory steady-state responses might serve as an objective measure for estimating behavioral thresholds.
Similar content being viewed by others

Introduction
Auditory neuropathy is a challenging clinical disorder accounting for ~10% of cases of sensorineural hearing loss (SNHL) in children1,2,3. The pathogenesis of auditory neuropathy encompasses a wide range of disease mechanisms, with pathologies localized to multiple sites along the auditory pathway, including inner hair cells4, synapses5, spiral ganglion neurons6, auditory nerve7, or brainstem auditory nuclei8. Many acquired and genetic factors can contribute to auditory neuropathy1,2,9,10. Acquired auditory neuropathy include infection during pregnancy, prematurity, kernicterus, and perinatal hypoxia1,9. Auditory neuropathy may also be associated with certain types of syndromic or non-syndromic hereditary hearing impairment9.
Genetic factors may contribute to approximately 40% of auditory neuropathy patients in whom clinical manifestations might present as components of specific syndromes or as isolated non-syndromic disorders9. A number of hereditary neurodegenerative syndromes have been associated with auditory neuropathy, including Charcot-Marie-Tooth disease, Friedreich’s ataxia, Leber’s hereditary optic neuropathy, autosomal dominant optic atrophy, autosomal recessive optic atrophy, Mohr-Tranebjaerg syndrome, Refsum disease, Wolfram syndrome, Pelizaeus-Merzbacher disease, and CAPOS syndrome9,11,12,13,14. Genetic causes of non-syndromic auditory neuropathy include autosomal dominant mutations in DIAPH3 and PCDH9; autosomal recessive mutations in OTOF, PJVK, and GJB2; and mitochondrial mutations in MT-RNR19,11.
Audiologically, auditory neuropathy is characterized by mild to profound SNHL in the presence of otoacoustic emissions (OAEs) and/or cochlear microphonics (CMs), but absent or abnormal auditory brainstem responses (ABRs) and absent acoustic reflexes2,9,10. Given the heterogeneous etiologies and diverse audiologic features, benefits derived from hearing aids or cochlear implants (CIs) vary significantly among patients2,10,15,16. This poses difficulties in the management of auditory neuropathy. Recent advances in molecular genetics and imaging technologies have revolutionized diagnosis, counseling for, and treatment of childhood SNHL by precisely defining the etiology and pathology17,18. This is especially relevant for pediatric auditory neuropathy, as treatment outcomes correlate closely with the underlying etiologies19. To explore practical factors important for the guidance of assessment and management, we revisited etiologies and hearing profiles in a large auditory neuropathy cohort using an integrative audiologic, genetic, and imaging approach.
Results
Determination of etiologies
One-hundred-and-one children with auditory neuropathy, including 57 (56.4%) boys and 44 (43.6%) girls, were enrolled in this study. Among them, forty-eight patients (47.5%) were considered to have acquired auditory neuropathy, including 20 with prematurity, 5 with kernicterus, 5 with perinatal hypoxia, and 18 with two or more pre-/perinatal events (Table 1). All patients underwent genetic testing, and 16 (15.8%) patients were considered to have genetic auditory neuropathy, including 13 non-syndromic patients (12 with bi-allelic OTOF mutations and 1 with a homoplasmic mitochondrial m.1555 A > G mutation), 1 with Wolfram syndrome (WFS1 p.A684V mutation), 1 with autosomal dominant optic atrophy (OPA1 p.C472R mutation), and 1 with Pelizaeus-Merzbacher disease (PLP1 duplication). Among the 12 patients with bi-allelic OTOF mutations, 4 were homozygotes for OTOF p.E1700Q mutations and 8 were compound heterozygotes (2 had p.E841K/p.E1700Q mutations, 1 had p.D1235fs/p.E1700Q mutations, 1 had p.R1344X/p.E1700Q mutations, 2 had p.E1700Q/p.R1735W mutations, 1 had p.E1700Q/p.H1779Y mutations, and 1 had p.E1700Q/p.R1856W mutations). Eleven (10.9%) patients were found to have cochlear nerve deficiency (CND) based on the imaging studies. In 26 (25.7%) of the 101 patients, the etiology of auditory neuropathy was indefinite.
Forty-six (95.8%) patients with acquired auditory neuropathy had hearing loss at birth, which was a higher percentage of patients than those in the other three groups (Table 2, p = 0.001; acquired vs. others: odds ratio, 10.2; 95% confidence interval, 2.2–47.4). Notably, 5 of the 26 patients in the indefinite group reported a family history of childhood-onset SNHL, indicating that auditory neuropathy in these patients might also be related to genetic factors.
Auditory neuropathy can be classified as presynaptic (i.e., lesions at inner hair cells or ribbon synapses) or postsynaptic (i.e., lesions central to the ribbon synapse in the auditory pathway) based on the site of pathology19. Reported causes of presynaptic auditory neuropathy include perinatal hypoxia, OTOF mutations, and mitochondrial mutations, while reported causes of postsynaptic auditory neuropathy include prematurity, kernicterus, autosomal dominant optic atrophy, Pelizaeus-Merzbacher disease, and CND14,19. In 75 patients with identifiable etiologies and risk factors, 19 could be considered to have presynaptic auditory neuropathy, and 39 have postsynaptic auditory neuropathy. The sites of pathology differed significantly among the four groups (p < 0.001), with genetic auditory neuropathy most likely to be presynaptic (81.3%) and CND-related auditory neuropathy exclusively associated with postsynaptic pathologies (100%).
Audiologic features
Distortion product otoacoustic emissions (DPOAEs) were present in 61 of the 101 patients (60.4%) at the time of diagnosis of auditory neuropathy (Table 3). These patients included 27 (56.3%), 14 (87.5%), 6 (54.5%), and 14 (53.8%) individuals with acquired, genetic, CND-related, and indefinite auditory neuropathy, respectively. The presence rate of DPOAEs was significantly higher in patients with genetic auditory neuropathy than in those in the other three groups (genetic vs. others: odds ratio, 10.7; 95% confidence interval, 1.3–85.4). The follow-up period of DPOAEs was 2.0 ± 2.0 years on average, and there was no significant difference among the four groups (p = 0.16). During the follow-up period, DPOAEs disappeared in 15 (55.6%), 5 (35.7%), 5 (83.3%) and 2 (14.3%) patients in the acquired, genetic, CND-related, and indefinite groups, respectively. On the other hand, CMs were identified in 95 of the 101 patients (94.1%) at the time of the first visit. These patients included 48 (100%), 13 (81.3%), 10 (90.9%), and 24 (92.3%) individuals with acquired, genetic, CND-related, and indefinite auditory neuropathy, respectively, showing no significant differences among the four groups (p = 0.059). In the 6 patients who did not have CMs (n = 4) or in whom they were unavailable (n = 2), the presence of DPOAEs enabled the diagnosis of auditory neuropathy.
There were no significant differences in the hearing thresholds, hearing loss patterns, or audiogram configurations among the four groups. Nineteen patients underwent cochlear implantation, including 11 patients with genetic causes (9 patients with OTOF mutations, 1 with Wolfram syndrome, and 1 with autosomal dominant optic atrophy), 2 patients with CND, and 6 patients with indefinite auditory neuropathy. The post-operative auditory and speech performance in the two CND patients were suboptimal, with Categories of Auditory Performance (CAP) score of 5 and Speech Intelligibility Rating (SIR) score of 1 in both patients, despite more than 3 years of rehabilitation. In contrast, all the other 17 patients without CND showed favorable outcomes with CI, with CAP scores of up to 6–7 and SIR scores of up to 3–5 within 3 years post-operatively.
Imaging findings
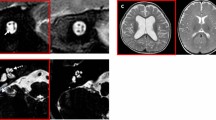
The imaging findings are summarized in Table 4. CND was identified in 11 patients, including 8 with cochlear nerve aplasia (Fig. 1a) and 3 with cochlear nerve hypoplasia (Fig. 1b). Inner ear anomaly was identified in 1 patient with acquired auditory neuropathy (prematurity-related), who had dysplastic changes in the vestibule and semicircular canals. Abnormalities of the central nervous system were identified in 10 patients with acquired neuropathy, including 1 with cerebral hypomyelination (Fig. 1c), 1 with diffuse cerebral parenchymal loss (Fig. 1d), 5 with thin corpus callosum, and 3 with both cerebral hypomyelination and thin corpus callosum. Moreover, abnormalities of the central nervous system were identified in 1 patient with genetic auditory neuropathy (Pelizaeus-Merzbacher disease, cerebral hypomyelination), in 1 patient with CND (cerebral hypomyelination and thin corpus callosum), and in 1 patient with indefinite auditory neuropathy (cerebral hypomyelination).

Representative imaging findings on posterior fossa magnetic resonance imaging T2-weighted images. (a) Sagittal view showing cochlear nerve aplasia. The arrow indicates the absence of the cochlear nerve. (b) Sagittal view showing cochlear nerve hypoplasia. The cochlear nerve (arrow) is thinner than the facial nerve (arrow head). (c) Axial view showing hypomyelination of the frontoparietal central parenchyma. (d) Axial view showing diffuse parenchymal loss.
Correlations of Behavioral Thresholds and Auditory Steady-State Response (ASSR) Thresholds
To objectively estimate hearing levels in the patients with auditory neuropathy, we compared the data obtained from different audiologic tests. We calculated the correlations between the thresholds of behavioral audiograms (i.e. visual reinforcement audiometry, conditioned play audiometry, and pure tone audiometry) and ASSR measured at the same testing time. The data were available to be analyzed in a total of 51 patients. These patients were tested at the age of 6 months to 7 years old, with 48 of them (94%) tested at younger than 3 years of age. Pearson correlation analyses revealed a moderate positive correlation between the behavioral and ASSR thresholds (r = 0.61, p < 0.001; Fig. 2a). The slope of the regression line between the behavioral and ASSR thresholds (0.73), however, did not approach 1, indicating that correction is required to estimate behavioral thresholds based on ASSR thresholds.

Correlations between behavioral thresholds and auditory steady-state response (ASSR) thresholds. (a) The correlation between behavioral and ASSR thresholds in all patients. (b,c) The correlations between behavioral and ASSR thresholds in patients with acquired and genetic auditory neuropathy, respectively, and representative behavioral and ASSR audiograms for the same patients from each group. (d,e) The correlations between behavioral and ASSR thresholds among prematurity-related auditory neuropathy patients and OTOF-related auditory neuropathy patients, respectively. (f,g) The correlations between behavioral and ASSR thresholds in patients with presynaptic and postsynaptic auditory neuropathy, respectively.
When classified according to the etiology, there was a moderate correlation between the behavioral and ASSR thresholds (r = 0.51, p = 0.01, slope = 0.76; Fig. 2b) in patients with acquired auditory neuropathy, whereas there was a strong correlation between the behavioral and ASSR thresholds (r = 0.82, p = 0.002, slope = 0.76; Fig. 2c) in patients with genetic auditory neuropathy. Specifically, there was a moderate correlation between the behavioral and ASSR thresholds (r = 0.66, p = 0.01, slope = 0.93; Fig. 2d) in patients with prematurity-related auditory neuropathy, and strong correlation between the behavioral and ASSR thresholds (r = 0.83, p = 0.012, slope = 0.44; Fig. 2e) in patients with OTOF mutations.
Similarly, when classified according to the site of pathology, we also observed moderate correlations between behavioral and ASSR thresholds both in patients with presynaptic auditory neuropathy (r = 0.67, p = 0.012, slope = 0.69; Fig. 2f) and those with postsynaptic auditory neuropathy (r = 0.74, p < 0.001, slope = 1.02; Fig. 2g).
Discussion
Despite the etiological heterogeneity, we were able to obtain information regarding the cause of pediatric auditory neuropathy in ~75% (75/101) of the patients using comprehensive history-taking, genetic examinations, and imaging studies. The major factors related to auditory neuropathy were acquired factors (i.e., pre-/perinatal events), followed by genetic and developmental (e.g., CND) factors. Prematurity, either isolated or in combination with other risk factors, was the most commonly identified pre-/perinatal event contributing to acquired auditory neuropathy in this study. Selective loss of inner hair cells and relative preservation of outer hair cells have been reported as causes of auditory neuropathy in premature infants4,20. In addition, prematurity is associated with several comorbid factors including hyperbilirubinemia and hypoxia, which might also contribute to auditory neuropathy2,21.
Secondary to the acquired factors, genetic etiologies constituted another important common cause of auditory neuropathy in our cohort. Recessive OTOF mutations were the predominant genetic cause among patients with non-syndromic auditory neuropathy in this study. Excluding the 48, 3 and 11 patients who were confirmed to have acquired, syndromic auditory neuropathy, and CND-related auditory neuropathy, respectively, recessive OTOF mutations accounted for 12 (30.8%) of the remaining 39 patients with non-syndromic non-acquired auditory neuropathy. This prevalence is similar to those reported in previous studies in other populations, wherein OTOF mutations were detected in approximately 30–50% of patients with non-syndromic auditory neuropathy22,23,24,25,26,27,28.
CND was detected using imaging studies in 11 of the patients without identified acquired or genetic factors. CND may be unilateral or bilateral and may occur in children with physiologically normal cochleae29,30. As a well-documented imaging abnormality associated with auditory neuropathy and poor CI outcomes, the pathogenesis of CND in humans remains largely unclear. CND may result from the nerve either partially (hypoplasia) or completely (aplasia or agenesis) development failure or as a consequence of post-developmental degeneration29. Animal studies indicate that the expression of certain neurotrophic factors such as brain-derived neurotrophic factor and neurotrophin-3 by the otocyst is required for early cell migration, neurite outgrowth, and survival of vestibular and cochlear ganglion cells31,32. Absence of these factors might lead to the loss of ganglion cells and agenesis of the cochlear nerve31,32.
While clarifying the pathology of the disease is crucial for designing the treatment plan, some patients may have more than one pathological site. For instance, certain disease entities, such as Wolfram syndrome, could involve both presynaptic and postsynaptic pathologies33. In addition, some patients could have more than one risk factors for auditory neuropathy at birth, such as having both kernicterus and perinatal hypoxia, which might also result in presynaptic and postsynaptic lesions simultaneously. Accordingly, among the 75 patients with identifiable etiologies and/or risk factors, the pathological sites could not be ascertained in 17 patients, as presynaptic and postsynaptic lesions might coexist. More sophisticated investigations, such as electrocochleography, are needed to delineate the actual pathological sites of auditory neuropathy in these patients.
Our results indicate that patients with auditory neuropathy with distinct etiologies exhibited different hearing profiles. For instance, more than 95% of patients with acquired auditory neuropathy presented with symptoms at birth immediately after pre-/perinatal insults, whereas about 30–40% of patients with genetic and indefinite causes developed auditory neuropathy later in life. Late-onset progressive auditory neuropathy has been reported in patients with OTOF or OPA1 mutations34,35, this is consistent with our observation. In addition, the presence rate of DPOAEs was higher in patients with genetic auditory neuropathy than in those in the other 3 groups. This might be attributed to the high percentage of patients with OTOF mutations in the genetic group. As the pathology of OTOF mutations is confined to the ribbon synapses of inner hair cells, the high presence rate of DPOAEs might reflect the relatively intact function of outer hair cells.
On the other hand, most of the patients presented with CMs, which was consistent among the four groups. In contrast to OAEs, which might disappear with time in patients with auditory neuropathy36,37, CMs usually remain stable with time38. Absent OAEs have been reported in some patients with auditory neuropathy despite the presence of CMs1,39. It has been postulated that in such patients, CMs are generated in both outer and inner hair cells, and that the outer hair cells are able to produce CMs, but are sufficiently impaired so as to fail in the generation of OAEs1.
Patients with auditory neuropathy may exhibit a range of hearing thresholds on subjective audiologic tests, ranging from normal to profound hearing loss, and hearing levels may change at different measurements1,2,19. Moreover, objective audiologic tests commonly used to estimate behavioral thresholds, such as the ABR and acoustic reflex, are of limited utility in children with auditory neuropathy due to the absence of responses in such patients. It is thus difficult to determine the appropriate amplification level when using hearing aids. In this study, we investigated the usefulness of ASSR to estimate the hearing levels in auditory neuropathy patients. Although ASSR has been confirmed as a reliable tool for the estimation of behavioral thresholds in cochlear SNHL40,41, previous studies on its utility in patients with auditory neuropathy have had inconsistent results. Some studies failed to find significant correlations between behavioral and ASSR thresholds41,42, whereas others have suggested that ASSR might serve as a valuable objective measure of behavioral thresholds in children with auditory neuropathy (e.g., Attias, et al.43). Our correlation analyses in a relatively large cohort of patients with pediatric auditory neuropathy revealed moderate to strong correlations (r = 0.51–0.83) between behavioral and ASSR thresholds in patients with different etiologies or sites of pathology. As such, in the absence of other reliable objective audiologic tests, the results of ASSR might offer some insights into hearing acuity in children with auditory neuropathy.
In addition to achieving a high diagnostic yield rate in the etiologic work-up, the integration of genetic and imaging studies into clinical assessments also had advantages for decision-making in the management of auditory neuropathy. The anatomic location of the pathology has been identified as a variable predictive of CI performance44,45, and both genetic and imaging studies are useful in pinpointing the site of pathology in auditory neuropathy. For instance, previous studies reported that patients with auditory neuropathy with OTOF mutations usually had excellent CI outcomes, likely because the pathology is confined to the synapse, and postsynaptic neurons and nerve fibers are intact for electrical stimulation of CI46,47,48. Cochlear implantation should thus be performed in patients with auditory neuropathy with OTOF mutations whenever indicated without unnecessary delay to achieve better CI outcome49. On the other hand, the presence of CND on imaging studies is indicative of unfavorable CI performance50. Although children with CND who have small nerves may benefit from cochlear implantation or amplification, such interventions are contraindicated in children with completely absent cochlear nerves51.
The major strength of this study lies in the comprehensive etiologic and audiologic analyses carried out in a large cohort of patients with pediatric auditory neuropathy. However, some limitations merit discussion. First, biases inherent in the retrospective nature of the study could have influenced our results. For instance, we only have limited data in our patients regarding the newborn hearing screening (NHS) results, speech perception, and language development. Many of our patients were diagnosed with auditory neuropathy before the implementation of the universal newborn hearing screening program by the Taiwan government in 2012. Patients who did not receive NHS and those who received NHS using OAEs might have delayed diagnosis of auditory neuropathy. Similarly, given the relatively young age of our patients and the compromised speech perception associated with auditory neuropathy, we were unable to obtain sufficient data on speech perception and language development for further analyses. Second, because our analyses focused only on children with auditory neuropathy, our findings cannot be generalized to adult patients with auditory neuropathy. Finally, although our results demonstrated some utility of ASSR in estimating the hearing level in pediatric patients with auditory neuropathy, the estimation was not optimal, as only moderate to strong correlations were present between behavioral and ASSR thresholds. Exploration and validation of other objective audiologic tests are thus warranted. Cortical auditory evoked potentials may be a potential candidate, as young patients with auditory neuropathy generally have recordable cortical potentials despite absent or abnormal brainstem responses52,53.
In conclusion, comprehensive history-taking, genetic examinations, and imaging studies can provide etiological clues in approximately 75% of children with auditory neuropathy. Different etiologies are associated with different audiologic features, likely due to different anatomic sites of the pathologies involved. In the absence of other reliable objective audiologic tests, ASSR might serve as an objective measure for the estimation of behavioral thresholds.
Materials and Methods
Patient recruitment
From 1997–2018, children diagnosed with bilateral auditory neuropathy at a tertiary university-affiliated medical center were included for the study. Diagnosis of auditory neuropathy was based on repeated findings of absent or abnormal ABR waveforms, and the presence of OAEs or CMs. Basic demographic data, family history, maternal history, birth history, and past medical history were ascertained for each patient. Patients older than 18 years, those with conductive or mixed-type hearing impairment, those with unilateral hearing impairment, and those without complete history records were excluded. This study was approved by the Research Ethics Committee of the National Taiwan University Hospital. Written informed consent was obtained from all patients and/or their parents. All methods were performed in accordance with the relevant guidelines and regulations.
Audiologic tests
Each patient underwent a battery of audiologic assessments appropriate for his/her age and neurological status, including DPOAEs, ABR, ASSR, and behavioral audiometries. The assessments were performed at least once every 6 months during the follow-up periods. DPOAEs were analyzed by using Titan – DPOAE440 (Interacoustics, Middelfart, Denmark). The f2/f1 ratio was 1.22, and the intensity ratio was set at 65/55 dBSPL. The frequency settings were 1, 1.5, 2, 3, 4, and 6 kHz. For each frequency, a signal-to-noise ratio of> 6 dB was determined as a positive DPOAEs response, and at least five frequencies with a positive DPOAEs response were considered to have presence of OAEs. The CMs were identified using ABR clicks at 95 dBnHL by comparing different types of stimulus polarities (rarefaction, condensation, and alternating). Additional control tests with the tubing clamped under same stimulus were applied to confirm the presence of artifacts. ASSR was performed with Bio-logic Multiple Auditory Steady-state Response (MASTER) II software system (Natus Medical Inc., IL, USA) running on a Navigator PRO hardware platform (Natus Medical Inc., IL, USA). The patients were either naturally asleep or sedated with chloral hydrate. Each ear was tested for four frequencies, including 0.5, 1, 2, and 4 kHz, simultaneously, and the modulation frequencies were set between 82–99 Hz with different carrier frequencies at both sides. Behavioral audiometries were performed using visual reinforcement audiometry, conditioned play audiometry, or pure tone audiometry, depending on the developmental level of the patient.
The audiologic data included hearing levels, onset of hearing loss, hearing loss patterns, and configurations of audiograms54,55. Hearing levels were determined by the four-frequency average thresholds (0.5, 1, 2, and 4 kHz). Hearing loss onset was categorized as at birth (i.e., those who failed newborn hearing screening), younger than 10 years, and 10 years or older. At least three consecutive audiologic tests during at a follow-up period were ascertained to determine the hearing loss patterns, which were categorized as stable, fluctuating, progressive, improving, or other. The audiogram configurations were classified as flat, sloping, or other54,55. The CAP score and SIR scale were evaluated in patients who received cochlear implantation, before and after the operation, by a questionnaire provided to the parents during follow-up to evaluate the outcomes of hearing performance46,56.
Genetic examinations
Genetic counseling was offered to all patients and/or their parents at the clinic. All subjects with informed consent for genetic testing had Sanger sequencing performed for four common deafness genes: GJB2 (Gene ID: 2706), SLC26A4 (Gene ID: 5172), OTOF (Gene ID: 9831), and the mitochondrial 12S rRNA gene (MTRNR1, Gene ID: 4549)34,57. Patients with bi-allelic GJB2, SLC26A4 or OTOF mutations, and those with homoplasmic or heteroplasmic mitochondrial mutations were considered to have a definite genetic diagnosis. Patients in whom we were unable to identify common mutations underwent comprehensive genetic examinations using a next-generation sequencing panel targeting 159 known deafness genes58,59,60.
Imaging studies
Non-contrast brain magnetic resonance imaging (MRI) with a resolution of 0.5-mm thickness was performed to investigate the central auditory pathway and cochlear nerve. Temporal bone high-resolution computed tomography (HRCT) with contiguous axial and coronal sections of 0.6-mm thickness was performed to investigate the structure of the inner ear. All images were reviewed according to previously published criteria61,62,63,64. CND was defined as a cochlear nerve diameter smaller than that of the facial nerve on the oblique sagittal MRI view of the internal auditory canal, or as a cochlear aperture smaller than 1.5 mm on an axial HRCT view61,63,64. Cerebral and brainstem abnormalities were determined with reference to the patient’s age62.
Patient grouping
The patients were divided into the following four groups based on medical history, genetic testing results, and imaging findings: genetic, acquired, CND-related, and indefinite. Patients documented to have prenatal or perinatal events, including prematurity of less than 34 weeks of gestational age, history of kernicterus with plasmapheresis, and perinatal hypoxia were considered to have acquired auditory neuropathy2,3,21. Patients with genetic mutations and those with phenotypes related to specific hereditary syndromes were considered to have genetic auditory neuropathy. Patients without identified acquired or genetic factors who had CND based on imaging studies were considered to have CND-related auditory neuropathy. Other patients were considered to have auditory neuropathy of indefinite causes. The hearing profiles and imaging findings were then compared among the four groups.
Statistical analysis
Statistical analyses were performed using SPSS for Windows version 22.0 (SPSS Inc., Chicago, IL, USA). Categorical data were analyzed using Pearson Chi-square test or Fisher’s exact test and summarized as number (%); continuous data were analyzed using ANOVA and summarized as mean ± standard deviation (SD). Pearson correlation analyses were performed to calculate correlations between hearing levels obtained using different audiologic tests. P values <0.05 (2-sided) were interpreted as statistically significant.
Data availability
The datasets generated during and/or analyzed during the current study are available from the corresponding authors on reasonable request.
References
Rance, G. Auditory neuropathy/dys-synchrony and its perceptual consequences. Trends in amplification 9, 1–43 (2005).
Berlin, C. I. et al. Multi-site diagnosis and management of 260 patients with auditory neuropathy/dys-synchrony (auditory neuropathy spectrum disorder). International journal of audiology 49, 30–43, https://doi.org/10.3109/14992020903160892 (2010).
Bielecki, I., Horbulewicz, A. & Wolan, T. Prevalence and risk factors for auditory neuropathy spectrum disorder in a screened newborn population at risk for hearing loss. International journal of pediatric otorhinolaryngology 76, 1668–1670, https://doi.org/10.1016/j.ijporl.2012.08.001 (2012).
Amatuzzi, M. G. et al. Selective inner hair cell loss in premature infants and cochlea pathological patterns from neonatal intensive care unit autopsies. Archives of otolaryngology–head & neck surgery 127, 629–636, https://doi.org/10.1001/archotol.127.6.629 (2001).
Starr, A. et al. Absence of both auditory evoked potentials and auditory percepts dependent on timing cues. Brain: a journal of neurology 114(Pt 3), 1157–1180, https://doi.org/10.1093/brain/114.3.1157 (1991).
Starr, A. et al. Pathology and physiology of auditory neuropathy with a novel mutation in the MPZ gene (Tyr145->Ser). Brain: a journal of neurology 126, 1604–1619, https://doi.org/10.1093/brain/awg156 (2003).
Starr, A., Picton, T. W., Sininger, Y., Hood, L. J. & Berlin, C. I. Auditory neuropathy. Brain: a journal of neurology 119(Pt 3), 741–753, https://doi.org/10.1093/brain/119.3.741 (1996).
Blegvad, B. & Hvidegaard, T. Hereditary dysfunction of the brain stem auditory pathways as the major cause of speech retardation. Scandinavian audiology 12, 179–187 (1983).
Norrix, L. W. & Velenovsky, D. S. Auditory neuropathy spectrum disorder: a review. Journal of speech, language, and hearing research: JSLHR 57, 1564–1576, https://doi.org/10.1044/2014_jslhr-h-13-0213 (2014).
Moser, T. & Starr, A. Auditory neuropathy–neural and synaptic mechanisms. Nature reviews. Neurology 12, 135–149, https://doi.org/10.1038/nrneurol.2016.10 (2016).
Manchaiah, V. K., Zhao, F., Danesh, A. A. & Duprey, R. The genetic basis of auditory neuropathy spectrum disorder (ANSD). International journal of pediatric otorhinolaryngology 75, 151–158, https://doi.org/10.1016/j.ijporl.2010.11.023 (2011).
Rendtorff, N. D. et al. Identification of p.A684V missense mutation in the WFS1 gene as a frequent cause of autosomal dominant optic atrophy and hearing impairment. American journal of medical genetics. Part A 155a, 1298–1313, https://doi.org/10.1002/ajmg.a.33970 (2011).
Han, K. H. et al. ATP1A3 mutations can cause progressive auditory neuropathy: a new gene of auditory synaptopathy. Scientific reports 7, 16504, https://doi.org/10.1038/s41598-017-16676-9 (2017).
Morlet, T. et al. Auditory function in Pelizaeus-Merzbacher disease. Journal of neurology 265, 1580–1589, https://doi.org/10.1007/s00415-018-8884-x (2018).
Hood, L. J. Auditory Neuropathy/Dys-Synchrony Disorder: Diagnosis and Management. Otolaryngologic clinics of North America 48, 1027–1040, https://doi.org/10.1016/j.otc.2015.06.006 (2015).
Walker, E., McCreery, R., Spratford, M. & Roush, P. Children with Auditory Neuropathy Spectrum Disorder Fitted with Hearing Aids Applying the American Academy of Audiology Pediatric Amplification Guideline: Current Practice and Outcomes. Journal of the American Academy of Audiology 27, 204–218, https://doi.org/10.3766/jaaa.15050 (2016).
Prosser, J. D., Cohen, A. P. & Greinwald, J. H. Diagnostic Evaluation of Children with Sensorineural Hearing Loss. Otolaryngologic clinics of North America 48, 975–982, https://doi.org/10.1016/j.otc.2015.07.004 (2015).
Korver, A. M. et al. Congenital hearing loss. Nature reviews. Disease primers 3, 16094, https://doi.org/10.1038/nrdp.2016.94 (2017).
Rance, G. & Starr, A. Pathophysiological mechanisms and functional hearing consequences of auditory neuropathy. Brain: a journal of neurology 138, 3141–3158, https://doi.org/10.1093/brain/awv270 (2015).
Amatuzzi, M., Liberman, M. C. & Northrop, C. Selective Inner Hair Cell Loss in Prematurity: A Temporal Bone Study of Infants from a Neonatal Intensive Care Unit. Journal of the Association for Research in Otolaryngology 12, 595–604, https://doi.org/10.1007/s10162-011-0273-4 (2011).
Beutner, D., Foerst, A., Lang-Roth, R., von Wedel, H. & Walger, M. Risk factors for auditory neuropathy/auditory synaptopathy. ORL; journal for oto-rhino-laryngology and its related specialties 69, 239–244, https://doi.org/10.1159/000101545 (2007).
Migliosi, V. et al. Q829X, a novel mutation in the gene encoding otoferlin (OTOF), is frequently found in Spanish patients with prelingual non-syndromic hearing loss. Journal of medical genetics 39, 502–506, https://doi.org/10.1136/jmg.39.7.502 (2002).
Rodriguez-Ballesteros, M. et al. Auditory neuropathy in patients carrying mutations in the otoferlin gene (OTOF). Human mutation 22, 451–456, https://doi.org/10.1002/humu.10274 (2003).
Varga, R. et al. OTOF mutations revealed by genetic analysis of hearing loss families including a potential temperature sensitive auditory neuropathy allele. Journal of medical genetics 43, 576–581, https://doi.org/10.1136/jmg.2005.038612 (2006).
Romanos, J. et al. Novel OTOF mutations in Brazilian patients with auditory neuropathy. Journal of human genetics 54, 382–385, https://doi.org/10.1038/jhg.2009.45 (2009).
Matsunaga, T. et al. A prevalent founder mutation and genotype-phenotype correlations of OTOF in Japanese patients with auditory neuropathy. Clinical genetics 82, 425–432, https://doi.org/10.1111/j.1399-0004.2012.01897.x (2012).
Jin, Y. J., Park, J., Kim, A. R., Rah, Y. C. & Choi, B. Y. Identification of a novel splice site variant of OTOF in the Korean nonsyndromic hearing loss population with low prevalence of the OTOF mutations. International journal of pediatric otorhinolaryngology 78, 1030–1035, https://doi.org/10.1016/j.ijporl.2014.03.033 (2014).
Zhang, Q. J. et al. High frequency of OTOF mutations in Chinese infants with congenital auditory neuropathy spectrum disorder. Clinical genetics 90, 238–246, https://doi.org/10.1111/cge.12744 (2016).
Buchman, C. A. et al. Auditory neuropathy characteristics in children with cochlear nerve deficiency. Ear and hearing 27, 399–408, https://doi.org/10.1097/01.aud.0000224100.30525.ab (2006).
Huang, B. Y., Roche, J. P., Buchman, C. A. & Castillo, M. Brain stem and inner ear abnormalities in children with auditory neuropathy spectrum disorder and cochlear nerve deficiency. AJNR. American journal of neuroradiology 31, 1972–1979, https://doi.org/10.3174/ajnr.A2178 (2010).
Fritzsch, B., Silos-Santiago, I., Bianchi, L. M. & Farinas, I. The role of neurotrophic factors in regulating the development of inner ear innervation. Trends in neurosciences 20, 159–164, https://doi.org/10.1016/s0166-2236(96)01007-7 (1997).
Bernd, P. The role of neurotrophins during early development. Gene expression 14, 241–250 (2008).
Cryns, K. et al. The WFS1 gene, responsible for low frequency sensorineural hearing loss and Wolfram syndrome, is expressed in a variety of inner ear cells. Histochemistry and cell biology 119, 247–256, https://doi.org/10.1007/s00418-003-0495-6 (2003).
Chiu, Y. H. et al. Mutations in the OTOF gene in Taiwanese patients with auditory neuropathy. Audiology & neuro-otology 15, 364–374, https://doi.org/10.1159/000293992 (2010).
Leruez, S. et al. Sensorineural hearing loss in OPA1-linked disorders. Brain: a journal of neurology 136, e236, https://doi.org/10.1093/brain/aws340 (2013).
Santarelli, R., Scimemi, P., Dal Monte, E. & Arslan, E. Cochlear microphonic potential recorded by transtympanic electrocochleography in normally-hearing and hearing-impaired ears. Acta otorhinolaryngologica Italica: organo ufficiale della Societa italiana di otorinolaringologia e chirurgia cervico-facciale 26, 78–95 (2006).
Kitao, K. et al. Deterioration in Distortion Product Otoacoustic Emissions in Auditory Neuropathy Patients With Distinct Clinical and Genetic Backgrounds. Ear and hearing 40, 184–191, https://doi.org/10.1097/aud.0000000000000586 (2019).
Mittal, R. et al. Auditory neuropathy spectrum disorder: its prevalence and audiological characteristics in an Indian tertiary care hospital. International journal of pediatric otorhinolaryngology 76, 1351–1354, https://doi.org/10.1016/j.ijporl.2012.06.005 (2012).
Shi, W. et al. Characteristics of cochlear microphonics in infants and young children with auditory neuropathy. Acta oto-laryngologica 132, 188–196, https://doi.org/10.3109/00016489.2011.630016 (2012).
Swanepoel, D., Hugo, R. & Roode, R. Auditory steady-state responses for children with severe to profound hearing loss. Archives of otolaryngology–head & neck surgery 130, 531–535, https://doi.org/10.1001/archotol.130.5.531 (2004).
Rance, G. et al. Hearing threshold estimation in infants using auditory steady-state responses. Journal of the American Academy of Audiology 16, 291–300 (2005).
Jafari, Z., Malayeri, S., Ashayeri, H. & Farahani, M. A. Adults with auditory neuropathy: comparison of auditory steady-state response and pure-tone audiometry. Journal of the American Academy of Audiology 20, 621–628 (2009).
Attias, J., Buller, N., Rubel, Y. & Raveh, E. Multiple auditory steady-state responses in children and adults with normal hearing, sensorineural hearing loss, or auditory neuropathy. The Annals of otology, rhinology, and laryngology 115, 268–276, https://doi.org/10.1177/000348940611500404 (2006).
Eppsteiner, R. W. et al. Prediction of cochlear implant performance by genetic mutation: the spiral ganglion hypothesis. Hearing research 292, 51–58, https://doi.org/10.1016/j.heares.2012.08.007 (2012).
Wu, C. C. et al. Identifying Children With Poor Cochlear Implantation Outcomes Using Massively Parallel Sequencing. Medicine 94, e1073, https://doi.org/10.1097/md.0000000000001073 (2015).
Wu, C. C. et al. Timing of cochlear implantation in auditory neuropathy patients with OTOF mutations: Our experience with 10 patients. Clinical otolaryngology: official journal of ENT-UK; official journal of Netherlands Society for Oto-Rhino-Laryngology & Cervico-Facial Surgery 43, 352–357, https://doi.org/10.1111/coa.12949 (2018).
Nishio, S. Y. & Usami, S. I. Outcomes of cochlear implantation for the patients with specific genetic etiologies: a systematic literature review. Acta oto-laryngologica 137, 730–742, https://doi.org/10.1080/00016489.2016.1276303 (2017).
Runge, C. L. et al. A novel otoferlin splice-site mutation in siblings with auditory neuropathy spectrum disorder. Audiology & neuro-otology 18, 374–382, https://doi.org/10.1159/000354978 (2013).
Park, J. H. et al. Outcome of Cochlear Implantation in Prelingually Deafened Children According to Molecular Genetic Etiology. Ear and hearing 38, e316–e324, https://doi.org/10.1097/AUD.0000000000000437 (2017).
Peng, K. A., Kuan, E. C., Hagan, S., Wilkinson, E. P. & Miller, M. E. Cochlear Nerve Aplasia and Hypoplasia: Predictors of Cochlear Implant Success. Otolaryngology–head and neck surgery: official journal of American Academy of Otolaryngology-Head and Neck Surgery 157, 392–400, https://doi.org/10.1177/0194599817718798 (2017).
Freeman, S. R. & Sennaroglu, L. Management of Cochlear Nerve Hypoplasia and Aplasia. Advances in oto-rhino-laryngology 81, 81–92, https://doi.org/10.1159/000485542 (2018).
Sharma, A. & Cardon, G. Cortical development and neuroplasticity in Auditory Neuropathy Spectrum Disorder. Hearing research 330, 221–232, https://doi.org/10.1016/j.heares.2015.06.001 (2015).
Apeksha, K. & Kumar, U. A. Cortical processing of speech in individuals with auditory neuropathy spectrum disorder. European archives of oto-rhino-laryngology: official journal of the European Federation of Oto-Rhino-Laryngological Societies (EUFOS): affiliated with the German Society for Oto-Rhino-Laryngology - Head and Neck Surgery 275, 1409–1418, https://doi.org/10.1007/s00405-018-4966-8 (2018).
Wu, C. C., Chen, Y. S., Chen, P. J. & Hsu, C. J. Common clinical features of children with enlarged vestibular aqueduct and Mondini dysplasia. The Laryngoscope 115, 132–137, https://doi.org/10.1097/01.mlg.0000150691.85387.3f (2005).
Lin, P. H. et al. Etiologic and Audiologic Characteristics of Patients With Pediatric-Onset Unilateral and Asymmetric Sensorineural Hearing Loss. JAMA otolaryngology–head & neck surgery 143, 912–919, https://doi.org/10.1001/jamaoto.2017.0945 (2017).
Wu, C. M. et al. Long-Term Cochlear Implant Outcomes in Children with GJB2 and SLC26A4 Mutations. Plos one 10, e0138575, https://doi.org/10.1371/journal.pone.0138575 (2015).
Wu, C. C. et al. Prospective mutation screening of three common deafness genes in a large Taiwanese Cohort with idiopathic bilateral sensorineural hearing impairment reveals a difference in the results between families from hospitals and those from rehabilitation facilities. Audiology & neuro-otology 13, 172–181, https://doi.org/10.1159/000112425 (2008).
Wu, C. C. et al. Application of massively parallel sequencing to genetic diagnosis in multiplex families with idiopathic sensorineural hearing impairment. Plos one 8, e57369, https://doi.org/10.1371/journal.pone.0057369 (2013).
Lin, Y. H. et al. A novel missense variant in the nuclear localization signal of POU4F3 causes autosomal dominant non-syndromic hearing loss. Scientific reports 7, 7551, https://doi.org/10.1038/s41598-017-08236-y (2017).
Lin, Y. H. et al. Targeted Next-Generation Sequencing Facilitates Genetic Diagnosis and Provides Novel Pathogenetic Insights into Deafness with Enlarged Vestibular Aqueduct. The Journal of molecular diagnostics: JMD 21, 138–148, https://doi.org/10.1016/j.jmoldx.2018.08.007 (2019).
Komatsubara, S., Haruta, A., Nagano, Y. & Kodama, T. Evaluation of cochlear nerve imaging in severe congenital sensorineural hearing loss. ORL; journal for oto-rhino-laryngology and its related specialties 69, 198–202, https://doi.org/10.1159/000099231 (2007).
Giedd, J. N. & Rapoport, J. L. Structural MRI of pediatric brain development: what have we learned and where are we going? Neuron 67, 728–734, https://doi.org/10.1016/j.neuron.2010.08.040 (2010).
Joshi, V. M. et al. CT and MR imaging of the inner ear and brain in children with congenital sensorineural hearing loss. Radiographics: a review publication of the Radiological Society of North America, Inc 32, 683–698, https://doi.org/10.1148/rg.323115073 (2012).
Sennaroglu, L. & Bajin, M. D. Classification and Current Management of Inner Ear Malformations. Balkan medical journal 34, 397–411, https://doi.org/10.4274/balkanmedj.2017.0367 (2017).
Acknowledgements
This work was supported by the Ministry of Science and Technology [grant number MOST 107-2314-B-002-137-MY3]. We thank for all subjects and their parents for participating in this study.
Author information
Authors and Affiliations
Contributions
P-H.L. and C-C.W. ascertained clinical data, analyzed data, and wrote the paper; Yin-Hung L. and Yi-Hsin L. performed genetic examinations and analyses; S-Y.Y. collected and analyzed audiological data; T-H.Y. collected clinical data; P-L.C. supervised the genetic examinations and analyses; T-C.L. and C-J.H. supervised the whole study and provided critical revision.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Lin, PH., Hsu, CJ., Lin, YH. et al. An integrative approach for pediatric auditory neuropathy spectrum disorders: revisiting etiologies and exploring the prognostic utility of auditory steady-state response. Sci Rep 10, 9816 (2020). https://doi.org/10.1038/s41598-020-66877-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-66877-y
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
