
Abstract
Determining the etiology of severe-to-profound sensorineural hearing loss (SP-SNHL) in pediatric subjects is particularly important in aiding the decision for auditory rehabilitation. We aimed to update the etiologic spectrum of pediatric SP-SNHL by combining internal auditory canal (IAC)-MRI with comprehensive and state-of-the-art genetic testings. From May 2013 to September 2020, 119 cochlear implantees under the age of 15 years with SP-SNHL were all prospectively recruited. They were subjected to genetic tests, including exome sequencing, and IAC-MRI for etiologic diagnosis. Strict interpretation of results were made based on ACMG/AMP guidelines and by an experienced neuroradiologist. The etiology was determined in of 65.5% (78/119) of our cohort. If only one of the two tests was done, the etiologic diagnostic rate would be reduced by at least 21.8%. Notably, cochlear nerve deficiency (n = 20) detected by IAC-MRI topped the etiology list of our cohort, followed by DFNB4 (n = 18), DFNB1 (n = 10), DFNB9 (n = 10) and periventricular leukomalacia associated with congenital CMV infection (n = 8). Simultaneous application of state-of-the-art genetic tests and IAC-MRI is essential for etiologic diagnosis, and if lesions of the auditory nerve or central nerve system are carefully examined on an MRI, we can identify the cause of deafness in more than 65% of pediatric SP-SNHL cases.
Similar content being viewed by others
Introduction
Even though pediatric patients without etiologic confirmation could receive the proper auditory rehabilitation based on the hearing status, identifying the etiology of severe-to-profound sensorineural hearing loss (SP-SNHL) puts us obviously in a better position to predict the natural course of the hearing loss and outcome of each hearing rehabilitation in pediatric patients, leading to the most appropriate auditory rehabilitation. Awareness of the etiology of SP-SNHL sometimes, helps to make a decision of timely cochlear implantation (CI). In some cases, parents hesitate before deciding on CI for their child, especially when the cause of SP-SNHL remains unknown. This could result in prolonged auditory deprivation and interference with language development1. Knowing the etiology—either molecular genetically or radiologically—will help convince parents and prevent unnecessary delay of hearing rehabilitation. For example, SLC26A4 and GJB2 variants are the major cause of SP-SNHL with different progressive features from each other, so when they are confirmed, hearing rehabilitation can be planned accordingly2,3. And it has been reported that subjects with OTOF-related auditory neuropathy spectrum disorder could be managed effectively with timely CI after molecular etiologic confirmation, which would have been difficult without genetic confirmation4,5. In some cases etiologic diagnosis of hearing loss enables early recognition of comorbidities such as visual loss in Usher syndrome, potential thyroid issues in Pendred syndrome, and kidney disease in Alport syndrome6,7. At least, knowing the etiology can help better inform parents of the possible options and prognosis.
Like this, there have been many reports of the etiologic spectrum using genetic tests or imaging tests, respectively. Non-syndromic and syndromic genetic causes are known to account for about 45% of bilateral SNHL in pediatric patients8. Congenital CMV infection (cCMV) reportedly accounts for about 17% of total bilateral SNHL with varying degrees, however this figure could reach upto 33% among those exclusively with an unknown etiology, bringing up the issue of importance of scanning the lesions of central nervous system (CNS) or cochlear enhancement that could suggest cCMV related deafness8. DFNB1 due to GJB2 variants is the most common non-syndromic hearing loss accounting for about 30% of confirmed genetic cases and Pendred, Usher and Waardenburg syndromes are known to be the most common syndromic hearing loss9. A genetic study in 459 adult and child CI subjects revealed that top five causative genes were GJB2 (16%), TMPRSS3 (10%), SLC26A4 (8%), MYO7A (7%), and MT-RNR1 (5%)10. However, to the best of our knowledge, there is paucity of reports clarifying the etiologic spectrum of pediatric CI candidates manifesting bilateral SP-SNHL by internal auditory canal (IAC)-MRI in combination with state-of-the-art genetic tests including chromosome microarray analysis (CMA) in the single SP-SNHL cohort. Therefore, it was not feasible to compare the relative prevalence of specific imaging abnormalities and molecular genetic abnormalities in the bilateral SP-SNHL group. Further, in previous studies using IAC-MRI, cohorts were mixed with both unilateral and bilateral hearing loss, which precludes these previous analyses from isolating the etiologic diagnostic yield of IAC-MRI to only bilateral pediatric SP-SNHL. Conversely, in most studies addressing the molecular genetic etiologies of pediatric hearing loss patients11,12,13,14,15,16, comprehensive IAC-MRI findings were not provided in tandem, leading to failure in elucidating the full etiologic spectrum in a single cohort.
Recently, our group has identified and published the etiologic spectrum of mild to moderate SNHL in children through state-of-the-art genetic tests17,18. While imaging tests contribute relatively little to the elucidation of etiology of mild to moderate SNHL in children, their contribution is much larger in pediatric SP-SNHL. We believed that it was necessary to elucidate the full etiologic spectrum of pediatric SP-SNHL again through combination of genetic tests and imaging tests. Given this, we aim to establish the full etiologic spectrum of pediatric bilateral SP-SNHL by combining both IAC-MRI and state-of-the-art genetic tests, thereby, to the best of our knowledge, providing the highest diagnostic yield of pediatric bilateral SP-SNHL in literature.
Results
Overall etiologic spectrum of pediatric SP-SNHL
The etiology of SNHL was convincingly confirmed in 65.5% (78/119) of subjects (Table 1). Following strict criteria for interpretation of pathogenic potential of candidate variants, molecular genetic and radiologic diagnoses were made in 39.4% and 43.7% of subjects, respectively. The genetic tests and IAC-MRI found an overlapping etiology in 17.6% (21/119) of cases. If one of the two tests were performed independently, the etiologic diagnostic rate would be reduced by at least 21.8% without genetic tests, and 26.1% without IAC-MRI.
Documented molecular genetic etiology
Molecular genetic testing was omitted in 12 subjects due to identification of non-genetic etiologies either from MRI or additional clinical information (seven subjects with CMV infections, three with cochlear nerve deficiency and one each with diffuse brain atrophy and neonatal intracranial hemorrhage).
Of the 107 subjects who underwent genetic testing, 47 were genetically diagnosed (Tables 1, 2A). The etiology in 26 of these would not be elucidated without genetic testing, as they presented with normal IAC-MRI results (Table 1). The molecular genetic etiologies of the 47 genetically diagnosed subjects were all point variants. The details of the genetic test results are shown in Table 2A and Supplementary Table S1A. SLC26A4 was the most frequent gene that harbors causative recessive variants in 18 subjects, followed by GJB2 variants (n = 10) and OTOF variants (n = 10). Other causative genes were TMC1, MYO15A, NLRP3, ATP1A3, KCNQ1, PAX3, POU3F4, PDZD7 and CHD7 (n = 1 each).
Additionally, there were 10 subjects who are ‘genetically suspected,” or with candidate variants with high probability of causality (Table 2B, Supplementary Table S1B). With the inclusion of these cases, the diagnostic yield of genetic tests could increase up to 47.9% (57/119). There were also three cases where genetic variants possibly associated with SNHL were identified but the criteria were not satisfied (Supplementary Table S1C).
There were 11 subjects manifesting syndromic deafness, and five of them were identified to carry a causative genetic variant (Table 2A). In the case of CHARGE syndrome (n = 4), only one subject carried a detectable CDH7 variant; likewise, in the case of Waardenburg syndrome (n = 2), only one subject carried a detectable PAX3 variant. A causative genetic abnormality was detected from the three other syndromic subjects (CINCA syndrome, CAPOS syndrome and Jervell and Lange-Nielsen syndrome, n = 1 each). In the ‘genetically suspected’ cases, Chromosome 4p16.3 deletion was detected in one subject suspected with Wolf-Hirschhorn syndrome (Table 2B).
None of the four chromosomal deletions detected were able to satisfy the ACMG/AMP criteria. Three subjects with chromosomal deletions (Chr. 4p16.3 deletion, Chr. 18q deletion, and Chr. 22q13.3 deletion) were thus classified as cases with ‘genetically suspected’ variants (Table 2B). However, the association between 3q deletion (SB318-627) and SNHL was doubtful.
Radiologically documented etiology based on IAC-MRI findings
IAC-MRI revealed radiologic abnormalities that were either direct or indirectly related to SP-SNHL in 52 (43.7%) out of 119 subjects (Tables 1, 3). Among these, pathologies in the inner ear (cochlea/vestibular aqueduct) were the most common, followed by auditory nerve and central nervous system (CNS) lesions (42.3% (22/52) vs 38.5% (20/52) vs 19.2% (10/52), providing a roughly 1:1:0.5 ratio and emphasizing the importance of taking a careful look at the auditory nerve and CNS rather than limiting ourselves to the inner ear.
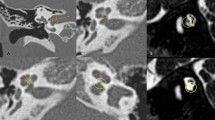
In our pediatric cohort, cochlear nerve deficiency (CND) was the most prevalent radiologic abnormality (n = 20), accounting for 16.8% of our entire cohort, followed by enlarged vestibular aqueduct (EVA) (n = 18 (15.1%)) and periventricular leukomalacia (PVLM) (n = 8 (6.7%)) (Table 3, Fig. 1). CND was detected not only in an idiopathic form, but also in various syndromes, such as CHARGE syndrome (n = 4) and Waardenburg type IV syndrome (n = 1).

Three major abnormalities related to pediatric SP-SNHL as seen in IAC-MRI. Images on the left side show abnormal findings, while normal findings are on the right. (a) Cochlear nerve deficiency: Cochlear nerve is not observed in the internal auditory canal (arrow). (b) Enlarged vestibular aqueduct with incomplete partition type 2: Vestibular aqueduct is enlarged and conical in shape (arrowhead), and the interscalar septum is not present (dotted arrow). (c) Periventricular leukomalacia: Ventriculomegaly with irregular margins of the bodies of the lateral ventricles and loss of periventricular white matter are observed.
In our cohort with 119 subjects, the significant CNS lesions were observed from 19 subjects (16.0%). Notably, CNS lesions found on IAC-MRI were considered related to pediatric SP-SNHL in 10 subjects with documented or strongly suspected congenital CMV infection based on audiological phenotypes, CMV viral culture and CMV-PCR testing, making CNS lesion an important radiological finding in pediatric SP-SNHL (Table 3).
The other nine subjects with CNS lesions consist of four cases with PVLM without any evidence suggesting congenital CMV infection and one each of ‘diffuse brain atrophy’, ‘cystic cerebromalacia’, ‘T2 high signal lesions in both cerebral subcortex’, ‘asymmetric size of lateral ventricle with tinning of corpus callosum’ and ‘T2 high signal lesions in both globus pallidus’ (Supplementary Table S2). In our present study, these MRI abnormalities did not qualify as the etiologic diagnosis of SP-SNHL.
Interpretation of SP-SNHL without genetic variants or abnormal MRI findings (n = 41)
We suspected that at least five of the 41 subjects might have congenital CMV infection because of either positive CMV culture and CMV-PCR results taken just after 3 weeks (n = 4), or the progressive and asymmetric character of SNHL (n = 1). However, we were not able to confirm the etiology of hearing loss of these subjects as congenital CMV infection.
Discussion
Herein, we elucidated the etiologic spectrum of 119 prospectively recruited pediatric SP-SNHL subjects and were able to establish a diagnostic rate of 65.5% (78/119) using a protocol that combined IAC-MRI and genetic tests. To the best of our knowledge, this etiologic diagnostic rate in pediatric SP-SNHL subjects is the highest among studies of this type.
Notably, CND turned out to be the most frequent IAC-MRI abnormality (16.8%), even outnumbering EVA. A retrospective study of 207 child patients with bilateral SNHL revealed there were 10 cases (4.8%) of CND and 20 cases (9.7%) of EVA in the cohort19. It could be due to the cohort criteria, difference of region and image diagnosis criteria. Recently, a couple of candidate genes that might be associated with CND or other cochlear anomalies have been proposed through a trio study20; however, none of these candidate genes were causally linked to pediatric subjects in our cohort. Therefore, taking IAC-MRI from pediatric SP-SNHL patients in a timely manner is of paramount importance.
Detection of CNS lesions such as PVML in IAC-MRI from ten subjects (8.4% of the entire cohort) also supported the etiologic diagnosis among cases with suspected or already diagnosed congenital CMV infection. This figure was not significantly different from 6.6% of severe to profound SNHL previously reported by Lin et al.21.
The diagnostic yield of the temporal bone CT had been previously reported to be approximately 18 to 20 percent21,22,23, while very recent studies suggested 30 to 37 percent24,25. A similar range was noted for IAC-MRI, ranging from 24 to 42.7%21,24,25,26,27. However, most of these studies included both bilateral and unilateral SNHL cases in their analysis. The present study is unique in that we included purely bilateral SP-SNHL cases and we were able to achieve a diagnostic yield of 43.7% for IAC-MRI.
Up to 60% of congenital hearing loss is thought to be due to genetic etiologies28,29. With the improvement of genetic diagnosis technology and the accumulation of genetic information for hearing loss, the molecular genetic diagnostic rate has been significantly increasing30,31. In accordance with this, a total of 12 kinds of monogenic autosomal genes accounted for SP-SNHL in our pediatric cohort. Without extensive use of exome sequencing, ATP1A332, NLRP312,13, and PDZD716 could not have been screened, leading to a decrease in diagnostic rate by 2.5%. In the literature, the diagnostic yield of SNHL with genetic consideration has been reported to be between 19 and 39%21,23,24,27,33. The most common causative gene was GJB2, accounting for approximately 6–10%—but as high as 20–25% in a certain population—of congenital hearing loss22,24,27. Recently, our group reported that 54.8% of pediatric SP-SNHL were genetically diagnosed through rigorous genetic tests34. In our present study 47 of 119 (39.5%) subjects were found to have a genetic etiology. This lower diagnostic rate could be attributed to the significantly stricter criteria for inclusion to the ‘genetically diagnosed’ group in our present study. In this study, only the autosomal dominant with ‘pathogenic’ or ‘likely pathogenic’ and the autosomal recessive cases with both ‘likely pathogenic’ variants based on ACMG/AMP guideline35,36 were eligible as ‘genetically diagnosed’ cases. If we included the seven cases suspected to have potentially causative genetic variants for calculation of the diagnostic rate, then the proportion of pediatric SP-SNHL subjects with the molecular genetic diagnosis and the etiologic diagnosis could even reach up to 45.3% (54/119) and 71.4% (85/119), respectively. Actually, before the ACMG/AMP guideline was issued, these seven cases would have been considered ‘genetically diagnosed’.
Additionally, it is notable that our study included FISH into the molecular genetic diagnostic battery, thus identifying chromosomal abnormalities that were suspected to have a causal relationship with SP-SNHL in 2.5% (3/119) of the cohort. Although the causality between these three large genomic deletions and SP-SNHL deafness is not fully established due to nonfulfillment of the ACMG/AMP guideline criteria, we believe that these abnormalities are associated with SNHL for the following reasons: Chr.18q deletion was reported to be associated with SNHL37,38 and congenital hypothyroidism itself frequently accompanying 18q deletion could also serve as a risk factor for SNHL39. PVLM is also a common sign of 18q deletion40 and the developmental insult itself causing PVLM could cause hearing loss41, also providing causality between 18q deletion and hearing loss. The hemizygous 4p16.3 deletion has also been proposed to be a molecular genetic cause of SNHL42 and Wolf–Hirschhorn syndrome43. Lastly, an etiologic diagnosis of hearing loss from SB418-819 with a VUS, hemizygous 22q13.3 deletion, mild CND and thinning of the corpus callosum was challenging. However, some children carrying 22q13.3 deletion showed either auditory neuropathy or central auditory processing disorder44, which was partially compatible with the presence of mild CND and thinning of corpus callosum. If these three subjects carrying chromosomal deletions that were likely to be associated with SP-SNHL were also included, total diagnostic rates would increase potentially up to 73.9% (88/119).
Notably, Lin et al.21 pointed out that genetically diagnosed DFNB1 subjects carrying variants in GJB2 were nearly nonoverlapping compared with children etiologically diagnosed through imaging. This raised the possibility that comprehensive diagnostic battery incorporating both genetic tests and imaging study may significantly improve the etiologic diagnostic rate. However, studies evaluating the combination of genetic tests and imaging study are scarce in literature. Among these studies, the diagnostic yield ranged from 33 to 40%22,23. These did not test the genes beyond m.1555A > G, GJB2 and SLC26A4, and also did not report the CNS lesions or CND in detail, leaving room for further investigations. Regardless of the necessity of exome sequencing, the first line screening for prevalent variants of GJB2, OTOF and SLC26A4 using previously reported diagnostic kits45,46 in tandem with IAC-MRI could possibly lead to better etiologic diagnosis, as high as 58.0% in pediatric SP-SNHL (Fig. 2).

Systematic etiologic diagnostic flowchart used in our study. First line screening using internal auditory canal MRI in tandem with molecular screening of prevalent variants enables etiologic identification in 58.0% of subjects. Subsequent in-depth genetic testing elucidates molecular etiology in 9 more subjects (7.6%), leaving 41 subjects etiologically undiagnosed.
Even with rigorous genetic tests and imaging studies, about 30% of etiology was still not clarified. Undiagnosed cases might be related to perinatal problems such as mostly CMV infection, hypoxemia, hyperbilirubinemia and sepsis, ototoxic medication exposure, autoimmune etiologies and other congenital infections including syphilis, rubella47,48,49. So, in addition to genetic and imaging studies, family and medical history taking, evaluation of comorbidities, neonatal CMV screening and testing for congenital syphilis or rubella should also be considered for evaluation of congenital hearing loss50.
Consensus statement of the International Pediatric Otolaryngology Group recommended including genetic tests in etiology study for pediatric bilateral SNHL patients51. In the era of precision medicine, this etiologic information could serve as an important basis for decision making in auditory rehabilitation, prediction of prognosis after cochlear implantation, and potentially future gene therapies. Based on what we observed from the present study, the etiologic diagnostic rate would pave the way for future efforts to uncover the hidden causative genes, especially related to CND, and to find diagnostic methods which make it possible to confirm congenital CMV deafness even after the current golden time of diagnosis (within 3 weeks after birth) has passed.
In this study, we have clearly identified the cause of hearing loss in at least 65.5% of pediatric SP-SNHL subjects. Increasing etiologic diagnostic rate of pediatric SP-SNHL is expected to provide the effective hearing rehabilitation and help spread genetic tests in subject with hearing loss. The ratio of abnormal findings on IAC-MRI and genetic tests among our cohort is almost the same, but the overlapping rate is only about 17.6%. So if both tests are not performed, the causative etiology can be missed in more than 20% of patients. Analysis of imaging findings revealed CND as the most common cause among the entire SP-SNHL cohort, and CNS lesions qualifying for diagnostic clues are also observed in 8.4% of subjects. Based on this, it seems necessary to always consider the possibility of CND or CNS lesions when interpreting IAC-MRI findings.
Methods
Recruitment of study participants
We prospectively established a cohort exclusively comprised of children under the age of 15 years, who were all treated by a single surgeon (B.Y.C) in the same tertiary referral hospital between May 2013 and September 2020 for SP-SNHL. In total, 119 patients meeting the criteria of bilateral SP-SNHL with 500, 1000 and 2000 Hz averaging hearing thresholds exceeding 70 dB HL were included. Subjects with single sided deafness or asymmetric hearing loss were excluded. This study was carried out in accordance with the Declaration of Helsinki. The study was approved by the Seoul National University Bundang Hospital Institutional Review Board (IRB Number B-2108-705-103). All participants and their legal guardians were given written informed consent before participating in this study.
Data collection
Demographic data, IAC-MRI findings, and genetic test results were collected and analyzed52. The mean age of children at recruitment after rigorous evaluation of hearing thresholds was 27.96 months (± 27.432, 6–178 months). Male-to-female ratio of subjects was 75:44. IAC-MRI was carried out in all 119 cases. Abnormal IAC-MRI findings related to SP-SNHL were divided into those from the inner ear, the cochlear nerve, and the central nervous system (CNS). If the IAC-MRI results were consistent with the syndrome with SP-SNHL as one of the symptoms, it was determined to account for the SP-SNHL.
At least one of the four genetic tests: U-Top screening kit45,46, panel sequencing53,54, exome sequencing18,55 or fluorescent in situ hybridization (FISH), was carried out in 107 subjects. For those without any confirmatory molecular diagnosis by U-Top screening kit, panel sequencing or exome sequencing were performed. Unless panel sequencing clearly revealed the causative variants, exome sequencing was performed. FISH was performed only for syndromic deafness with significant co-morbid abnormality, of which causative variants had not been elucidated after exome sequencing56. In all cases, genomic DNA was extracted from the peripheral blood. Obtained reads from panel or exome sequencing were mapped onto the University of California–Santa Cruz (UCSC) hg19 reference genome assembly, using the Lasergene 14 software package (DNASTAR, Madison, WI, USA). Several global minor allele frequency (MAF) databases were consulted for checking minor allele frequency, such as Exome Aggregation Consortium (ExAC), 1000 Genomes Project (1000 Genomes), National Heart, Lung, and Blood Institute (NHLBI), as well as the Grand Opportunity Exome Sequencing Project (GO-ESP) and the Korean Reference Genome Database (KRGDB). Single-nucleotide polymorphisms (SNP) that were incompatible with the autosomal recessive pattern were ruled out. To predict the pathogenicity of each missense variant, we referred to diverse in silico prediction software, such as Sorting Intolerant from Tolerant (SIFT), PolyPhen-2, MutationTaster, Combined Annotation Dependent Depletion (CADD), and Rare Exome Variant Ensemble Learner (REVEL) analyses. Whenever possible, the remaining SNPs were validated in other family members by Sanger sequencing for segregation study.
Description of pathogenic potential of candidate variants were made according to the American College of Medical Genetics and Genomics (ACMG) 2015 guidelines, the newly specified ACMG/Association for Molecular Pathology (AMP) hearing loss rules36, and ‘Expert specification of the ACMG/AMP variant interpretation guidelines for genetic hearing loss’35. In autosomal recessive (AR) cases, it was notated as ‘genetically diagnosed’ only when both variants were classified as at least ‘likely pathogenic (LP)’. In autosomal dominant (AD) cases, it was indicated as ‘genetically diagnosed’ when there was a variant of ‘LP’ or ‘pathogenic’ classification. Cases with candidate genetic variants of AD inheritance classified as ‘variant of unknown significance (VUS)’ and of AR inheritance with one variant as ‘LP’ or ‘pathogenic’ but the other variant as ‘VUS’ were notated as ‘genetically suspected’. When a large genomic deletion with high probability of relationship with deafness was identified but not 100% certain, it was also notated as ‘genetically suspected’. Notably, genetic tests were omitted in 12 cases where etiological diagnosis had already been established through IAC-MRI.
Diagnostic yield of each modality, categorization of the subjects
The diagnostic yield of IAC-MRI and genetic tests for our pediatric SP-SNHL cohort was calculated. Subjects with ‘genetically suspected’ candidate variants were not considered ‘genetically diagnosed’.
Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request. Some data may not be made available because of privacy or ethical restrictions.
References
Karltorp, E. et al. Cochlear implants before 9 months of age led to more natural spoken language development without increased surgical risks. Acta Paediatr. 109, 332–341. https://doi.org/10.1111/apa.14954 (2020).
Wu, C. M. et al. Long-term cochlear implant outcomes in children with GJB2 and SLC26A4 Mutations. PLoS ONE 10, e0138575. https://doi.org/10.1371/journal.pone.0138575 (2015).
Lee, S. Y. et al. Natural course of residual hearing with reference to GJB2 and SLC26A4 genotypes: Clinical implications for hearing rehabilitation. Ear Hear. 42, 644–653. https://doi.org/10.1097/AUD.0000000000000965 (2021).
Zheng, D. & Liu, X. Cochlear implantation outcomes in patients with OTOF mutations. Front. Neurosci. 14, 447. https://doi.org/10.3389/fnins.2020.00447 (2020).
Lee, S. Y. et al. Central auditory maturation and behavioral outcomes after cochlear implantation in prelingual auditory neuropathy spectrum disorder related to OTOF variants (DFNB9): Lessons from pilot study. PLoS ONE 16, e0252717. https://doi.org/10.1371/journal.pone.0252717 (2021).
Bozanic Urbancic, N., Battelino, S., Tesovnik, T. & Trebusak Podkrajsek, K. The importance of early genetic diagnostics of hearing loss in Children. Medicina (Kaunas) 56, 471. https://doi.org/10.3390/medicina56090471 (2020).
Parker, M. & Bitner-Glindzicz, M. Genetic investigations in childhood deafness. Arch. Dis. Child 100, 271–278. https://doi.org/10.1136/archdischild-2014-306099 (2015).
van Beeck Calkoen, E. A. et al. The etiological evaluation of sensorineural hearing loss in children. Eur. J. Pediatr. 178, 1195–1205. https://doi.org/10.1007/s00431-019-03379-8 (2019).
Kilic, S. et al. Comprehensive medical evaluation of pediatric bilateral sensorineural hearing loss. Laryngosc. Investig. Otolaryngol. 6, 1196–1207. https://doi.org/10.1002/lio2.657 (2021).
Seligman, K. L. et al. Genetic causes of hearing loss in a large cohort of cochlear implant recipients. Otolaryngol. Head Neck Surg. 166, 734–737. https://doi.org/10.1177/01945998211021308 (2022).
Han, R. et al. Efficiency of microarray and SNPscan for the detection of hearing loss gene in 71 cases with nonsyndromic hearing loss. Medicine (Baltimore) 96, e7149. https://doi.org/10.1097/MD.0000000000007149 (2017).
Kim, B. J. et al. Outcome of cochlear implantation in NLRP3-related autoinflammatory inner ear disorders. Otol. Neurotol. 42, e168–e171 (2021).
Kim, B. J. et al. Otological aspects of NLRP3-related autoinflammatory disorder focusing on the responsiveness to anakinra. Rheumatology 60, 1523 (2020).
Lynch, E. D. et al. Nonsyndromic deafness DFNA1 associated with mutation of a human homolog of the Drosophila gene diaphanous. Science 278, 1315–1318 (1997).
Mujtaba, G. et al. A mutation of MET, encoding hepatocyte growth factor receptor, is associated with human DFNB97 hearing loss. J. Med. Genet. 52, 548–552. https://doi.org/10.1136/jmedgenet-2015-103023 (2015).
Schneider, E. et al. Homozygous disruption of PDZD7 by reciprocal translocation in a consanguineous family: A new member of the Usher syndrome protein interactome causing congenital hearing impairment. Hum. Mol. Genet. 18, 655–666. https://doi.org/10.1093/hmg/ddn395 (2009).
Kim, B. J. et al. Significant Mendelian genetic contribution to pediatric mild-to-moderate hearing loss and its comprehensive diagnostic approach. Genet. Med. 22, 1119–1128. https://doi.org/10.1038/s41436-020-0774-9 (2020).
Kim, N. K. et al. Whole-exome sequencing reveals diverse modes of inheritance in sporadic mild to moderate sensorineural hearing loss in a pediatric population. Genet. Med. 17, 901–911. https://doi.org/10.1038/gim.2014.213 (2015).
van Beeck Calkoen, E. A. et al. Evaluation of the outcome of CT and MR imaging in pediatric patients with bilateral sensorineural hearing loss. Int. J. Pediatr. Otorhinolaryngol. 108, 180–185. https://doi.org/10.1016/j.ijporl.2018.02.022 (2018).
Kari, E. et al. Genes implicated in rare congenital inner ear and cochleovestibular nerve malformations. Ear Hear. 41, 983–989. https://doi.org/10.1097/AUD.0000000000000819 (2020).
Lin, J. W. et al. Comprehensive diagnostic battery for evaluating sensorineural hearing loss in children. Otol. Neurotol. 32, 259 (2011).
Yaeger, D. et al. Outcomes of clinical examination and genetic testing of 500 individuals with hearing loss evaluated through a genetics of hearing loss clinic. Am. J. Med. Genet. A 140, 827–836. https://doi.org/10.1002/ajmg.a.31179 (2006).
Ramos, P. Z. et al. Etiologic and diagnostic evaluation: Algorithm for severe to profound sensorineural hearing loss in Brazil. Int. J. Audiol. 52, 746–752. https://doi.org/10.3109/14992027.2013.817689 (2013).
Funamura, J. L. Evaluation and management of nonsyndromic congenital hearing loss. Curr. Opin. Otolaryngol. Head Neck Surg. 25, 385–389. https://doi.org/10.1097/MOO.0000000000000398 (2017).
Shah, J. et al. Evaluating diagnostic yield of computed tomography (CT) and magnetic resonance imaging (MRI) in pediatric unilateral sensorineural hearing loss. Int. J. Pediatr. Otorhinolaryngol. 115, 41–44. https://doi.org/10.1016/j.ijporl.2018.09.003 (2018).
McClay, J. E. et al. Evaluation of pediatric sensorineural hearing loss with magnetic resonance imaging. Arch. Otolaryngol. Head Neck Surg. 134, 945–952 (2008).
Mehta, D. et al. Outcomes of evaluation and testing of 660 individuals with hearing loss in a pediatric genetics of hearing loss clinic. Am. J. Med. Genet. A 170, 2523–2530. https://doi.org/10.1002/ajmg.a.37855 (2016).
Alford, R. L. et al. American College of Medical Genetics and Genomics guideline for the clinical evaluation and etiologic diagnosis of hearing loss. Genet. Med. 16, 347–355. https://doi.org/10.1038/gim.2014.2 (2014).
Raymond, M., Walker, E., Dave, I. & Dedhia, K. Genetic testing for congenital non-syndromic sensorineural hearing loss. Int. J. Pediatr. Otorhinolaryngol. 124, 68–75. https://doi.org/10.1016/j.ijporl.2019.05.038 (2019).
Sun, Y. et al. Increased diagnostic yield by reanalysis of data from a hearing loss gene panel. BMC Med. Genomics 12, 76. https://doi.org/10.1186/s12920-019-0531-6 (2019).
Wright, C. F. et al. Making new genetic diagnoses with old data: Iterative reanalysis and reporting from genome-wide data in 1,133 families with developmental disorders. Genet. Med. 20, 1216–1223. https://doi.org/10.1038/gim.2017.246 (2018).
Han, K.-H. et al. ATP1A3 mutations can cause progressive auditory neuropathy: A new gene of auditory synaptopathy. Sci. Rep. 7, 1–11 (2017).
Sloan-Heggen, C. M. et al. Comprehensive genetic testing in the clinical evaluation of 1119 patients with hearing loss. Hum. Genet. 135, 441–450. https://doi.org/10.1007/s00439-016-1648-8 (2016).
Park, J. H. et al. Exploration of molecular genetic etiology for Korean cochlear implantees with severe to profound hearing loss and its implication. Orphanet J. Rare Dis. 9, 1–14 (2014).
Oza, A. M. et al. Expert specification of the ACMG/AMP variant interpretation guidelines for genetic hearing loss. Hum. Mutat. 39, 1593–1613. https://doi.org/10.1002/humu.23630 (2018).
Richards, S. et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17, 405–424. https://doi.org/10.1038/gim.2015.30 (2015).
Perry, B. P. et al. Sensorineural hearing loss in people with deletions of 18q. Otol. Neurotol. 35, 782 (2014).
Cody, J. D. et al. American Journal of Medical Genetics Part C Seminars in Medical Genetics 265–280 (Wiley, 2021).
Braga, H. et al. Congenital hypothyroidism as a risk factor for hearing and parents’ knowledge about its impact on hearing. J. Otol. 16, 71 (2020).
Loevner, L. A., Shapiro, R. M., Grossman, R. I., Overhauser, J. & Kamholz, J. J. A. White matter changes associated with deletions of the long arm of chromosome 18 (18q-syndrome): A dysmyelinating disorder? Am. J. Neuroradiol. 17, 1843–1848 (1996).
Zaghloul, N. & Ahmed, M. J. Pathophysiology of periventricular leukomalacia: What we learned from animal models. Neural Regen. Res. 12, 1795 (2017).
Xiang, Y. B. et al. Next-generation sequencing identifies rare pathogenic and novel candidate variants in a cohort of Chinese patients with syndromic or nonsyndromic hearing loss. Mol. Genet. Genomic Med. 8, e1539 (2020).
Mekkawy, M. K. et al. Clinical and genetic characterization of ten Egyptian patients with Wolf-Hirschhorn syndrome and review of literature. Mol. Genet. Genomic Med. 9, e1546 (2020).
Group, R. C. D. S. (2019).
Han, K. H. et al. Establishment of a flexible real-time polymerase chain reaction-based platform for detecting prevalent deafness mutations associated with variable degree of sensorineural hearing loss in Koreans. PLoS ONE 11, e0161756. https://doi.org/10.1371/journal.pone.0161756 (2016).
Lee, S. Y. et al. Flexible real-time polymerase chain reaction-based platforms for detecting deafness mutations in Koreans: A proposed guideline for the etiologic diagnosis of auditory neuropathy spectrum disorder. Diagnostics (Basel). https://doi.org/10.3390/diagnostics10090672 (2020).
Korver, A. M. et al. Congenital hearing loss. Nat. Rev. Dis. Primers 3, 16094. https://doi.org/10.1038/nrdp.2016.94 (2017).
Weng, W., Reid, A., Thompson, A. & Kuthubutheen, J. Evaluating the success of a newly introduced feed and wrap protocol in magnetic resonance imaging scanning of the temporal bone for the evaluation of congenital sensorineural hearing loss. Int. J. Pediatr. Otorhinolaryngol. 132, 109910. https://doi.org/10.1016/j.ijporl.2020.109910 (2020).
Billings, K. R. & Kenna, M. A. Causes of pediatric sensorineural hearing loss: Yesterday and today. Arch. Otolaryngol. Head Neck Surg. 125, 517–521. https://doi.org/10.1001/archotol.125.5.517 (1999).
Lieu, J. E. C., Kenna, M., Anne, S. & Davidson, L. Hearing loss in children: A review. JAMA 324, 2195–2205. https://doi.org/10.1001/jama.2020.17647 (2020).
Liming, B. J. et al. International Pediatric Otolaryngology Group (IPOG) consensus recommendations: Hearing loss in the pediatric patient. Int. J. Pediatr. Otorhinolaryngol. 90, 251–258. https://doi.org/10.1016/j.ijporl.2016.09.016 (2016).
Han, J. J. et al. Prediction of the outcome of cochlear implantation in the patients with congenital cytomegalovirus infection based on magnetic resonance imaging characteristics. J. Clin. Med. https://doi.org/10.3390/jcm8020136 (2019).
Kim, B. J. et al. Targeted exome sequencing of deafness genes after failure of auditory phenotype-driven candidate gene screening. Otal. Neurotol. 36, 1096–1102 (2015).
Choi, B. Y. et al. Diagnostic application of targeted resequencing for familial nonsyndromic hearing loss. PLoS ONE 8, e68692. https://doi.org/10.1371/journal.pone.0068692 (2013).
Lee, S. Y. et al. Severe or profound sensorineural hearing loss caused by novel USH2A variants in Korea: Potential genotype-phenotype correlation. Clin. Exp. Otorhinolaryngol. 13, 113–122. https://doi.org/10.21053/ceo.2019.00990 (2020).
Rincon, A., Paez-Rojas, P. & Suarez-Obando, F. 8q222q223 microdeletion syndrome associated with hearing loss and intractable epilepsy. Case Rep. Genet. 2019, 7608348. https://doi.org/10.1155/2019/7608348 (2019).
Acknowledgements
The authors acknowledge Pf. Seung Ha Oh, Pf. Jun Ho Lee and Dr. Doo Yi Oh for their technical support.
Funding
This research was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (2018R1A2B2001054 to B.Y.C.) (2021R1A2C2092038 to B.Y.C.) and the research funds of Seoul National University Bundang Hospital (16-2019-006 to B.Y.C) (13-2018-015 to B.Y.C). The funding bodies had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
The final manuscript has been seen and approved by all the authors, and they have given necessary attention to the manuscript to ensure the integrity of the work. Y.K., H.W.J., Y.S.K. and B.Y.C. designed and performed experiments, analyzed data and wrote the paper; S.Y.L., N.Y., J.H.H., M.Y.K., B.H.K., Y.J.B., B.J.K. and B.Y.C. collected and analyzed data including genetic profile; H.W.J. performed the statistical analysis; H.Y.C., M.C. and J.W.K. provided critical revision. These data have not been previously published and are not submitted elsewhere for publication.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kim, Y.S., Kim, Y., Jeon, H.W. et al. Full etiologic spectrum of pediatric severe to profound hearing loss of consecutive 119 cases. Sci Rep 12, 12335 (2022). https://doi.org/10.1038/s41598-022-16421-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-022-16421-x
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
