
Abstract
Tuberous sclerosis complex (TSC) is an autosomal dominant disorder characterized by hamartomas in the skin and other organs, including brain, heart, lung, kidney and bones. TSC is caused by mutations in TSC1 and TSC2. Here, we present the TSC1 and TSC2 variants identified in 168 Danish individuals out of a cohort of 327 individuals suspected of TSC. A total of 137 predicted pathogenic or likely pathogenic variants were identified: 33 different TSC1 variants in 42 patients, and 104 different TSC2 variants in 126 patients. In 40 cases (24%), the identified predicted pathogenic variant had not been described previously. In total, 33 novel variants in TSC2 and 7 novel variants in TSC1 were identified. To assist in the classification of 11 TSC2 variants, we investigated the effects of these variants in an in vitro functional assay. Based on the functional results, as well as population and genetic data, we classified 8 variants as likely to be pathogenic and 3 as likely to be benign.
Similar content being viewed by others
Introduction
Tuberous sclerosis complex (TSC) is an autosomal dominant disorder of high penetrance with an incidence of 1:6,000–1:10,000 and an estimated prevalence of 1:14,000–1:25,0001,2. TSC is characterized by the presence of mainly benign tumors that can affect multiple organ systems e.g. the central nervous system, heart, kidney, lung, bone and skin. TSC patients are phenotypically and genetically heterogeneous and there is considerable variation in the number, location and size of the different TSC-associated lesions. Mutations in one of two genes, TSC1 (OMIM#191100) and TSC2 (OMIM#191092), cause TSC3,4.
TSC1 is located on chromosome 9q34 and consists of 23 exons, which encode the 130 kDa TSC1 protein, hamartin. TSC2 is located on chromosome 16p13.3 and consists of 42 exons which encode the 200 kDa TSC2 protein, tuberin. TSC1 and TSC2, together with a third subunit, TBC1D75, form a stable protein complex, the TSC complex. The TSC complex is a GTPase-activating protein (GAP) specific for the small GTPase, Ras homologue enriched in brain (RHEB)6. Active RHEB is involved in the activation of the mechanistic target of rapamycin (mTOR) complex 1 (mTORC1), a critical regulator of anabolic processes such as protein and lipid synthesis7. The TSC complex inactivates RHEB to down-regulate mTORC1 signaling and inhibit cell growth. TSC-associated tumors are characterized by increased phosphorylation of S6, elongation factor 4E binding protein 1 (4E-BP1), p70 S6 kinase (S6K) and other downstream targets of mTORC1 (Fig. 1).

Tuberous Sclerosis Complex signaling. The TSC complex is a central node in mTORC1 signaling and receives inputs from multiple cellular pathways that influencing TSC complex activity. mTORC1 also responds to amino acids through the RAG GTPases (not shown). However, the amino acid dependent regulation of mTORC1 is independent of the TSC complex. Inhibitory and activating phosphorylation events are indicated.
Approximately 2/3 of TSC cases are due to sporadic de novo germline mutations2. TSC2 mutations are identified in the majority of TSC patients and, in general, cause a more severe phenotype than TSC1 mutations8,9. Exceptions to this rule are however observed10,11.
Large genomic deletions that affect both TSC2 and the adjacent PKD1 (OMIM# 601313) locus are associated with a subset of patients with TSC and severe, early-onset autosomal dominant polycystic kidney disease.
While a pathogenic TSC1 or TSC2 variant can be identified in most TSC patients, in 10–15% of affected individuals conventional molecular testing fails to identify the causative mutation. Recent studies indicate that this is most likely because these individuals are either mosaic for a pathogenic TSC1 or TSC2 variant, or have a pathogenic variant in a region of TSC1 or TSC2 that is not routinely screened12,13,14. In addition, it is not always clear whether an identified TSC1 or TSC2 variant is disease-causing. In such cases, functional assessment can help establish pathogenicity15.
In this report, we present the molecular test results of a cohort of 327 Danish patients suspected of TSC. Furthermore, the effects of eleven variants on the ability of the TSC complex to inhibit mTORC1 activity, were investigated using an in vitro functional assay.
Material and methods
Subjects
The project was performed according to the Declaration of Helsinki. Agreement was obtained from all the participants or, if under 18, from a parent, prior to molecular genetic testing. Between 2003 and 2018, 327 individuals suspected of TSC were identified in pediatric and clinical genetic departments in Denmark and referred to Copenhagen University Hospital for molecular diagnosis. Some patients fulfilled the clinical criteria for definite TSC16, whereas others only had one of the major features of TSC. In a large number of patients (approximately 80%) only very limited clinical information was provided. A total of 6 prenatal cases in which rhabdomyomas were revealed by ultrasound scanning were also included. Genomic DNA was prepared by standard methods from peripheral blood, or tissue, as described previously17.
Screening for pathogenic variants
Screening for mutations in TSC1 and TSC2 was performed either by denaturing gradient gel electrophoresis (DGGE) (before 2006) as described previously17, by direct Sanger sequencing of PCR products of all coding exons plus 20 bp of flanking intronic sequences (in the period 2006–2017), or since 2017, by Next Generation Sequencing (NGS) on a MiSeq Benchtop Sequencer (Illumina) following HaloPlex Custom Region Enrichment (Agilent). NGS data was analyzed in SureCall software (Agilent) using a BWA MEM aligner and SNPPET SNP caller. At least 99% of the target region (exon sequences as well as 20 base pairs of flanking intron sequences) had a read depth >20. Variants identified by DGGE or NGS and selected for reporting were verified by Sanger sequencing. The primers used for PCR amplification of the individual exons are listed in Supplementary Tables 1 and 2. Single and multiple exon deletions and duplications were detected by multiplex ligation probe amplification (MLPA) using the SALSA MLPA P124-TSC1 and P046-TSC2 probe-mixes (MRC Holland).
Nomenclature
The nomenclature of the identified mutations is given according to HGVS (www.hgvs.org) guidelines. Nucleotide numbering for TSC1 is according to reference transcript NM_000368.4, and for TSC2 according to NM_000548.3. In both cases, c.1 is the A of the ATG translation initiation codon, and p.M1 is the initiation codon. Genomic reference sequences were NG_012386.1 for TSC1 and NG_005895.1 for TSC2. TSC1 contains two non-coding exons (exon 1 and exon 2). TSC2 contains one non-coding exon (exon 1).
Functional investigation
We derived expression constructs for TSC2 variants by site-directed mutagenesis (SDM) of a wild-type TSC2 expression construct18. All constructs were verified by sequencing of the complete TSC2 open reading frame. Each variant was tested in at least 3 separate transfection experiments in 3H9-1B1 (TSC2:/TSC1 double knockout HEK 293 T) cells19. Cells expressing the variants were compared to cells expressing wild-type TSC2, the pathogenic TSC2 p.Arg611Gln variant, and cells not expressing TSC2 (TSC1/S6K only). A S6K reporter and TSC1 expression constructs (both encoding a myc epitope tag) were included in each transfection mixture. Transfections were performed in 24-well dishes (1 × 105–2 × 105 cells per well). Cells were lysed 18 hours after transfection in 50 mM Tris-HCl (pH 7.6), 100 mM NaCl, 50 mM NaF, 1% Triton-X-100, 1 mM EDTA and Complete protease inhibitors (Roche, Basel, Switzerland). After centrifugation (10 000 x g for 10 minutes at 4 °C), the cleared cell lysates were separated by SDS-PAGE and transferred to nitrocellulose filters. The levels of the expressed TSC2, TSC1, total S6K and T389-phosphorylated S6K were estimated by immunoblotting using the following antibodies: 1A5 anti-Thr389 phospho- p70 S6 kinase (S6K) mouse monoclonal, 9B11 anti-myc tag mouse monoclonal, anti-myc tag rabbit polyclonal (Cell Signaling Technology, Danvers, MA, USA), and anti-TSC1 and anti-TSC2 rabbit polyclonal20. Secondary antibodies were from Li-Cor Biosciences (Lincoln, NE) and blots were scanned using the Odyssey scanner (Li-Cor Biosciences, Lincoln, NE). Signal intensities were measured and normalized to the signals corresponding to wild-type TSC2.
Predicting pathogenicity
Identified sequence variations were classified into five categories: class 5 (pathogenic), class 4 (likely pathogenic), class 3 (variant of unknown significance), class 2 (likely benign), and class 1 (benign), according to the guidelines of ACMG21. Variants were classified as pathogenic based on allelic frequency, and the predicted effect of the variant on TSC1 or TSC2. Variants that occur relatively often in the general population (gnomAD:>1:5000), are unlikely to cause TSC and were classified as benign and only reported if the variant had been previously categorized as pathogenic in the HGMD database. Information obtained from the Leiden Open Variation Database (LOVD) (http://chromium.lovd.nl/LOVD2/TSC/home.php?select_db=TSC1) was used to help variant classification. Rare (gnomAD: <1:5000) variants which led to a frameshift, and/or created a stop codon were classified as pathogenic or likely pathogenic. Determining the pathogenicity of rare missense variants and in-frame duplications or deletions, is often difficult. In addition to allele frequency, these variants were classified according to the results of in vitro functional assessment. To investigate possible effects of the identified variants on splicing, we used several web-based tools (MaxEntScan22, NNSPLICE23, and Human Splice Finder24) combined in Alamut Visual biosoftware (http://www.interactive-biosoftware.com/alamut-visual/). Rare variants resulting in a 99–100% reduction in the prediction score were classified as pathogenic. Otherwise we classified the variant as a variant of uncertain clinical significance (VUS).
Ethics statement
This study is approved by the local institutional review board, Pactius (P-2019-304). No other permission was required. Written informed consent was waived. All methods were carried out in accordance with the Copenhagen University Hospital’s, Rigshospitalets, guidelines.
Results
Identification of sequence variants
Molecular testing of TSC1 and TSC2 in 327 Danish individuals suspected of TSC resulted in the identification of 137 different variants in a total of 168 individuals. The TSC1 and TSC2 variants identified in our cohort are summarized in Supplementary Tables 3 and 4.
The majority of the variants had been reported previously in other TSC cohorts but 45 were novel, as defined by their absence from the HGMD (http://www.hgmd.cf.ac.uk/ac/index.php), LOVD (Leiden Open Variation Database (http://chromium.lovd.nl/LOVD2/TSC/home.php?select_db=TSC1)), and Clin Var (https://www.ncbi.nlm.nih.gov/clinvar/). The 8 novel TSC1 variants and 37 novel TSC2 variants are listed in Tables 1 and 2.
Most of the new variants lead to formation of a premature stop codon. This was the case for 20 of the novel TSC2 variants and for six of the novel TSC1 variants. Five TSC2 variants were predicted to lead to an amino acid substitution or an in-frame deletion/insertion.
Classification of variants
Unlike variants leading to premature termination of translation which can mostly be classified as pathogenic or likely pathogenic, classification of missense and in-frame deletion/insertion variants can be difficult. Functional investigation of several of the identified TSC1 and TSC2 variants had been performed previously. The TSC1 p.(Leu50Pro) variant15, and the TSC2 p.(Arg611Gln)25, p.(Phe897Ser)26, p.(Arg905Trp)26, p.(Arg1032Pro)15, p.(Gln1554His)17, p.(Arg1570Trp)17, p.(Gly1642Asp)15, p.(Ser1653Phe)27, p.(Pro1675Leu)27, p.(Pro1709Leu)27, p.(Arg1743Gln)27, p.(Arg1743Trp)28 and p.(His1746_Arg1751del)27 variants have all been found to disrupt TSC complex function (Supplementary Tables 3 and 4).
For the TSC2 p.(Leu292Pro), p.(Glu1558Lys), p.(His1620Tyr), p.(Lys1638del), p.(Asn1681Lys) and p.(Pro1675Leu) variants, de novo occurrence was noted in LOVD, indicating pathogenicity.
To help classify the remaining missense variants and in-frame deletions/insertions identified in our cohort we performed in vitro functional assessment. We derived expression constructs for the following TSC2 variants: c.815C > A, p.(Ala272Asp), c.1283_1285del, p.(Ser428del), c.1699_1701dup, p.(Leu568dup), c.2326T > G, p.(Tyr776Asp), c.1292C > T, p.(Ala431Val) and c.1915C > T, p.(Arg639Trp). The c.1291C > T, p.(Ala431Val) and c.1915C > T, p.(Arg639Trp) variants were inherited in cis on the paternal allele. Therefore, we derived an expression construct containing both variants, referred to as p.(Ala431Val/Arg639Trp). Furthermore, expression constructs were generated for the previously identified TSC2 c.856A > G, p.(Met286Val), c.1220_1240del, p.(Tyr407_Arg413del), c.1853T > C, p.(Leu618Pro), c.4672G > A, p.(Glu1558Lys) and c.5043C > G, p.(Asn1681Lys) variants.
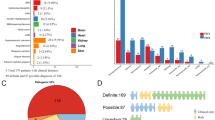
Variants were expressed in 3H9-1B1 (TSC2:TSC1 double knockout HEK 293 T) cells together with TSC1 and a S6K reporter construct. The levels of the exogenous TSC2, TSC1, total S6K and T389-phosphorylated S6K proteins were estimated by immunoblotting. The stability of the expressed TSC2 and the stability of the TSC complex were estimated from the TSC2 (Fig. 2A) and TSC1 (Fig. 2B) signals respectively. The total S6K signal was used to estimate the relative transfection efficiency (Fig. 2C) and the ratio of the signals for T389-phosphorylated S6K and total S6K (T389/S6K ratio) was used to estimate mTORC1 activity (Fig. 2D).

Functional assessment of missense variants, Functional assessment of the TSC2 (NM_000548.3) variants; c.815C > A, p.(Ala272Asp), c.856A > G, p.(Met286Val), c.1220_1240del, p.(Tyr407_Arg413del), c.1283_1285del, p.(Ser428del), c.1292C > T, p.(Ala431Val), c.1699_1701dup, p.(Leu598dup), c.1853T > C, p.(Leu618Pro), c.1915C > T, p.(Arg639Trp), c.1292C > T/c.1915C > T, p.(Ala431Val/Arg639Trp), c.2326T > G, p.(Tyr776Asp), c.4672G > A, p.(Glu1558Lys) and c.5043C > G, p.(Asn1681Lys). 3H9-1B1 (TSC2/TSC1 double knockout HEK 293 T) cells were transfected with the indicated combinations of expression constructs. Twenty-four hours after transfection the cells were harvested, and the cleared cell lysates analysed by immunoblotting. The signals for TSC2, TSC1, total S6K (S6K) and T389-phosphorylated S6K (T389) were determined per variant, relative to the wild-type control (TSC2) in 2 independent experiments. The mean TSC2 (A), TSC1 (B) and S6K (C) signals and mean T389/S6K ratio (D) are shown for each variant. In each case the dotted line indicates the signal/ratio for wild-type TSC2 (=1.0). Error bars represent the standard error of the mean; variants that were significantly different from wild-type TSC2 are indicated with an asterisk (P < 0.025; Student’s paired t-test). Amino acid changes are given according to TSC2 cDNA reference transcript sequence NM_000548.3.
The TSC2 p.(Ala272Asp), p.(Tyr407_Arg413del), p.(Ser428del), p.(Leu568dup), p.(Leu618Pro), p.(Tyr776Asp), p.(Glu1558Lys) and p.(Asn1681Lys) variants disrupted TSC complex function. In each case, mTORC1 activity, as estimated from the T389/S6K ratio, was significantly increased, compared to wild-type TSC2. In addition, the p.(Ala272Asp), p.(Tyr407_Arg413del), p.(Ser428del), p.(Leu568dup), p. (Leu618Pro) and p.(Tyr776Asp) variants were associated with significantly decreased TSC1 signals, most likely due to their inability to interact with and stabilize TSC1. The TSC2 p.(Met286Val), p.(Ala431Val), p.(Arg639Trp) and p.(Ala431Val;Arg639Trp) variants did not significantly disrupt the TSC complex dependent inhibition of mTORC1 activity in our assay, nor did they significantly affect TSC1 or TSC2 signals.
The p.(Met286Val), p.(Ala431Val), and p.(Arg639Trp) variants are all reported in gnomAD with an individual overall frequency of 0.18%, 0.038% and 0.0040% respectively. In comparison, none of the other missense or in-frame variants were present in gnomAD. For the p.(Met286Val) variant, conflicting conclusions regarding pathogenicity were registered. The variant was originally classified as pathogenic or most likely pathogenic in the HGMD database, but as neutral in the LOVD database. The gnomAD frequency for this variant is as high as 1.9% in the East Asian population. Based on the observed activity and stability of the p.(Met286Val) variant in our assay, combined with the gnomAD frequency, we classified the p.(Met286Val), p.(Ala431Val), p.(Arg639Trp) and p.(Ala431Val;Arg639Trp) variants as benign or most likely benign (Table 3).
Variants located in and around canonical splice sites can be difficult to classify. We identified five novel variants, including one in TSC1 and four in TSC2, that were absent from gnomAD and were predicted to be >99% likely to affect splicing according to web-based tools (MaxEntScan22, NNSPLICE23, and Human Splice Finder24) combined in Alamut Visual biosoftware. We classified these variants as pathogenic or likely pathogenic. Furthermore, we identified the TSC1 c.2042-5 A > G variant, which was predicted to affect splicing with 34% probability. This variant has been identified previously as a de novo change in an individual with TSC (http://chromium.lovd.nl/LOVD2/TSC/home.php?select_db=TSC1). Therefore, we classified the variant as likely to be pathogenic (Supplementary Tables 3 and 4).
In addition, we identified the novel TSC1 c.2626-4 T > G and TSC2 c.976-16 C > A, c.3284 + 3 G > A and c.5260-34_5260-10del variants, as well as the previously identified TSC2 c.3883 + 5 C > T variant. These variants are all predicted to affect splicing but at a probability significantly below 100% (between 1% and 68%). We classified all these variants as VUS. We classified also the novel TSC2 c.336 + 14 C > T variant, predicted to have no effect on splicing, as VUS (Supplementary Tables 3 and 4). Unfortunately it was not possible to investigate the effects of the variants on TSC1 and TSC2 pre-mRNA splicing in the corresponding affected individuals because no RNA was available from these individuals11.
In summary, seven novel predicted pathogenic variants were identified in TSC1 (Table 1) and 33 in TSC2 (Table 2). Furthermore, five variants predicted to be of uncertain pathogenicity were identified.
Discussion
We have reviewed the TSC1 and TSC2 variants identified in a cohort of Danish patients, we identified 137 different mutations in 168 TSC patients from a cohort of 327 Danish individuals suspected of TSC. In our cohort, 33 of the 137 different suspected pathogenic variants were identified in TSC1 (24%) while 104 were identified in TSC2 (76%) (Table 1). This distribution is in accordance with previous publications8,29,30.
In total, 33 different predicted pathogenic TSC1 variants were identified in 42 individuals and 106 different predicted pathogenic TSC2 variants were identified in 126 individuals. In addition to the predicted pathogenic variants, 8 variants with uncertain pathogenicity were identified (Supplementary Tables 3 and 4). Twenty variants, 7 in TSC1 and 13 in TSC2, were identified more than once in our cohort. The most common variants in TSC1 were the nonsense mutations c.1525C > T, p.(Arg509*) in exon 15 and c.2074C > T, p.(Arg692*) in exon 17, both identified in three unrelated patients. In TSC2, c.1832G > A, p.(Arg611Gln) located in exon 17 and c.5238_5255del, p.(His1746_Arg1751del) in exon 41 were the most common variants, identified in 5 unrelated patients each. In six cases molecular testing was performed on individuals in whom cardiac rhabdomyomas had been revealed by prenatal ultrasound scanning. Pathogenic variants were identified in five of these cases: TSC2 c.1832G > A, p.(Arg611Gln), c.4537G > T, p.(Glu1513*), c.4993C > T, p.(Gln1665*), c.5024C > T, p.(Pro1675Leu), and c.(1-?)_(975 + ?)del (del Ex2-10).
All the predicted pathogenic variants identified in TSC1 were small changes, involving a single base pair (30 cases), two base pairs (2 cases) or 23 base pairs (1 case). In 25 cases, the identified change created a premature stop codon, and in seven cases, the variant was in a region important for splicing. Only a single variant predicted to lead to an amino acid substitution was identified. In TSC2, 73 variants affected a single base-pair and 22 variants affected between two and 33 base-pairs. In 54 of these cases a premature stop codon was created, in 15 cases the variant was in a region important for splicing and in 24 cases the variant was predicted to change the amino acid sequence. Furthermore, 9 variants leading to large deletions of one or more exons of TSC2 were identified.
Most of the identified variants had been identified previously in other TSC patients, but a total of 33 novel predicted pathogenic variants were identified in TSC2 and 7 novel predicted pathogenic variants were identified in TSC1.
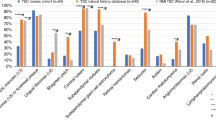
The observed distributions of pathogenic TSC1 and TSC2 variants, shown in Fig. 3, are similar to previous studies8,29,30. TSC2 variants were scattered all over the gene and TSC1 variants were most often identified in exons 15 and 18. Although most of the variants were either nonsense changes or deletions, missense mutations were often found in TSC2. In contrast, only a single missense variant was identified in TSC1.

Structural and functional domains of TSC1 and TSC2 and the distribution of the TSC1 and TSC2 variants identified in this study. TSC1 consists of a conserved N-terminal domain (NTD) and a large domain that is critical for the interaction with the N-terminal of TSC2, and a domain at the C-terminus important for interacting with the TBC1D7. TSC2 is the catalytic subunit of the TSC complex and acts as a GTPase activating protein towards RHEB. The active site is indicated. The types and frequencies of the TSC1 and TSC2 variants identified in in our cohort are illustrated. In total, 33 different pathogenic variants in TSC1 and 104 different pathogenic variants in TSC2 were identified in 168 patients. The variants were categorized as either missense, nonsense, splicing, small deletions, small insertions, or small delins. Gross deletions (9 in TSC2) are not shown. The figure is modified from previous publications34,35,36.
Missense and small in-frame indel variants encode proteins that only differ from the wild-type proteins by a few amino acids. If a single amino acid substitution is pathogenic, then it is likely that that amino acid and/or the surrounding peptide sequence is functionally important. The only missense variant in TSC1 identified in this study, p.(Leu50Pro), affects the N-terminal domain (NTD) of TSC1, resulting in destabilization of the TSC complex31. A high proportion, (11/19; ~60%) of the TSC2 missense variants identified map to exons 36–41, encoding amino acids 1555–1704, that contain the GAP domain (amino acids 1533–1722), even though this region only accounts for ~11% of the total coding region. Furthermore, two pathogenic missense variants affecting this region were identified in multiple cases. These results indicate that the NTD region of TSC1 and the GAP domain of TSC2 are critical for TSC complex function. Our functional analysis of the p.(Glu1558Lys) and p.(Asn1681Lys) variants is in line with this hypothesis.
Previous studies report a mutation detection rate of 74–83% in TSC8,29,30,32. In the present study we identified a mutation in only 52% of the patients. This is in contrast to a previous publication from our laboratory17, where 65 Danish patients who had been clinically diagnosed with TSC, were investigated and pathogenic mutations were identified in 51 patients (78%). In the present study only limited clinical information was available, whereas all the patients included in our previous study fulfilled the diagnostic criteria for TSC. At that time NGS was still not available and TSC1 and TSC2 molecular screening was difficult and time consuming. This might have forced the clinician to carefully evaluate their patient for signs of TSC prior to referral for molecular genetic investigation. Today, with NGS, the laboratory work is reduced, and the turn-around time faster. This might lead to increased numbers of patients being referred who do not fulfill the clinical diagnostic criteria for TSC. The large number of cases without identification of a pathogenic TSC1 or TSC2 variant does not exclude the possibility that these individuals have TSC. The variant could be located in a region not tested in any of our set ups, like deep within an intron, or the variants might be present in mosaic form, in a limited number of patient cells. Indeed, recent studies indicate that at least 50% of TSC cases who fulfill the clinical diagnostic criteria and do not have a mutation identified by standard molecular testing will have a pathogenic TSC1 or TSC2 variant in mosaic form12,13. Only a minor fraction of the cases presented here were screened using NGS. So far, we have identified mosaicism in one case. The TSC2 c.1283_1285del variant was identified in 84 out of 1082 reads (8%) and was verified by PCR using deletion specific primers. The further application of NGS should lead to an increase in the number of clarified cases. Also, re-investigation of mutation-negative cases might reveal additional pathogenic variants in mosaic form.
Careful re-assessment of all the previously published mutations identified in our cohort revealed conflicting interpretations of pathogenicity. The release of the genome aggregation database (gnomAD) which is comprised of data from 123,136 individuals and whole genome sequencing from 15,496 individuals33 has increased our knowledge about the frequencies of many single nucleotide variants (SNPs), and led us to re-classify some variants as unlikely to be disease causing. Furthermore, assessment of pathogenicity using functional studies helped support the genetic and clinical data. For example, re-classification of the TSC2, p.(Met286Val) variant as benign was supported by both the frequency data and the functional assessment.
Reliable classification of identified variants is critically important. Functional in vitro investigation is an important contribution to classification of variants leading to missense changes and in frame deletions and insertions. Routine investigation of potential splice-site mutations by reverse-transcription (RT)-PCR performed on RNA isolated from the affected individuals might also help improve the classification of variants, particularly those located in splice site regions.
References
Hallett, L., Foster, T., Liu, Z., Blieden, M. & Valentim, J. Burden of disease and unmet needs in tuberous sclerosis complex with neurological manifestations: systematic review. Curr. Med. Res. Opin. 27, 1571–1583 (2011).
Osborne, J. P., Fryer, A. & Webb, D. Epidemiology of Tuberous Sclerosis. Ann. N. Y. Acad. Sci. 615, 125–127 (1991).
The European Chromosome 16 Tuberous Sclerosis Consortium. Identification and characterization of the tuberous sclerosis gene on chromosome 16. Cell 75, 1305–1315 (1993).
van Slegtenhorst, M. et al. Identification of the tuberous sclerosis gene TSC1 on chromosome 9q34. Science 277, 805–8 (1997).
Gai, Z. et al. Structure of the TBC1D7–TSC1 complex reveals that TBC1D7 stabilizes dimerization of the TSC1 C-terminal coiled coil region. J. Mol. Cell Biol. 8, 411–425 (2016).
Inoki, K., Li, Y., Xu, T. & Guan, K.-L. Rheb GTPase is a direct target of TSC2 GAP activity and regulates mTOR signaling. Genes Dev. 17, 1829–34 (2003).
Laplante, M. & Sabatini, D. M. MTOR signaling in growth control and disease. Cell 149, 274–293 (2012).
Dabora, S. L. et al. Mutational analysis in a cohort of 224 tuberous sclerosis patients indicates increased severity of TSC2, compared with TSC1, disease in multiple organs. Am. J. Hum. Genet. 68, 64–80 (2001).
Au, K. S. et al. Genotype/phenotype correlation in 325 individuals referred for a diagnosis of tuberous sclerosis complex in the United States. Genet. Med. 9, 88–100 (2007).
Peron, A., Au, K. S. & Northrup, H. Genetics, genomics, and genotype–phenotype correlations of TSC: Insights for clinical practice. Am. J. Med. Genet. Part C Semin. Med. Genet. 178, 281–290 (2018).
Møller, L. B. et al. Development of hypomelanotic macules is associated with constitutive activated mTORC1 in tuberous sclerosis complex. Mol. Genet. Metab. 120, 384–391 (2017).
Tyburczy, M. E. et al. Mosaic and Intronic Mutations in TSC1/TSC2 Explain the Majority of TSC Patients with No Mutation Identified by Conventional Testing. PLOS Genet. 11, e1005637 (2015).
Nellist, M. et al. Targeted Next Generation Sequencing reveals previously unidentified TSC1 and TSC2 mutations. BMC Med. Genet. 16, 10 (2015).
Qin, W. et al. Ultra deep sequencing detects a low rate of mosaic mutations in tuberous sclerosis complex. Hum. Genet. 127, 573–582 (2010).
Hoogeveen-Westerveld, M. et al. Functional assessment of variants in the TSC1 and TSC2 genes identified in individuals with Tuberous Sclerosis Complex. Hum. Mutat. 32, 424–435 (2011).
Northrup, H. et al. Tuberous sclerosis complex diagnostic criteria update: Recommendations of the 2012 international tuberous sclerosis complex consensus conference. Pediatr. Neurol. 49, 243–254 (2013).
Rendtorff, N. D. et al. Analysis of 65 tuberous sclerosis complex (TSC) patients by TSC2 DGGE, TSC1 / TSC2 MLPA, and TSC1 long-range PCR sequencing, and report of 28 novel mutations. Hum. Mutat. 26, 374–383 (2005).
Ekong, R. et al. Variants Within TSC2 Exons 25 and 31 Are Very Unlikely to Cause Clinically Diagnosable Tuberous Sclerosis. Hum. Mutat. 37, 364–370 (2016).
Dufner Almeida, L. G. et al. Comparison of the functional and structural characteristics of rare TSC2 variants with clinical and genetic findings. Hum. Mutat., https://doi.org/10.1002/humu.23963 (2019).
van Slegtenhorst, M. et al. Interaction between hamartin and tuberin, the TSC1 and TSC2 gene products. Hum. Mol. Genet. 7, 1053–1057 (1998).
Richards, S. et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17, 405–423 (2015).
Yeo, G. & Burge, C. B. Maximum entropy modeling of short sequence motifs with applications to RNA splicing signals. J. Comput. Biol. 11, 377–394 (2004).
Reese, M. G., Eeckman, F. H., Kulp, D. & Haussler, D. Improved splice site detection in Genie. J. Comput. Biol. 4, 311–323 (1997).
Desmet, F.-O. et al. Human Splicing Finder: an online bioinformatics tool to predict splicing signals. Nucleic Acids Res. 37, e67 (2009).
Nellist, M. et al. TSC2 missense mutations inhibit tuberin phosphorylation and prevent formation of the tuberin-hamartin complex. Hum. Mol. Genet. 10, 2889–2898 (2001).
Jansen, A. C. et al. Unusually mild tuberous sclerosis phenotype is associated with TSC2 R905Q mutation. Ann. Neurol. 60, 528–539 (2006).
Hoogeveen-Westerveld, M. et al. Functional Assessment of TSC 2 Variants Identified in Individuals with Tuberous Sclerosis Complex. Hum. Mutat. 34, 167–175 (2013).
Hung, C.-C. et al. Molecular and clinical analyses of 84 patients with tuberous sclerosis complex. BMC Med. Genet. 7, 72 (2006).
Sancak, O. et al. Mutational analysis of the TSC1 and TSC2 genes in a diagnostic setting: genotype – phenotype correlations and comparison of diagnostic DNA techniques in Tuberous Sclerosis Complex. Eur. J. Hum. Genet. 13, 731–741 (2005).
Jones, A. C. et al. Comprehensive Mutation Analysis of TSC1 and TSC2—and Phenotypic Correlations in 150 Families with Tuberous Sclerosis. Am. J. Hum. Genet. 64, 1305–1315 (2002).
Sun, W. et al. Crystal structure of the yeast TSC1 core domain and implications for tuberous sclerosis pathological mutations. Nat. Commun. 4, 2135 (2013).
Lin, S. et al. Tuberous Sclerosis Complex in Chinese patients: Phenotypic analysis and mutational screening of TSC1/TSC2 genes. Seizure 71, 322–327 (2019).
Karczewski, K. J. et al. Variation across 141,456 human exomes and genomes reveals the spectrum of loss-of-function intolerance across human protein-coding genes. bioRxiv 531210, https://doi.org/10.1101/531210 (2019).
Rosset, C. et al. TSC1 and TSC2 gene mutations and their implications for treatment in Tuberous Sclerosis Complex: a review. Genet. Mol. Biol. 40, 69–79 (2017).
Santiago Lima, A. J. et al. Identification of Regions Critical for the Integrity of the TSC1-TSC2-TBC1D7 Complex. PLoS One 9, e93940 (2014).
Soucek, T. et al. Tuberous sclerosis causing mutants of the TSC2 gene product affect proliferation and p27 expression. Oncogene 20, 4904–4909 (2001).
Acknowledgements
We thank Susan Peters for proof-reading the manuscript and Pia Hougaard and Pia Skovgaard for excellent technical assistance. The research was funded by the Danish council for independent research (#12-127196) and Aase and Ejnar Danielsens Fond (Denmark #10-000748), the Michelle Foundation (The Netherlands), TS Association (UK)(2016-P07), TS Alliance (USA)(06-16) and CAPES (Brazil)(Process:88881.132401/2016-01).
Author information
Authors and Affiliations
Contributions
T.R., L.B.M., L.J.L. and C.A.B.H. performed the figures. L.B.M., M.D., J.E. and L.R. carried out the clinical investigation of the patients and collected samples. S.N., L.G.D. and M.N. performed the functional investigations. T.R., L.B.M., B.S.G., L.J.L. and M.N. drafted the manuscript and all authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Rosengren, T., Nanhoe, S., de Almeida, L.G.D. et al. Mutational analysis of TSC1 and TSC2 in Danish patients with tuberous sclerosis complex. Sci Rep 10, 9909 (2020). https://doi.org/10.1038/s41598-020-66588-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-66588-4
This article is cited by
-
Renal phenotypes correlate with genotypes in unrelated individuals with tuberous sclerosis complex in China
Orphanet Journal of Rare Diseases (2022)
-
An Israeli tuberous sclerosis cohort: the efficacy of different anti-epileptic strategies
Child's Nervous System (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
