
Abstract
Purpose
To examine the prevalence and spectrum of mosaic variant allele frequency (MVAF) in tuberous sclerosis complex (TSC) patients with low-level mosaicism and correlate genetic findings with clinical features and transmission risk.
Methods
Massively parallel sequencing was performed on 39 mosaic TSC patients with 170 different tissue samples.
Results
TSC mosaic patients (MVAF: 0–10%, median 1.7% in blood DNA) had a milder and distinct clinical phenotype in comparison with other TSC series, with similar facial angiofibromas (92%) and kidney angiomyolipomas (83%), and fewer seizures, cortical tubers, and multiple other manifestations (p < 0.0001 for six features). MVAF of TSC1/TSC2 pathogenic variants was highly variable in different tissue samples. Remarkably, skin lesions were the most reliable tissue for variant identification, and 6 of 39 (15%) patients showed no evidence of the variant in blood. Semen analysis showed absence of the variant in 3 of 5 mosaic men. The expected distribution of MVAF in comparison with that observed here suggests that there is a considerable number of individuals with low-level mosaicism for a TSC2 pathogenic variant who are not recognized clinically.
Conclusion
Our findings provide information on variability in MVAF and risk of transmission that has broad implications for other mosaic genetic disorders.
Similar content being viewed by others

INTRODUCTION
Mosaicism occurs in all humans,1,2 but its occurrence at low allele frequencies, variability among different tissues, and rate of transmission of new, clinically relevant alleles have not been studied in great detail. Mosaic variant allele frequencies (MVAF) range from extremely low (<1%) to relatively high (20–40%), and likely depend on developmental timing of the mutational event.2,3,4,5,6,7 Mosaicism can be either generalized, seen in multiple cell types and organs, or be restricted to a single cell lineage, including the germ cell lineage.5,6,7,8
Tuberous sclerosis complex (TSC, MIM 191100 and 613254) is a neurodevelopmental tumor suppressor gene syndrome characterized by seizures, intellectual disability, and autism, as well as tumors involving the skin, heart, brain, kidneys, lungs, and other organs.9,10,11 TSC is due to inactivating variants in either TSC1 or TSC2, and follows the Knudson model of tumor development.10 Mosaicism is well-known in TSC,10,11 but its clinical implications have not been studied in detail. Mildly affected TSC patients of childbearing age with no variant identified (NMI) by conventional genetic testing often desire to know their variant status to inform reproductive decision making, prompting the current study. Previously, we demonstrated that 26 of 45 (58%) TSC NMI patients had mosaic TSC1/TSC2 pathogenic variants.10 Considering that approximately 15% of TSC patients are NMI by conventional sequencing methods, and over half of those are mosaic, this represents a substantial cohort of TSC patients whose clinical phenotype, natural history, and transmission risk are poorly studied.
MATERIALS AND METHODS
Samples from all TSC mosaic patients reported here were collected between 2012 and 2018 (Supplementary Material). All subjects enrolled in our research study provided written informed consent and the research protocol was approved by our Institutional Review Board, the Partners Human Research Committee (2013P002667).
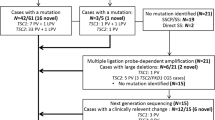
Thirty-nine subjects meeting TSC diagnostic criteria12 (14 male and 25 female; age range: 1–57 years, median: 27 years) with 0–10% MVAF in either TSC1 or TSC2 in blood, saliva, and/or normal skin are reported here (20 subjects were previously reported in more limited extent10) (Table S1). These individuals were referred to our lab with NMI status by prior negative conventional genetic testing. One hundred seventy DNA samples (range 1–11 per subject, median 5) from these 39 subjects were analyzed, including biopsies from TSC skin lesions (facial angiofibroma, toe ungual fibroma, shagreen patch, hypomelanotic macules), normal skin, blood, saliva, cultured cells (melanocytes, keratinocytes, fibroblasts from normal skin and/or ungual fibroma), urine, and semen (Table S2). Mosaic pathogenic variants were identified by deep coverage targeted massively parallel sequencing (MPS) (Supplementary Material) at 300–1200-fold read depth (Table S2, S3), and were then assessed and validated by amplicon MPS10 on all samples from each subject, at read depths of 25,000 to 1,000,000 (Table S4).
RESULTS
Three subjects (8%) had TSC1 (Table S2; subjects S6, S14, S34), and 36 (92%) had TSC2 pathogenic variants. Eleven subjects had additional TSC2 variants seen in only a single cutaneous lesion, consistent with a somatic second hit event.13 Two subjects had a TSC2 variant identified in a single facial skin lesion, not seen in any other tissues analyzed, consistent with the identified variant being a somatic second hit, and a first TSC2 variant (mosaic or not) refractory to detection by our methods (Table S2; subjects S38, S39). Alternatively, these two variants may have been the generalized mosaic pathogenic variant in these two subjects, which were not detected in other available samples.
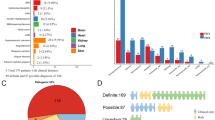
Comparison of the clinical features of 24 subjects in our mosaic cohort of age 19–34 years, median 26 (since many TSC clinical manifestations are age-dependent) with 240 TSC patients from the TSC Natural History Database who were age- and sex-matched showed many differences. Facial angiofibromas (92% in this cohort) and renal angiomyolipomas (83%) were both somewhat more common in our mosaic subjects (Fig. 1), though not statistically significant. In contrast, the majority of other TSC clinical features were much less common in our mosaic cohort, including hypomelanotic macules, ungual fibromas, shagreen patch, cortical tubers, subependymal nodules, subependymal giant cell astrocytomas, seizures, cardiac rhabdomyomas, and lymphangioleiomyomatosis (LAM) (many comparisons had p < 0.0001, false discovery rate (FDR) <0.05; Fig. 1, Table S5). Similar though smaller differences were also seen in comparison with a recently published set of TSC subjects who were NMI14 (Fig. 1, Table S6).

Comparison of clinical features of mosaic tuberous sclerosis complex (TSC) individuals reported here with those from the TSC Natural History Database, and a recent report of TSC no variant identified (NMI) patients.14 Twenty-four mosaic patients of age 19–34 years (median 26) were compared with 240 TSC Natural History Database patients of median age 26.5, and were matched 10:1 to the mosaic patients by gender and age +/- 1 year. The 22 TSC NMI patients were of median age 18 (ref. 14). P values are indicated for pairwise comparisons of the mosaic patient cohort with each of the other two cohorts, using Fisher’s exact test, and FDR-corrected q values. Multiple clinical features are less common in the mosaic cohort in comparison with one or both of the other two cohorts (9 of 14, 64%); while two features, facial angiofibromas/forehead plaque and angiomyolipomas, were slightly more common in this mosaic cohort. Differences were larger in comparison with the TSC Natural History Database cohort in general.
Interestingly, TSC patients from our cohort with the lowest MVAF (<0.63%, lowest tertile) in normal tissues had fewer TSC clinical features in comparison with patients with higher MVAF (0.67–7.2%; p = 0.02, one-tailed Mann–Whitney U test, Fig. 2a).

Comparison of mosaic variant allele frequencies (MVAFs) in different tuberous sclerosis complex (TSC) subjects and tissues. a Comparison of number of TSC clinical features in 32 TSC mosaic patients of age >14 years with low MVAF (<10%) in normal tissues (median of blood, saliva, and/or normal skin), divided into the lowest MVAF tertile (n = 11) and the upper 2 MVAF tertiles (n = 21). P = 0.02, Wilcoxon rank sum test; horizontal line, median value. b,c Graph of predicted (red symbols) and observed (blue squares) distribution of median MVAF from normal tissues (blood, saliva, and/or normal skin), considering the current 39 subjects with MVAF <10%, and 5 with MVAF 15–34%, previously reported.10 Numbers next to blue squares indicate the number of observed affected individuals, and number of predicted affected individuals (italics) are listed next to red symbols, for each MVAF bin that corresponds to serial twofold reduction in MVAF (Table S7). The latter are based on the function 2n, where n = number of cell divisions. Scale: b log scale, c linear scale.
We were interested in examining whether there were consistent differences in MVAF according to the tissue sample examined, to guide suitable tissue choice for molecular testing for other patients with suspected mosaicism. Remarkably, the MVAF varied widely across different tissues and lesions for each subject (Figure S1a–e). The median MVAF in TSC skin lesion biopsies (range 0.77–14%, median 4.01%) was >2-fold higher than what was seen in blood (range: 0–8.10%, median: 1.7%, p < 0.0001), saliva (range: 0–7.17%, median: 2.04%, p < 0.0001), and normal skin (range 0–10.71%, median 0.63%, p = 0.0004; two-tailed nonparametric paired t test) (Figure S2a–c). Among all TSC skin lesions analyzed, shagreen patch and ungual fibromas showed the highest median MVAF (Figure S1a), suggesting that these lesions had a higher tumor cell purity. Note that these lesions are due to clonal fibroblast tumor cell proliferation, but biopsies are poorly enriched in tumor cells due to the overlying keratinocyte-rich epidermis.13 In contrast, there was no significant difference in MVAF among any pair of normal tissues (skin, saliva, and blood DNA) assessed in these mosaic subjects (Figure S2e, f, two-tailed nonparametric paired t test). There were six (15%) subjects in this series for whom a mosaic TSC variant was identified in two or more samples, but was not detected (<0.07% MVAF, and not significantly different from control samples) at an appreciable MVAF in blood (Table S2; subjects S30–S34, S37).
Recent genome-wide studies suggest that the chance of mutation during early development is constant for each individual cell.15,16 Because the number of cells increases exponentially during early development, this model predicts that in our mosaic cohort there should be an exponential increase in the number of subjects with progressively lower levels of mosaicism (Fig. 2b, c). Notably, at higher levels of mosaicism (MVAF 3–12%), our cohort matches this prediction fairly well. However, at MVAF <3%, the number of cases in our cohort progressively falls below the predicted distribution, particularly at MVAF <0.8%.
Five male subjects of reproductive age in our cohort were interested to know the risk of disease transmission of their germline mosaic pathogenic TSC allele to potential offspring, leading us to perform DNA analysis on semen. Three men with TSC2 median MAFs of 0.63%, 4.45%, and 1.28% in normal tissues had no evidence of the TSC2 pathogenic variant in two different semen samples analyzed (separated by >6 months). One fathered a healthy male infant and the other two subjects have each fathered fetuses that are negative for the paternal TSC2 mosaic variant (Table S1, S2; subjects S25, S28, and S29). Two other men had a TSC2 variant with median MVAFs of 2.3% and 4.3% in normal tissues, and had semen median MVAFs of 2.6% and 14.3%, respectively (Table S1, S2; subjects S26 and S27). In addition, two female subjects in our cohort with MVAF 0.2% and 5.9% in blood DNA gave birth to children with TSC with the same TSC2 heterozygous variant (Table S1, S2; subjects S11 and S36). Remarkably, these two mildly affected women were diagnosed with TSC only after they gave birth to affected children, whereas two other mosaic subjects, diagnosed at ages 40 and 54, have healthy offspring without TSC (Table S1, S2; subjects S17 and S35 with two and three children, respectively).
DISCUSSION
In the present study, we describe the clinical and molecular features of 39 TSC patients with low frequency mosaicism (<10%). We found that mosaic TSC individuals have a milder phenotype with distinctive clinical features in comparison with a large population study of TSC patients, the TSC Natural History Database. These differences must be considered in the context of how these patients were identified and entered our study. The majority were of childbearing age, had normal intellectual ability and limited TSC-related morbidity, and were seeking genetic information to guide personal family planning. On the other hand, our mosaic subjects all met standard TSC diagnostic criteria12 meaning that they had to have at least two clinical features,12 of which facial angiofibroma and renal angiomyolipoma were the most common. Thus, our cohort is enriched for TSC patients with a milder phenotype, through this ascertainment effect, and it is not surprising that they had low-level mosaic pathogenic variants.
These results broaden our understanding of the spectrum of clinical features seen in mosaic TSC, and should enhance both clinical suspicion and appropriate analysis for mosaicism detection. Furthermore, these findings provide information on which tissues should be sampled to provide the best chance at identifying a pathogenic variant. TSC cutaneous lesions consistently contain higher levels of the mosaic pathogenic allele, due to the presence of clonal tumor fibroblasts, and thus biopsy of such lesions enhances pathogenic variant identification, while testing of multiple samples provides validation of findings, and helps to distinguish generalized mosaic variants from lesion-specific somatic second hit variants. Facial angiofibroma, a common cutaneous lesion that is easily accessible, is quite suitable for genetic analysis when TSC mosaicism is suspected. Mosaic allele frequencies were consistently higher in these and other TSC lesions than in blood, saliva, or normal skin, including six subjects in whom the mosaic pathogenic allele was not detectable in blood DNA. Absence of the pathogenic TSC2 variant in blood in these six TSC mosaic patients suggests that there was no contribution of variant-containing cells to the hematopoietic cell lineage, in contrast to other tissues (e.g., nervous system, mesenchymal) in which pathogenic variants lead to TSC manifestations.
We found that there is a deficit in the observed number of TSC individuals at very low MVAF, in comparison with the number predicted by genome-wide studies and theoretical modeling (Fig. 2b, c). This suggests that there is a substantial number of individuals in the population with low-level mosaicism for a pathogenic variant in TSC2 who are not recognized clinically due to minimal or no TSC clinical manifestations. Unrecognized low-level mosaic individuals may contribute to the known recurrence risk for parents of a sporadic TSC child, usually estimated as 1–2% (ref. 17).
As shown here, the frequency of germ cell lineage (egg or sperm) involvement by a mosaic TSC2 pathogenic variant is highly variable, and the transmission risk is not predictable based on MVAF levels in other tissues. Semen analysis is an effective method for determination of transmission risk in mosaic TSC men.
We note that a parallel study on mosaicism in TSC, also reported in this issue, came to similar conclusions on the prevalence and clinical significance of mosaicism.18 These two sets of mosaic TSC patients have many similarities as well as some differences, which we suspect are due in part to the methods by which they were ascertained.
Although our analyses have focused on TSC, these observations are applicable to many other genetic diseases, including other neurocutaneous disorders (e.g., NF1, NF2), and megalencephaly and somatic overgrowth syndromes,19,20,21 in which new mutational events occur spontaneously, and mosaicism affects multiple tissues. Hence, these data may provide information valuable for the care of many patients with other mosaic human genetic disorders with respect to variability in mosaicism levels in different tissues and risk of disease transmission.
Change history
04 December 2020
This Article was originally published without the accompanying Supplementary Material in the Excel format. This file is now available in the HTML version of the Article; the PDF was correct from the time of publication.
22 December 2020
A Correction to this paper has been published: https://doi.org/10.1038/s41436-020-01066-w
References
Forsberg LA, Gisselsson D, Dumanski JP. Mosaicism in health and disease - clones picking up speed. Nat Genet. 2017;18:128–142.
De S. Somatic mosaicism in healthy human tissues. Trends Genet. 2011;27:217–223.
Halvorsen M, Petrovski S, Shellhaas R, et al. Mosaic mutations in early-onset genetic diseases. Genet Med. 2016;18:746–749.
Lim YH, Moscato Z, Choate KA. Mosaicism in Cutaneous Disorders. Annu Rev Genet. 2017;51:123–141.
Campbell IM, Shaw CA, Stankiewicz P, Lupski JR. Somatic mosaicism: implications for disease and transmission genetics. Trends Genet. 2015;31:382–392.
Rahbari R, Wuster A, Lindsay SJ, et al. Timing, rates and spectra of human germline mutation. Nat Genet. 2016;48:126–133.
Rose VM, Au KS, Pollom G, et al. Germ-line mosaicism in tuberous sclerosis: how common? Am J Hum Genet. 1999;64:986–992.
Campbell IM, Yuan B, Robberecht C, et al. Parental somatic mosaicism is underrecognized and influences recurrence risk of genomic disorders. Am J Hum Genet. 2014;95:173–182.
de Kock L, Wang YC, Revil T, et al. High-sensitivity sequencing reveals multi-organ somatic mosaicism causing DICER1 syndrome. J Med Genet. 2016;53:43–52.
Tyburczy ME, Dies KA, Glass J, et al. Mosaic and Intronic Mutations in TSC1/TSC2 Explain the Majority of TSC Patients with No Mutation Identified by Conventional Testing. PLoS Genet. 2015;5:1–17.
Henske EP, Jozwiak S, Kingswood JC, et al. Tuberous sclerosis complex. Nat Rev Dis Primers. 2016;2:16035.
Northrup H, Krueger DA, International Tuberous Sclerosis Complex Consensus Group. Tuberous sclerosis complex surveillance and management: recommendations of the 2012 International Tuberous Sclerosis Complex Consensus Conference. Pediatr Neurol. 2013;49:243–254.
Tyburczy ME, Wang JA, Li S, et al. Sun exposure causes somatic second-hit mutations and angiofibroma development in tuberous sclerosis complex. Hum Mol Genet. 2014;23:2023–2029.
Peron A, Vignoli A, Briola F, et al. Deep phenotyping of patients with Tuberous Sclerosis Complex and no mutation identified in TSC1 and TSC2. Eur J Med Genet. 2018;61:403–410.
Jacobs KB, Yeager M, Zhou W, et al. Detectable clonal mosaicism and its relationship to aging and cancer. Nat Genet. 2012;44:651–658.
Machiela MJ, Zhou W, Sampson JN, et al. Characterization of large structural genetic mosaicism in human autosomes. Am J Hum Genet. 2015;96:487–497.
Verhoef S, Bakker L, Tempelaars AM, et al. High rate of mosaicism in tuberous sclerosis complex. Am J Hum Genet. 1999;64:1632–1637.
Treichel AM, Hamieh L, Nathan NR, et al. Phenotypic distinctions between mosaic forms of tuberous sclerosis complex. Genet Med. 2019. https://doi.org/10.1038/s41436-019-0520-3.
Campbell IM, Stewart JR, James RA, et al. Parent of origin, mosaicism, and recurrence risk: probabilistic modeling explains the broken symmetry of transmission genetics. Am J Hum Genet. 2014;95:345–359.
Keppler-Noreuil KM, Parker VE, Darling TN, Martinez-Agosto JA. Somatic overgrowth disorders of the PI3K/AKT/mTOR pathway & therapeutic strategies. Am J Med Genet C Semin Med Genet. 2016;172:402–421.
Riviere JB, Mirzaa GM, O’Roak BJ, et al. De novo germline and postzygotic mutations in AKT3, PIK3R2 and PIK3CA cause a spectrum of related megalencephaly syndromes. Nat Genet. 2012;44:934–940.
Acknowledgements
The authors thank all subjects in this study; the clinicians who referred and evaluated the patients; Karthik V. Karnik, Rachel E. Yan, and Edward R. Kwiatkowski for their work on customized code for MPS analysis; and the Tuberous Sclerosis Alliance and the TSC Natural History Database Consortium. Supported by The Engles Family Fund for Research in TSC and LAM. The views expressed in this article are those of the authors and do not necessarily reflect the opinion of the Tuberous Sclerosis Alliance or the Tuberous Sclerosis Complex Natural History Database Consortium.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Disclosure
The authors declare no conflicts of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Giannikou, K., Lasseter, K.D., Grevelink, J.M. et al. Low-level mosaicism in tuberous sclerosis complex: prevalence, clinical features, and risk of disease transmission. Genet Med 21, 2639–2643 (2019). https://doi.org/10.1038/s41436-019-0562-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41436-019-0562-6
Keywords
This article is cited by
-
Clinical practice recommendations for kidney involvement in tuberous sclerosis complex: a consensus statement by the ERKNet Working Group for Autosomal Dominant Structural Kidney Disorders and the ERA Genes & Kidney Working Group
Nature Reviews Nephrology (2024)
-
VHL mosaicism: the added value of multi-tissue analysis
npj Genomic Medicine (2022)
-
Subependymal giant cell astrocytomas are characterized by mTORC1 hyperactivation, a very low somatic mutation rate, and a unique gene expression profile
Modern Pathology (2021)
-
Genetics of tuberous sclerosis complex: an update
Child's Nervous System (2020)
