
Abstract
The clusioid clade of Malpighiales is comprised of five families: Bonnetiaceae, Calophyllaceae, Clusiaceae, Hypericaceae and Podostemaceae. Recent studies have found the plastome structure of Garcinia mangostana L. from Clusiaceae was conserved, while plastomes of five riverweed species from Podostemaceae showed significant structural variations. The diversification pattern of plastome structure of the clusioid clade worth a thorough investigation. Here we determined five complete plastomes representing four families of the clusioid clade. Our results found that the plastomes of the early diverged three families (Clusiaceae, Bonnetiaceae and Calophyllaceae) in the clusioid clade are relatively conserved, while the plastomes of the other two families show significant variations. The Inverted Repeat (IR) regions of Tristicha trifaria and Marathrum foeniculaceum (Podostemaceae) are greatly reduced following the loss of the ycf1 and ycf2 genes. An inversion over 50 kb spanning from trnK-UUU to rbcL in the LSC region is shared by Cratoxylum cochinchinense (Hypericaceae), T. trifaria and Ma. foeniculaceum (Podostemaceae). The large inversed colinear block in Hypericaceae and Podostemaceae contains all the genes in the 50-kb inversed colinear block in a clade of Papilionoideae, with two extra genes (trnK-UUU and matK) at one end. Another endpoint of both inversions in the two clusioids families and Papilionoideae is located between rbcL and accD. This study greatly helped to clarify the plastome evolution in the clusioid clade.
Similar content being viewed by others

Introduction
Plastomes of heterotrophic plants are generally highly rearranged1, while plastomes of autotrophic angiosperms seem to be relatively conserved2. Most autotrophic angiosperm plastomes are characterized by a copy of Inverted Repeat (IR) regions, one Large Single Copy (LSC) region and one Small Single Copy (SSC) region, with the average size of 153 kb, generally include 101–118 unique genes that primarily participating in photosynthesis, transcription, and translation3,4. The advent of high-throughput sequencing has facilitated rapid progress in the field of comparative plastid genomics5,6.
Several distinct autotrophic angiosperms clades have substantial variations in plastome size and gene order. Large variation of plastome size is often associated with IR expansion or contraction7, but could also be influenced to some extent by gene and intron losses. The loss of two hypothetical open reading frames ycf1 and ycf2, two largest plastid genes, could significantly reduce the plastome size6. Multiple independent losses of some plastid genes and introns have been reported8,9, some of these genes have transferred to the nucleus9. Successful gene transfers from the plastid to the nuclear genome during angiosperm evolution have been documented for rpl22, rpl32 and infA9,10.
Inversions play an important role in plastid genome structural variations and have been fully characterized in a number of plastomes. Large inversions have been found in plastomes of many plant lineages, such as Onagraceae11, Asteraceae12, and Fabaceae13. In Fabaceae, multiple large inversions have been reported, including a 50-kb inversion shared by most Papilionoids except a few early-diverging clades, a 78-kb inversion in Phaseolinae of Phaseoleae, inversions of 23-kb, 24-kb, or 36-kb in the Genistoid clade, a 39-kb inversion in Robinia of Papilionoideae, and a 38-kb inversion in Tylosema of Cercidoideae13,14,15. Recent studies have found short Inverted Repeat (sIR) meditated flip-flop recombination event could induce large inversions13,15,16.
The clusioid clade (Malpighiales) contains five families (Bonnetiaceae, Calophyllaceae, Clusiaceae, Hypericaceae, and Podostemaceae) represented by 94 genera and ~1900 species17. Their distribution is nearly cosmopolitan, with the greatest species diversity in the tropics18. Species in this clade include large tropical rainforest trees, temperate and high-altitude tropical herbs and shrubs, and even aquatic plants (Podostemaceae) growing in swift-flowing rivers and streams18. Many species are economically important, such as tropical fruits including the mangosteen (Garcinia mangostana L.) and the mammey apple (Mammea americana L.), timber (Calophyllum brasiliense Cambess., Mesua ferrea L.), and medicine (Hypericum perforatum L.).
Previous studies found that the plastome of Garcinia mangostana L. from Clusiaceae was relatively conserved19, while plastomes of five riverweed species from Podostemaceae had highly variable structure20. Why closely related families have so diverged plastome structure? What is the plastome structural divergence pattern of this economically and ecologically important clade? The diversification pattern of plastome structure of the clusioid clade worth a further investigation. Here we determined five complete plastomes in the clusioid clade: Bonnetia paniculata Spruce ex Benth. (Bonnetiaceae), Me. ferrea (Calophyllaceae), Cratoxylum cochinchinense (Lour.) Blume. (Hypericaceae), Tristicha trifaria (Bory ex Willd.) Spreng. and Marathrum foeniculaceum Bonpl. (Podostemaceae). Comparison of the plastomes in this clade unveils significantly reduced IR regions in the plastomes of T. trifaria and Ma. foeniculaceum following the loss of ycf1 and ycf2. A large inversion over 50 kb spanning from trnK-UUU to rbcL in the LSC region is shared by C. cochinchinense, T. trifaria and Ma. foeniculaceum.
Results and Discussion
Plastome sequencing and general characteristics
Raw reads were all obtained through whole-genome sequencing. Due to the differences in plant materials and experimental procedures, the average coverage depth of plastomes varies from 89 to 1823 (Tables 1, Table S1). All five new plastomes of the clusioid clade exhibit a typical quadripartite structure. The plastome size among the sampled clusioids species ranges from 130,967 bp in T. trifaria to 161,473 bp in Me. ferrea. The length of IR ranges from 19,916 bp in Ma. foeniculaceum to 27,614 bp in Me. ferrea. The GC content of Ma. foeniculaceum is slightly lower. The plastome size and IR length of the two species from the Podostemaceae are significantly smaller than those of the other three clusioids families.
Phylogenetic relationships
A maximum likelihood tree was constructed using an 82-gene matrix. The clusioid clade was strongly supported with a bootstrap value (BS) of 100%. Previous studies such as Ruhfel et al. (2011) using three plastid and one mitochondrial loci and Xi et al. (2012) using broad-range sampling plastome data also strongly supported the clusioid clade17,21. Our results are congruent with previous studies, which resolved a well-supported (Bonnetiaceae, Clusiaceae) clade as the early diverged lineage, and strongly supported Calophyllaceae being the sister to the strongly supported (Hypericaceae, Podostemaceae) clade17,21,22 (Fig. 1). There are also many morphological characteristics of species in this clade supporting these phylogenetic relationships. Though the position of the wholly aquatic Podostemaceae has been very difficult to be determined owing to their highly atypical morphology, the terrestrial members of this clade (i.e., Bonnetiaceae, Calophyllaceae, Clusiaceae, and Hypericaceae) have long been considered closely related17,18,21. Bonnetiaceae and Clusiaceae share staminal fascicles opposite the petals. Hypericaceae and Podostemaceae share tenuinucellate ovules17. Additionally, some members of Hypericaceae and Podostemaceae have papillate stigmas. Besides, Hypericaceae, Calophyllaceae, and some Podostemaceae share resin-containing glands or canals that are especially visible in the leaves17. The phylogeny of the clusioid clade provides a framework for understanding the evolutionary history of this morphologically and ecologically diverse clade.

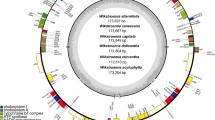
The plastid phylogeny of the clusioid clade. Maximum-likelihood (ML) tree inferred from the 82-gene (76 protein-coding and 4 rRNA genes) matrix. The number at each node indicates the ML bootstrap values. Species in the clusioid clade were color-coded according to family. A part of their plastome organization is shown on the right. The gray blocks indicate the IR regions. Gene lengths are not to scale. Gene arrow tips indicate the direction of transcription.
Plastome evolution
Plastomes structure of the early diverged three families (Clusiaceae, Bonnetiaceae and Calophyllaceae) are relatively conserved with only a few gene losses or pseudogenes. The infA gene and the second intron of ycf3 are lost in the plastid genome of G. mangostana, B. paniculata and Me. ferrea. The ndhK gene is pseudogene due to the presence of an internal stop codon in B. paniculata. Other gene losses include the rps16 gene in B. paniculata and the rpl32 gene in G. mangostana. However, the other two families (Hypericaceae and Podostemaceae) show more considerable plastome structural variations. The plastomes of C. cochinchinense, T. trifaria and Ma. foeniculaceum have lost the infA and rps16 genes, the second intron of the clpP gene, the second intron of the ycf3 gene, and the intron of the rps12 gene. The rpl32 gene in T. trifaria and Ma. foeniculaceum and the rps7 gene in Ma. foeniculaceum are pseudogenes due to the presence of premature stop codons. Additional gene losses in T. trifaria and Ma. foeniculaceum include plastid hypothetical ORFs (ycf genes), the ycf1 and ycf2 genes. As a result of the two ycf genes losses, the plastomes of the two Podostemaceae species are significantly smaller than the other four sequenced plastomes of the clusioid clade. IRs have expanded approximately 800 bp at the IR/SSC boundary in Ma. foeniculaceum, resulted in the relocation of the rps15 gene from SSC to IR.
The two identical copies of IR provide a template for error correction when a mutation occurs in one of the copies, and hence likely suppress the substitution rate in the IR3. Previous studies have reported the increased substitution rate of genes relocated from IR into SC23, and the decreased substitution rate of genes relocated from SC into IR24. However, relocation of ycf2 from IR into SC did not followed by an accelerated substitution rate, which has been explained by a recently occurred event in ginkgo evolution25. Studies in Pelargonium plastomes also found that expansion of IR does not result in decreased substitution rates of the relocated genes, suggesting the lineage- and locus-specific rate heterogeneity may have a larger effect that the IR on the substitution rate variation in plastid genes3,24. In our study, the relocated rps15 gene didn’t show decreased substitution rate (LRT p-value: 0.21, df = 1, details in Table S2). Since the relocation of rps15 did not accumulate significant mutations, we hypothesize that this relocation occurred recently or the rps15 gene is simply too short for the substitution rate to be detected. Our study supplies another case that the gene relocated into IR does not show decreased substitution rates. Patterns of molecular evolution in the IR and SC regions differ, most notably by a reduced rate of nucleotide substitution in the IR compared to the SC region, but the evolutionary consequences may be more complex than previous suggested3,24.
The loss of ycf1 and ycf2 genes have been documented in the plastomes of Poaceae26, Geraniaceae27, and Ericaceae28. The functions of both ycf genes are still controversial. Studies in tobacco (Nicotiana tabacum) and green algae (Chlamydomonas reinhardtii) suggested the ycf1 and ycf2 genes should not be related to photosynthesis, but encode products that are essential for cell survival29. These two genes have been inferred to be involved in cell division, DNAs/mRNA binding, protein assembly and transport, etc29. One essential function of the ycf1 and ycf2 genes might be linked to expression, assembly, or function of the accD gene product28,30. Some Poaceae species have lost both ycf genes in addition to the accD gene31. Plants that have lost the accD gene have divergent ycf1 and ycf2 sequences30. The plastome of T. trifaria and Ma. foeniculaceum, which have lost ycf1 and ycf2, have highly divergent accD sequences with only 51.8% and 51.1% identical sites, respectively, comparing with that of the early diverged three families (Clusiaceae, Bonnetiaceae and Calophyllaceae). Interestingly, the plastome of C. cochinchinense, which contains the two ycf genes, also has highly variable accD sequences with only 51.8% identical sites comparing with the firstly diverged three families. Further investigation is required to clarify the coevolution of accD and two ycf genes. Why plastomes of these taxa lost two ycf genes remains unclear, and they are also worth further explorations.
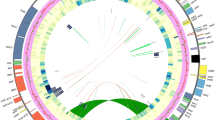
Inversions have been fully characterized in a number of plastomes and represent an essential mechanism for plastome rearrangements2. A Large inversion spanning from trnK-UUU to rbcL in the LSC region is shared by three plastomes of Hypericaceae and Podostemaceae (56 kb and 52 kb respectively; Fig. 2). The inversions are about 4 kb shorter in T. trifaria and Ma. foeniculaceum than that in C. cochinchinense, mainly due to the loss of some intergenic sequences in the Podostemaceae plastomes. Parallel inversions utilizing the same endpoints in distantly related taxa are extremely rare6. Within Fabaceae, a 50-kb inversion occurs in most Papilionoideae except a few basal lineages14,32. Interestingly, the large inversion of Hypericaceae and Podostemaceae contains all the genes in the 50-kb inversion of Papilionoideae, with two extra genes (trnK-UUU and matK) at one breakpoint. Another breakpoint of this inversion is located between rbcL and accD, being identical to that of the 50-kb inversion of Papilionoideae. Earlier studies have demonstrated a strong correlation between repetitive sequences and the incidence of inversions. In several cases, dispersed repeats have been inferred to promote inversions through intramolecular recombination2,15. The distribution of repeats was found to be strongly associated with breakpoints in the rearranged plastomes of Geraniaceae27. Studies confirmed that a specific plastomic inversion of a 34-kb fragment in Calocedrus macrolepis was likely to be mediated by an 11-bp IR. A 36-kb inversion in Lupinus and a 39-kb inversion in Robinia are probably mediated by a pair of 29-bp sIRs situated in the 3′-ends of two trnS genes13. No repeats have been found in boundary regions of the inversions in T. trifaria and Ma. foeniculaceum. While a pair of 76-bp sIRs are found at the breakpoints of the 56-kb inversion in C. cochinchinense (Fig. S1), which probably mediated this inversion.

Synteny and rearrangements detected in eight plastomes using the progressiveMauve. Light green-colored regions represent the large inversion shared by the plastomes of Cratoxylum cochinchinense (Hypericaceae), Tristicha trifaria and Marathrum foeniculaceum (Podostemaceae).
Methods
Taxon sampling and DNA sequencing
The previously published plastome of G. mangostana19 (NC_036341) in this clade was included in comparative analyses. Illumina sequencing data of two species were obtained from the NCBI Sequence Read Archive (accession no. SRR7121482 and SRR7121944) representing two families in clusioids: Me. ferrea (Calophyllaceae) and C. cochinchinense (Hypericaceae). Three species were sampled to represent the other two families of this clade: B. paniculata (Bonnetiaceae), T. trifaria and Ma. foeniculaceum (Podostemaceae). Two representative species, Licania heteromorpha (NC_024062) from Malpighiales and Averrhoa carambola (NC_033350) from Oxalidales were included as outgroups.
Total genomic DNA of B. paniculata, T. trifaria, and Ma. foeniculaceum was isolated from specimens using the DNeasy Plant Mini Kit, then fragmented to construct short-insert (350 bp) library following manufacturer’s manual (Illumina). Paired-end sequencing was performed on Illumina HiSeq X TEN at Plant Germplasm and Genomics Center (Kunming Institute of Botany, Chinese Academy of Sciences). Details of sample collection are listed in Table S1.
Genome assembly, annotation and analyses
The paired-end reads were filtered and assembled into complete plastome using GetOrganelle v1.6.1a33,34,35,36 under default settings, with kmers set dependent on the sequenced read length: -k 21,35,45,55,65,75,85,95,105,115,121 were used for 150-bp reads, while -k 21,45,65,85,89,91,95,99 were used for 100-bp reads. Final assembly graphs were checked in Bandage37. Two configurations of each plastome caused by the flip-flop recombination mediated by the IR were obtained, and one of the them was arbitrarily selected for downstream analysis since the plastome exists in two equimolar states38. All plastomes were initially annotated using PGA39 and GeSeq40, with annotated plastome from Amborella trichopoda (NC_005086) selected as the reference. For confirmation, all annotations were compared with the previously published plastome of G. mangostana and exon boundaries were manually adjusted in Geneious Prime41. All newly sequenced plastomes were deposited in GenBank under the accession nos. MK995178-MK995182. [Note to Reviewers: deposited sequences will be released immediately upon acceptance]
The 82 shared protein-coding and rRNA genes were extracted from the plastomes of eight species using “get_annotated_regions_from_gb.py” (https://github.com/Kinggerm/PersonalUtilities/)42, then aligned with prank v.14060343. Phylogenetic analysis was performed using maximum likelihood methods with 1000 bootstrap replicates on RAxML version 8.2.1144. We used codeml implemented in PAML45 to estimate nucleotide substitution rates of the rps15 gene in Ma. foeniculaceum under the null model (1 dN/dS ratios for all branches) and alternative model (2 or more dN/dS ratios for branches). The codon frequencies were determined using F3 × 4 model. The 2-rate model was tested against the 1-rate model by LRT using chi2 in PAML. One copy of IR was removed from each plastome and the remaining genome sequences were aligned using the progressiveMauve algorithm in Mauve v2.3.146. In order to identify and discard small or insignificant genome rearrangements, the minimum LCB weight was set as 1588. Repeats were identified using the Find Repeats implanted in Geneious Prime. The criteria used were set as follows: minimum repeat length: 30 bp, maximum mismatches: 3%, exclude repeats up to 10 bp longer than contained repeat and exclude contained repeats when longer repeats has frequency at least 3.
Data availability
The complete plastome sequences of Marathrum foeniculaceum, Tristicha trifaria, Cratoxylum cochinchinense, Mesua ferrea and Bonnetia paniculata sequenced in this study has been submitted to GenBank database under accession numbers MK995178-MK995182.
References
Wicke, S. et al. Mechanisms of functional and physical genome reduction in photosynthetic and nonphotosynthetic parasitic plants of the broomrape family. Plant Cell 25, 3711–3725 (2013).
Mower, J. P. & Vickrey, T. L. Structural diversity among plastid genomes of land plants in Advances in botanical research Vol. 85 (ed. Jansen, R.K. Chaw, S.M.) 263-292 (Academic Press, 2018).
Weng, M. L., Ruhlman, T. A. & Jansen, R. K. Expansion of inverted repeat does not decrease substitution rates in Pelargonium plastid genomes. New Phytol 214, 842–851 (2016).
Daniell, H., Lin, C. S., Yu, M. & Chang, W. J. Chloroplast genomes: diversity, evolution, and applications in genetic engineering. Genome Biol 17, 134 (2016).
Sabir, J. et al. Evolutionary and biotechnology implications of plastid genome variation in the inverted-repeat-lacking clade of legumes. Plant Biotechnol J 12, 743–754 (2014).
Rabah, S. O. et al. Passiflora plastome sequencing reveals widespread genomic rearrangements. J Syst Evol 57, 1–14 (2019).
Chumley, T. W. et al. The complete chloroplast genome sequence of Pelargonium × hortorum: organization and evolution of the largest and most highly rearranged chloroplast genome of land plants. Mol Biol Evol 23 (2006).
Jansen, R. K. et al. Analysis of 81 genes from 64 plastid genomes resolves relationships in angiosperms and identifies genome-scale evolutionary patterns. P Natl Acad Sci USA 104, 19369–19374 (2007).
Magee, A. M. et al. Localized hypermutation and associated gene losses in legume chloroplast genomes. Genome Res 20, 1700–1710 (2010).
Park, S., Jansen, R. K. & Park, S. Complete plastome sequence of Thalictrum coreanum (Ranunculaceae) and transfer of the rpl32 gene to the nucleus in the ancestor of the subfamily Thalictroideae. BMC Plant Biol 15, 40 (2015).
Hupfer, H. et al. Complete nucleotide sequence of the Oenothera elata plastid chromosome, representing plastome I of the five distinguishable Euoenothera plastomes. Mol Gen Genet 263, 581–585 (2000).
Walker, J. F., Zanis, M. J. & Emery, N. C. Comparative analysis of complete chloroplast genome sequence and inversion variation in Lasthenia burkei (Madieae, Asteraceae). Am J Bot 101, 722–729 (2014).
Wang, Y. H. et al. Plastid genome evolution in the early-diverging legume subfamily Cercidoideae (Fabaceae). Front Plant Sci 9, 138 (2018).
Doyle, J. J., Doyle, J. L., Ballenger, J. A. & Palmer, J. D. The distribution and phylogenetic significance of a 50-kb chloroplast DNA inversion in the flowering plant family Leguminosae. Mol Phylogenet Evol 5, 429–438 (1996).
Keller, J. et al. The evolutionary fate of the chloroplast and nuclear rps16 genes as revealed through the sequencing and comparative analyses of four novel legume chloroplast genomes from Lupinus. DNA Res 24, 343–358 (2017).
Qu, X. J., Wu, C. S., Chaw, S. M. & Yi, T. S. Insights into the existence of isomeric plastomes in Cupressoideae (Cupressaceae). Genome Biol Evol 9, 1110–1119 (2017).
Ruhfel, B. R. et al. Phylogeny of the clusioid clade (Malpighiales): evidence from the plastid and mitochondrial genomes. Am J Bot 98, 306–325 (2011).
Ruhfel, B. R., Bove, C. P., Philbrick, C. T. & Davis, C. C. Dispersal largely explains the Gondwanan distribution of the ancient tropical clusioid plant clade. Am J Bot 103, 1117–1128 (2016).
Jo, S. et al. The complete plastome of tropical fruit Garcinia mangostana (Clusiaceae). Mitochondrial DNA B 2, 722–724 (2017).
Bedoya, A. M. et al. Plastid genomes of five species of riverweeds (Podostemaceae): structural organization and comparative analysis in Malpighiales. Front Plant Sci 10 (2019).
Xi, Z. X. et al. Phylogenomics and a posteriori data partitioning resolve the Cretaceous angiosperm radiation Malpighiales. P Natl Acad Sci USA 109, 17519–17524 (2012).
Li, H. T. et al. Origin of angiosperms and the puzzle of the Jurassic gap. Nat Plants 5, 461–470 (2019).
Perry, A. S. & Wolfe, K. H. Nucleotide substitution rates in legume chloroplast DNA depend on the presence of the inverted repeat. J Mol Evol 55, 501–508 (2002).
Li, F. W., Kuo, L. Y., Pryer, K. M. & Rothfels, C. J. Genes translocated into the plastid inverted repeat show decelerated substitution rates and elevated GC content. Genome Biol Evol 8, 2452–2458 (2016).
Lin, C. P., Wu, C. S., Huang, Y. Y. & Chaw, S. M. The complete chloroplast genome of Ginkgo biloba reveals the mechanism of inverted repeat contraction. Genome Biol Evol 4, 374–381 (2012).
Guisinger, M. M., Chumley, T. W., Kuehl, J. V., Boore, J. L. & Jansen, R. K. Implications of the plastid genome sequence of Typha (Typhaceae, Poales) for understanding genome evolution in poaceae. J Mol Evol 70, 149–166 (2010).
Weng, M. L., Blazier, J. C., Govindu, M. & Jansen, R. K. Reconstruction of the ancestral plastid genome in Geraniaceae reveals a correlation between genome rearrangements, repeats, and nucleotide substitution rates. Mol Biol Evol 31, 645–659 (2014).
Braukmann, T. W. A., Broe, M. B., Stefanovic, S. & Freudenstein, J. V. On the brink: the highly reduced plastomes of nonphotosynthetic Ericaceae. New Phytol 216, 254–266 (2017).
Drescher, A., Ruf, S., Calsa, T. Jr., Carrer, H. & Bock, R. The two largest chloroplast genome-encoded open reading frames of higher plants are essential genes. Plant J 22, 97–104 (2000).
Delannoy, E. & Fujii, S. Colas des Francs-Small, C., Brundrett, M. & Small, I. Rampant gene loss in the underground orchid Rhizanthella gardneri highlights evolutionary constraints on plastid genomes. Mol Biol Evol 28, 2077–2086 (2011).
Doyle, J. J., Davis, J. I., Soreng, R. J., Garvin, D. & Anderson, M. J. Chloroplast DNA inversions and the origin of the grass family (Poaceae). Proc Natl Acad Sci USA 89, 7722–7726 (1992).
Cardoso, D. et al. Revisiting the phylogeny of papilionoid legumes: new insights from comprehensively sampled early-branching lineages. Am J Bot 99, 1991–2013 (2012).
Camacho, C. et al. BLAST+: architecture and applications. BMC Bioinformatics 10, 421 (2009).
Bankevich, A. et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol 19, 455–477 (2012).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat Methods 9, 357–359 (2012).
Jin, J.J. et al. GetOrganelle: a fast and versatile toolkit for accurate de novo assembly of organelle genomes. bioRxiv 256479, https://doi.org/10.1101/256479 (2019).
Wick, R. R., Schultz, M. B., Zobel, J. & Holt, K. E. Bandage: interactive visualization of de novo genome assemblies. Bioinformatics 31, 3350–3352 (2015).
Walker, J. F., Jansen, R. K., Zanis, M. J. & Emery, N. C. Sources of inversion variation in the small single copy (SSC) region of chloroplast genomes. Am J Bot 102, 1751–1752 (2015).
Qu, X. J., Moore, M. J., Li, D. Z. & Yi, T. S. PGA: a software package for rapid, accurate, and flexible batch annotation of plastomes. Plant Methods 15, 50 (2019).
Tillich, M. et al. GeSeq - versatile and accurate annotation of organelle genomes. Nucleic Acids Res 45, W6–W11 (2017).
Kearse, M. et al. Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28, 1647–1649 (2012).
Zhang, R. et al. Exploration of plastid phylogenomic conflict yields new insights into the deep relationships of Leguminosae. Syst Biol, https://doi.org/10.1093/sysbio/syaa013 (2020).
Loytynoja, A. & Goldman, N. Phylogeny-aware gap placement prevents errors in sequence alignment and evolutionary analysis. Science 320, 1632–1635 (2008).
Stamatakis, A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313 (2014).
Yang, Z. PAML 4: phylogenetic analysis by maximum likelihood. Mol Biol Evol 24, 1586–1591 (2007).
Darling, A. E., Mau, B. & Perna, N. T. progressiveMauve: multiple genome alignment with gene gain, loss and rearrangement. PLoS One 5, e11147 (2010).
Acknowledgements
We thank the Missouri Botanical Garden for providing specimen materials, BGI researchers for confirmation of the SRA data and specimens, Cheng Liu from the laboratory of the Germplasm Bank of Wild Species, Kunming Institute of Botany for identification of the specimen, and Molecular Biology Experiment Center, Germplasm Bank of Wild Species in Southwest China for skillful laboratory assistance. This work was supported by grants from the Large-scale Scientific Facilities of the Chinese Academy of Sciences (No. 2017-LSF-GBOWS-02); the Science and Technology Basic Resources Investigation Program of China (2019FY100900); the Strategic Priority Research Program of Chinese Academy of Sciences (XDB31010000); and the National Natural Science Foundation of China [key international (regional) cooperative research project No. 31720103903]; the open research project for “Cross-Cooperative Team” of the Germplasm Bank of Wild Species, Kunming Institute of Botany, Chinese Academy of Sciences to Jian-Jun Jin.
Author information
Authors and Affiliations
Contributions
D.M.J., J.J.J. and T.S.Y. conceived and designed the study; D.M.J. and J.J.J. analyzed data. D.M.J. wrote the manuscript, with contributions from all of the authors; All authors critically reviewed the paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Jin, DM., Jin, JJ. & Yi, TS. Plastome Structural Conservation and Evolution in the Clusioid Clade of Malpighiales. Sci Rep 10, 9091 (2020). https://doi.org/10.1038/s41598-020-66024-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-66024-7
This article is cited by
-
Decoding the complete chloroplast genome of Cissus quadrangularis: insights into molecular structure, comparative genome analysis and mining of mutational hotspot regions
Physiology and Molecular Biology of Plants (2023)
-
Gene loss, genome rearrangement, and accelerated substitution rates in plastid genome of Hypericum ascyron (Hypericaceae)
BMC Plant Biology (2022)
-
Characterization of the plastid genome of Cratoxylum species (Hypericaceae) and new insights into phylogenetic relationships
Scientific Reports (2022)
-
Calophyllaceae plastomes, their structure and insights in relationships within the clusioids
Scientific Reports (2021)
-
The evolutionary fate of rpl32 and rps16 losses in the Euphorbia schimperi (Euphorbiaceae) plastome
Scientific Reports (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
