
Abstract
Two new indolizidine alkaloids crepidatumines A (1) and B (2) together with the stereoisomer of dendrocrepidine B (3) and known analog dendrocrepine (4) were isolated from D. crepidatum. Their structures were determined by HR-ESI-MS, NMR, and Electronic Circular Dichroism (ECD) experiments together with comparison of analogues. Compound (1) possess a (5/6/6/5) tetra-hetero-cyclic ring, whereas compound (2) contains a tricyclic system with an unusual bridged ring, which are the first report in Nature. The biological evaluation revealed that dendrocrepine (4) displayed a potent hypoglycemic effect in vitro.
Similar content being viewed by others
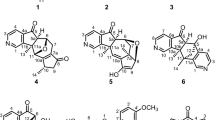
Introduction
Dendrobium genus, a large member of Orchid plant with more than 1500 species, is distributed mainly in tropical and subtropical areas of Asia and Oceania1. Seventy-six species including two variants are found in the southern region of the Qin Ling mountain and Huai He river, China2. As one of the most famous Traditional Chinese Medicine (TCM), Dendrobium genus has a wide range of biological effects, for example, maintaining gastric tonicity, enhancing production of body fluid, relieving and curing symptoms of dryness, and of body heat3, and it also displays anti-tumor, antioxidation, immuno-enhancement and anti-inflammatory bioactivities4,5,6. In ancient time of China, this genus is named as one of “Nine Fairy Grasses”, which is very precious and rare. More than 300 compounds including polysaccharides, sesquiterpenoids, alkaloids, flavonoids, bibenzyls and phenanthrenes have been isolated from different Dendrobium species7,8,9,10. D. crepidatum is a unique species with very beautiful flowers growing in warm and humid environments at an altitude of 500~1500 meters, which has diverse functions including moistening the lung, eliminating asthenia heat, and diminishing inflammation11,12. So far, there were only several indolizine-type alkaloids and two stilbene derivatives isolated from this medicinal plant13,14,15. Indolizidine alkaloids can be divided into two major groups including phenanthroindolizidines and simple indolizidines. This member of alkaloids displays a large range of biological effect such as glycosidase inhibition, immune modulation, anti-viral and anti-cancer activities16,17,18. During our ongoing research to mine new/novel bioactive secondary metabolites from TCM plants, two indolizidine alkaloids crepidatumines A (1) and B (2) with new skeletons together with the stereoisomer of dendrocrepidine B (3) and dendrocrepine (4) were isolated from D. crepidatum (Fig. 1). Compound (1) possesses a novel (5/6/6/5) tetra-hetero-cyclic structure, whereas compound (2) contains a unique bridged-ring system. In this report, the isolation, structural elucidation, and biological activities of these compounds were present.

Dendrobium crepidatum and two new indolizidine alkaloids crepidatumines A (1) and B (2). Fig. 1. Structures of 1–4.
The 1H, and 13C of compound 3 were similar with those of dendrocrepidine B, but the result from X-ray diffraction data determined its structure to be the stereoisomer of dendrocrepidine B, and this established its absolute configuration of 3 as 1R, 2R, 3S, 5R, 6S, 7R, and 9S (Fig. 2). The X-ray data of 3 was provided in Table 1.

X-Ray crystal structure of 3.
Results and Discussion
The molecular formula of crepidatumine A (1) was determined to be C21H29NO3 (8 degrees of unsaturation) on the basis of HRESI MS (m/z 344.2230 [M + H]+; Calcd 344.2226). The 1H, 13C, (Table 1) and HMQC NMR data of 1 (Table 2) revealed the presence of three methyls, four methylenes, five methines, three oxygenated carbons (one of which was suggested to be a hemiacetal carbon with chemical shift value δc = 104.6), and a mono-substituted phenyl ring. In addition, there are two singlet protons as free hydroxyl or amino groups. These data accounted for all the 1H and 13C-NMR resonance signals. Based on the degrees of unsaturation, it implied that 1 possessed a tetracyclic system. The 1H-1H COSY correlations established three isolated proton spin-systems including a mono-substituted phenyl unit, two fragments corresponding to –C-13-C-7-C-8-C-9-C-10-C-11-C-1-C-2–, and –C-4-C-5– (Fig. 3). The remaining connections were characterized by HMBC correlations. The correlations from 13-Me to C-6, from H-5 and H-7 to C-6 and C-1′, and from H-2′ (H-6′) to C-5, C-6 and C-7 confirmed that the C-6 was linked with C-5, C-7 and C-1′; correlations of H-5 with C-1 and C-9, and H-9 with C-1 established an indolizidine ring system; the HMBC cross peaks from 3-OH to C-2, C-3, C-4 and C-12 determined the connection of C-3 with C-2, C-4 and C-12, and a hydroxyl group was anchored at C-3; the correlations of 15-Me with C-4, C-14, and of 14-OH with C-4, C-14 and C-15 established the linkage of C-14 with C-4 and C-15, and of C-14 with an hydroxyl group. Considering the chemical shift value of C-14 (δc = 104.6), degrees of unsaturation and molecular formula, an ether bond must be shaped between C-6 and C-14 to form a unique hemiacetal group. Thus, the planar structure of 1 was characterized.

1H-1H COSY, Key HMBC and NOESY correlations of 1 and 2.
The relative configuration of 1 was determined on the basis of analysis of NOESY correlations and coupling constants (Fig. 3). The correlations from H-5 with H-1, 3-OH, H-7, H-9, and H-2′ (H-6′) implied that H-1, 3-OH, H-5, H-7, and H-9 possessed pesudo-axial bonds, whereas the mono-substituted phenyl and 12-Me possessed pesudo-equatorial bonds; the coupling constant between H-4/H-5 was 3.0 Hz, which revealed that H-4 possessed pesudo-equatorial bond; H-2b was a broad triplet (t, J = 13.2 Hz) and H-2a was a doublet doublet (dd, J = 13.2, 3.6 Hz) confirmed that H-2b possessed pesudo-axial bond and H-2a possessed pesudo-equatorial bond; 15-Me had NOESY correlations with 12-Me, and H-2b had NOESY correlations with 14-OH implied that these groups were close each other in space (Fig. 3). Thus, the relative configuration of 1 was determined. Electronic circular dichroism (ECD) experiments combined with quantum-chemical calculations adopting time-dependent density functional theory (TDDFT) was used to establish the stereochemistry of crepidatumine A (1). The theoretical calculation of ECD was conducted in MeOH using Time-dependent Density functional theory (TD-DFT) at the B3LYP/6-311 + g (d, p) level for all conformers of compounds 1. To get the final spectra, the simulated spectra of the conformers were averaged according to the Boltzmann distribution theory and their relative Gibbs free energy (ΔG). By comparing the experiment spectrum with the calculated ECD spectra, the stereochemistry for 1 was determined to be 1R, 3S, 4R, 5R, 6S, 7R, 9S, 14R (Fig. 4) same as that of its analogue 3, of which stereochemistry was determined by X-ray diffraction experiment.

ECD of structure of 1 and 2.
The HRESIMS (m/z 270.1857 [M + H]+, Calcd 270.1858) gave the molecular formula of crepidatumine B (2) to be C18H23NO. Analysis of the NMR spectra revealed that compound 2 (Table 2) also possessed an indolizidine ring coupled a mono-substituted phenyl unit same as found in structures 1 and 3 (Fig. 3). The 1H-1H COSY spectrum linked C-7 and C-8; the HMBC correlations from 11-CH2- to C-4 and C-5, from 5-CH2- to C-6 and C-7 established the fragment of–C-4-C-5-C-6-C-7–, which formed a bridged-ring fused with a indolizine ring to shape the tetrahydro-1H-5,8a-ethanoindolizin-7(8H)-one carbon skeleton. Thus, the planar structure of 2 was characterized. The relative configuration of 2 was established based on the coupling constant analysis and NOESY correlations. The coupling constant of H-9 (J = 4.20, 12.0 Hz) together with the NOESY correlation from H-8 and H-9 confirmed that H-9, H-10 and –C-8-C-7– possessed pesudo-axial bonds, and H-8 possessed pesudo-equatorial bond; the NOESY correlation between H-10 with 5-CH2 revealed that –C-4-C-5– possessed pesudo-axial bond. The absolute configuration was determined to be 4 S, 8 S, 9 S, and 10 R on the basis of comparison of CD and ECD spectra of 2 (Fig. 4).
Crepidatumine A (1) possesses an unusual dodecahydrofuro[2,3,4-ij] pyrrolo[2,1,5-de] quinolizine skeleton, which is formed by a decahydro-1H-pyrrolo[2,1,5-de] quinolizine ring coupled a tetrahydrofuran ring. More importantly, there are eight stereocenters (six carbons C-3-C-4(C-14)-C-5-C-6-C-7 are continued) and a hemiacetal hydroxyl group in the unique (5/6/6/5) tetra-hetero-cyclic system of crepidatumine A (1), which will be a potential star molecule to organic synthesis. Crepidatumine B (2) possess a unique tricyclic indolizine-type skeleton with an unusual bridged ring, which is the first report in Nature (Fig. 5).

Key skeletons of 1 and 2.
Compounds 1–4 were tested the anti-inflammatory activity in RAW 264.7 cells without biological effects, whereas the total alkaloids of D. crepidatum (TAD) inhibited the NO production induced by LPS in RAW 264.7 cells in a dose-dependent fashion without cytotoxicity (Fig. 6)19. Thus, it is merited to go to mine bioactive natural products from these total alkaloids. In addition, compounds 1–4 were evaluated the cytotoxic activities against three cancer cell lines A459, and HCT116, and gram-positive bacteria Bacillus subtilis 63501, Staphylococcus aureus 29213, and gram-negative bacterium Escherichia coli 25922 without bioactivities. The high glucose model of HepG2 cells was used to evaluate the hypoglycemic effect of compound 4. As a result, compared with the model group, this compound significantly increased the glucose consumption by 29% at the concentrations of 50 μmol/L without cytotoxicity, which implying that this compound might be a potent molecule as a hypoglycemic agent (Fig. 7).

Effect of TAD on the viability of RAW 264.7 macrophages. Data are expressed as mean ± SD (n = 3). **p < 0.01 compared with the normal group; **p < 0.01 compared with the model (LPS-treated) group.

Effect of compound 4 (C4) on cell viability (A) and glucose consumption (B) in HepG2 cell. Data are shown as the mean ± SD (n = 6). **p < 0.01, ***p < 0.001 versus control (A) or model (B).
Conclusion
Four indolizidine alkaloids including two new compounds crepidatumines A (1) and B (2) together with the stereoisomer (3) of dendrocrepidine B and known analog dendrocrepine (4) were isolated from D. crepidatum Compound (1) possess a unusual (5/6/6/5) tetra-hetero-cyclic ring, whereas compound (2) contains a unique tricylic system with an unusual bridged ring, which are the first report in Nature. Compounds 1–4 did not display anti-inflammatory, anti-microbial and cytotoxic activities, but compound 4 could increase glucose consumption, implying that 4 might be a potent molecule as a hypoglycemic agent.
Methods
General experimental procedures
Optical rotations were measured on a PerkinElmer 241 polarimeter, and UV data were determined on a ThermoGenesys-10S UV-vis spectrometer19. IR data were recorded using a Nicolet IS5FT-IR spectrophotometer. CD spectra were obtained on a JASCO J-810 spectrometer. 1H and 13C NMR data were acquired with a Bruker 600 spectrometer using solvent signals (DMSO-d6; δH 2.50/δc 39.5) as references. the HMQC and HMBC experiments were optimized for 145.0 and 8.0 Hz, respectively. HRESIMS were obtained using a TOF-ESI-MS (Waters Synapt G2, USA). Semipreparative HPLC separation was carried out using a Lumtech instrument packed with a YMC-Pack ODS-A column (5 μm, 250 × 10 mm). Sephadex LH-20 (Pharmacia Biotech AB, Uppsala, Sweden) and silica gel (200–300 mesh) (Qingdao Marine Chemical Plant, Qingdao, China) were used.
Plant material
The stems of D. crepidatum were collected from Ruili Resource Nursery of Dendrobium Germ Plasm and Resources, the Ministry of Agriculture and Rural Affairs of the People’s Republic of China in August 201719. The sample was identified by one of the co-authors Ze-Sheng Li from Yunnan Dehong Institute of Tropical Agricultural Science. A voucher specimen was deposited in the herbarium of the Institute of Medicinal Plant Development, Chinese Academy of Medical Sciences.
Extraction and isolation
The dried stems of D. crepidatum (9.0 kg) were extracted under reflux with 95% ethanol (50 L × 3 h, three times)19. The combined extract was suspended with water, and extracted with petroleum ether and CH2Cl2 three times separately. The fraction of CH2Cl2 was concentrated into extractum, and dissolved in 5% hydrochloric acid filtered, then adjusted to pH 10 with ammonia water. Finally, it was extracted by CH2Cl2 three times at room temperature. The CH2Cl2 extract was obtained the total alkaloids 90.0 g of crude extract. The original extract was fractionated on a silica gel CC eluted with petroleum ether- acetone (50:1, 40:1, 30:1, 20:1, 15:1, 10:1, 5:1, 2:1 and 0:1, v/v, each 6.6 L) to give five fractions (Fr.1 to Fr.5). Fr.2 (10.0 g) was fractionated on a silica gel column chromatography (CC) using petroleum ether-acetone isocratic elution (30:1) to afford six fractions (Fr.2.1-Fr.2.6) and the crystal compound 4 (4.3 g). Separation of Fr.2.4 (2.0 g) was separated over Sephadex LH-20 (CH2Cl2: MeOH/1:1) to give four fractions (Fr.2.4.1-Fr.2.4.4). Fr.2.4.2 (500.0 mg) was further purified by semi-preparative HPLC (60–100% MeOH-H2O for 30 min, v/v, 2 mL/min) to yield 1 (5.9 mg, tR 29.8 min). Fr.2.4.3 (1.0 g) was purified by semi-preparative HPLC (60–100% MeOH-H2O for 30 min, v/v, 2 mL/min) to yield 2 (15.0 mg, tR 26.0 min). Fr.2.5 (2.0 g) was further purified by semi-preparative HPLC (60–100% MeOH-H2O for 30.0 min, v/v, 2 mL/min) to yield 3 (4.0 mg, tR 33.0 min).
Crepidatumine A (1): white powder; [α]D25 + 4.44 (c 0.1, MeOH); UV (MeOH) λmax (log ε) 205 (3.70); IR (neat) νmax 3433, 2965, 1209, 1032, 935, 755 cm−1; for 1H NMR and 13C NMR data see Table 1; Positive HR-ESI-MS: m/z 344.2230 (calcd. for C21H30NO3 [M + H]+, 344.2226).
Crepidatumine B (2): orange oil; [α]D25-26.0 (c 0.1, MeOH); UV (MeOH) λmax (log ε) 205 (4.03); IR (neat) νmax 2924, 2874, 1708, 1056, 1031, 704 cm−1; for 1H NMR and 13C NMR data see Table 1; Positive HR-ESI-MS: m/z 270.1857 (calcd. for C18H24NO [M + H]+, 270.1858).
X-ray crystallographic analysis of 3
Upon crystallization from n-Hexane–CH2Cl2 (10:1) using the vapor diffusion method, colorless crystals were obtained for 3 C20H27NO4, M = 345.42, orthorhombic, a = 6.6679(4) Å, b = 10.7681(4) Å, c = 24.2111(9) Å, U = 1738.38(13) Å3, T = 107.75(10), space group P212121 (no. 19), Z = 4, μ (Cu Kα) = 0.737, 5698 reflections measured, 3256 unique (Rint = 0.0271) which were used in all calculations. The final wR (F2) was 0.1106 (all data).Crystallographic data for the structure of 3 has been deposited in the Cambridge Crystallographic Data Centre [deposition number: CCDC 1908235].
Cell culture and sample treatment
The murine macrophage RAW 264.7 cell line was obtained from American Type Culture Collection (ATCC, Rockville, MD, USA)20. The cells were cultured at 37 °C in DMEM (Invitrogen, California, USA) supplemented with 10% heat-inactivated FBS (Hyclone, Utah, Logan, USA), 2 m ML - glutamine, penicillin G (100 IU/mL) and streptomycin (100 mg/mL) in a humidified atmosphere containing 5% CO2 and 95% air. The cells were incubated with total alkaloids of D. crepidatum (TAD) at different concentrations and then stimulated with LPS (10 ng/mL) for the indicated time. The stock solutions of TAD were prepared in dimethyl sulfoxide (DMSO), and the final concentration of DMSO was less than 0.5%.
The level of NO production was monitored by measuring the nitrite level in the culture medium. This was performed by mixing the medium with Griess reagent (1% sulfanilamide in 5% phosphoric acid and 0.1% N-1-naphthylethylenediamine dihydrochloride in water). The absorbance was measured at a wavelength of 540 nm after incubation for 10 min. The nitrite concentration was calculated with reference to a standard curve of sodium nitrite generated from known concentrations. Cell viability was assessed using an MTT assay.
In Vitro Evaluation of Compound 4
Cell culture: Human hepatoma cells HepG2 were cultured in Dulbecco’s modified Eagle’s medium (DMEM, HyClone)19. DMEM was supplemented with 10% fetal bovine serum (Gibco) and 1% penicillin/streptomycin (HyClone) in a humidified atmosphere of 5% CO2 and 37 °C.
Assay for cell viability: Before the experiment, the assay for cell viability was determined with the cell counting kit-8 (CCK-8). HepG2 cells were seeded in 96-well plates as 2.5 × 103 cells each well. Afterculturing for 24 h,the control group was added with serum-free medium, while the experimental groups were with the medium containingdifferent concentrations (50and 100 μmol/L) of compound 4 or 200 μmol/Lof metformin for another 24 h. Then the cells were treated with CCK-8 for 3 h. Finally, the absorbance was measured at 450 nm. The cell viability was calculated as the absorbance of each treated well divided by the control.
Assay for hypoglycemic activity: For the experiment, the cells were seeded in 96-well plates as 1 × 104 cells each well. After culturing for 24 h, the medium containing different concentrations (25 and 50 μmol/L) of compound 4 were added for 24 h. The cells with 200 μmol/L metformin treatment were taken as positive control and the cells with phenol red-free DMEM as control. After the drug treatment, the glucose concentrations of the medium were determined with the glucose oxidase method. The glucose consumption of each well was obtained by subtracting the glucose concentrations of the experimental medium from the control group.
ECD calculations of 1 and 2
Monte Carlo conformational searches were carried out by means of the Spartan’s 10 software using Merck Molecular Force Field (MMFF)19. The conformers with Boltzmann-population of over 5% were chosen for ECD calculations, and then the conformers were initially optimized at B3LYP/6-31 + g (d, p) level in MeOH using the CPCM polarizable conductor calculation model. The theoretical calculation of ECD was conducted in MeOH using Time-dependent Density functional theory (TD-DFT) at the B3LYP/6-311 + g (d, p) level for all conformers of compounds 1 and 2. Rotatory strengths for a total of 50 excited states were calculated. ECD spectra were generated using the program SpecD is 1.6 (University of Würzburg, Würzburg, Germany) and GraphPad Prism 5 (University of California San Diego, USA) from dipole-length rotational strengths by applying Gaussian band shapes with sigma = 0.3 eV.
References
Li, Q., Ding, G., Li, B. & Guo, S. X. Transcriptome Analysis of genes involved in Dendrobine biosynthesis in Dendrobium nobile Lindl. infected with Mycorrhizal Fungus MF23 (Mycena sp.). Scientific Reports. 7(1), 316 (2017).
Zhang, Y. B. et al. Current approaches for the authentication of medicinal Dendrobium species and its products. Plant Genetic Resources. 3(2), 144–8 (2005).
Ng, T. B. et al. Review of research on Dendrobium, a prized folk medicine. Appl Microbiol Biotechnol. 93(5), 1795–803 (2012).
Zheng, Q. P. et al. Extraction of anti-tumor constituents derived from Dendrobium officinale. Modern Food Science. 30(5), 12–7 (2014).
Zha, X. Q. et al. Immunoregulatory activities of Dendrobium huoshanense polysaccharides in mouse intestine, spleen and liver. International Journal of Biological Macromolecules. 64(2), 377–82 (2014).
Wang, J. H., Zuo, S. R. & Luo, J. P. Structural analysis and immuno-stimulating activity of an acidic polysaccharide from the stems of Dendrobium nobile Lindl. Molecules. 22(4), 611–21 (2017).
Meng, L. et al. Effects of polysaccharides from different species of Dendrobium (Shihu) on macrophage function. Molecules. 18(5), 5779–91 (2013).
Tang, H. et al. Dendrobium officinale Kimura et Migo: A review on its ethnopharmacology, phytochemistry, pharmacology, and industrialization. Evidence-Based Complementray Alternative Medicine. 2017(5), 1–19 (2017).
Yang, L., Wang, Z. & Xu, L. Simultaneous determination of phenols (bibenzyl, phenanthrene, and fluorenone) in Dendrobium species by high-performance liquid chromatography with diode array detection. Journal of Chromatography A. 1104(1), 230–7 (2006).
Ye, Q., Qin, G. & Zhao, W. Immunomodulatory sesquiterpene glycosides from Dendrobium nobile. Phytochemistry. 61(8), 885–90 (2002).
Hu, Y. et al. Protective effects of total alkaloids from Dendrobium crepidatum against LPS-induced acute lung injury in mice and its chemical components. Phytochemistry. 149, 12–23 (2018).
Li, Q. et al. Molecular analysis of polysaccharide accumulation in Dendrobium nobile infected with the mycorrhizal fungus Mycena sp. Rsc Advances. 7(42), 25872–84. (2017).
Elander, M. et al. Studies on orchidaceae alkaloids. XXXII. Crepidine, crepidamine and dendrocrepine, three alkaloids from Dendrobium crepidatum Lindl. Acta Chemica Scandinavica. 27, 1907–13. (1973).
Hu, Y. et al. (±)-Homocrepidine A, a Pair of anti-inflammatory enantiomeric octahydroindolizine alkaloid dimers from Dendrobium crepidatum. Journal of Natural Products. 23(4), 319–22 (2015).
Li, C. B. & Zhou, J. Chemical components of Dendrobium crepidatum and their neurite outgrowth enhancing activities. Natural Products Bioprospecting. 3(2), 70–3 (2013).
Michalik, A. et al. Steviamine, a new indolizidine alkaloid from Stevia rebaudiana. Phytochemistry Letters. 3, 136–38. (2010).
Watson, A. A., Fleet, G. W. J. & Nolyneux, N. Polyhydroxylated alkaloids—natural occurrence and therapeutic applications. Phytochemistry. 56, 265–95. (2001).
Gellert, E. The indolizidine alkaloids. Journal of Natural Products 45(1), 50–73 (1982).
Xu, X. L. et al. Crepidatumines C and D, Two new indolizidine alkaloids from Dendrobium crepidatum Lindl. ex Paxt. Molecules 24(17), 3071 (2019).
Li, M. et al. Lipid-soluble extracts from Salvia miltiorrhiza inhibit production of LPS-induced inflammatory mediators via NF-κB modulation in RAW 264.7 cells and perform antiinflammatory effects in vivo. Phytotherapy Research Ptr. 26(8), 1195–204 (2012).
Acknowledgements
We gratefully acknowledge financial support from the National Key R&D Program of China (2018YFC1706200 for L. B), We acknowledge financial support from The National Key Research and Development Program of China “Research and Development of Comprehensive Technologies on Chemical Fertilizer and Pesticide Reduction and Synergism” (2017YFD0201402 to D.G.), Species and Varieties Resource Protection (Tropical Crops) Pro-gram “Germ Plasm and Resources Protection of Dendrobium” from the Ministry of Agriculture and Rural Affairs of the People’s Republic of China (151821301354052710 for L. Z. S); CAMS Initiative for Innovative Medicine (CAMS-2016-I2M-2-003 for L. B) and the National Natural Science Foundation of China (81473331).
Author information
Authors and Affiliations
Contributions
X.L.X., B.L. and G.D. conceived and designed the experiments. X.L.X. performed the experiments, analyzed the data. R.M.Y. did the biological evaluation, Z.S.L., H.G.Z. and Y.B.B. provided and identified the experimental materials. M.Y. did the Mass experiment. X.LX. and Z.S.L. drew all the figure in the paper, G.D., B.L., X.L.X. and X.Y.C. wrote the paper. G.D. provided technological guidance for the chemistry and edited the paper. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Xu, X., Chen, X., Yang, R. et al. Crepidtumines A and B, Two Novel Indolizidine Alkaloids from Dendrobium crepidatum. Sci Rep 10, 9564 (2020). https://doi.org/10.1038/s41598-020-66552-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-66552-2
This article is cited by
-
Recent advances and new insights in biosynthesis of dendrobine and sesquiterpenes
Applied Microbiology and Biotechnology (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
