
Abstract
The Zygophyllum and Tetraena genera are intriguingly important ecologically and medicinally. Based on morphological characteristics, T. hamiensis var. qatarensis, and T. simplex were transferred from Zygophyllum to Tetraena with the least genomic datasets available. Hence, we sequenced the T. hamiensis and T. simplex and performed in-depth comparative genomics, phylogenetic analysis, and estimated time divergences. The complete plastomes ranged between 106,720 and 106,446 bp—typically smaller than angiosperms plastomes. The plastome circular genomes are divided into large single-copy regions (~ 80,964 bp), small single-copy regions (~ 17,416 bp), and two inverted repeats regions (~ 4170 bp) in both Tetraena species. An unusual shrinkage of IR regions 16–24 kb was identified. This resulted in the loss of 16 genes, including 11 ndh genes which encode the NADH dehydrogenase subunits, and a significant size reduction of Tetraena plastomes compared to other angiosperms. The inter-species variations and similarities were identified using genome-wide comparisons. Phylogenetic trees generated by analyzing the whole plastomes, protein-coding genes, matK, rbcL, and cssA genes exhibited identical topologies, indicating that both species are sisters to the genus Tetraena and may not belong to Zygophyllum. Similarly, based on the entire plastome and proteins coding genes datasets, the time divergence of Zygophyllum and Tetraena was 36.6 Ma and 34.4 Ma, respectively. Tetraena stem ages were 31.7 and 18.2 Ma based on full plastome and protein-coding genes. The current study presents the plastome as a distinguishing and identification feature among the closely related Tetraena and Zygophyllum species. It can be potentially used as a universal super-barcode for identifying plants.
Similar content being viewed by others
Introduction
Accurately identifying plant species is necessary for the long-term use and conservation of biological resources1,2. Traditional taxonomy is focused on morphological characteristics, requiring taxonomists with extensive taxonomy knowledge and careful examination of specimens3. However, when there are many specimens in a study4, morphological analysis can be time-consuming, leading to a significant decrease in the authenticity of species identification. Furthermore, if available specimens belong to a complex genus, accurate identification using the standard method may be inefficient1,5. Because of the limited and uncertain diagnostic attributes, the morphology-based process of identifying plant species has yet to be discovered. Molecular markers are very useful in plant identification and have played a pivotal role in systematics for a long time. However, the currently available loci (DNA) only work for the closely related species, not all6.
Short, standardized DNA segments are used in DNA barcoding as an additional tool for morphological taxonomy. Paul Hebert first suggested it in 2003, and it was soon recognized as a valuable method for identifying and discovering new species7. Multiple genes, such as three plastid regions (e.g., rbcL, matK, and trnH- psbA) and nuclear DNA (Internal Transcribe Spacer—ITS) are usually recognized as universal DNA barcodes for terrestrial plants. However, only some barcodes can successfully identify most species8,9. DNA barcodes may be ineffective for complex taxonomic groups, especially in newly evolved and rapidly expanded taxa, for subspecies discrimination10. Therefore, there is a critical need to create new, reliable methods to meet the requirements of classifying complicated plant species. With the current advancement in next-generation sequencing (NGS) technologies, the accuracy and quality of DNA sequencing have considerably improved the species discrimination for complex genera11,12.
The Chloroplast genome (plastome) is an excellent resource for resolving the tree of life and identifying taxa11,13 Numerous studies have demonstrated that the chloroplast genome is a robust and effective method for uncovering plant phylogeny and evolutionary history more accurately14,15. Unlike the four standard barcodes, due to their significant genetic variety, DNA super-barcode, which employs the entire chloroplast genome sequence, can distinguish between closely related species11,16,17. Despite this, DNA super-barcodes face criticism and challenges in species identification, such as high sequencing costs, lack of a comprehensive cp-genome database, and difficulty tracking species boundaries18. In recent years, as the cost of NGS sequencing has decreased and analysis methods for genomes have improved, large plastomes datasets have become available in GenBank. This leads to several genera having corresponding reference genomes for species identification and phylogenomics12,19. Similarly, certain plastome regions identified as unique barcodes are tested and employed in problematic taxa, such as accD and rrn16-rrn23 for yew species11 and psbE-psbL and ndhA intron for Fagopyrum species20.
Plastome has more variation with a significantly higher resolution of phylogenies than the most often used and predicted genus-specific DNA barcodes, which is beneficial for revealing phylogenetic connections between closely related species21. Plastome has been widely applied in plant identification22, phylogenetic analyses16,22,23,24,25 and plant population studies25. Even with reference genomes or specific fragments, most taxa, such as Zygophyllum and Tetraena, can build a comprehensive database. The genus Zygophyllum L., located in the Arabian Peninsula, has received much interest because of its morphological and anatomical characteristics26,27,28,29,30,31,32,33. Most Saudi Arabian Zygophyllum taxa were reassigned to Tetraena, according to Beier et al.34 most recent Tetraena and Zygophyllum taxonomic proposal. In Zygophyllum and Tetraena, the growth habits, leaf features, floral traits, and fruit shapes are morphologically identical. Beier et al.34 employed fruit dehiscence and staminal appendages to distinguish Zygophyllum and Tetraena. With six genera: Zygophyllum, Fagonia, Augea, Roepera, Tetraena, and Melocarpum, the Zygophylloideae subfamily is currently the largest34,35. Sheahan and Chase36 examined rbcL and trnL-F sequences and discovered that Tetraena is nested within the large and diverse Zygophyllum.
Tetraena qatarensis (Hadidi), vernacular ‘Haram’ in Arabic, is an element of the flora of Eastern Saudi Arabia30 and a constituent specie of coastal lowland vegetation of Saudi Arabia37. El Hadidi26 reported the Z. qatarense as a new species of Qatar. Hosny29 also identified three varieties and thirteen Zygophyllum species, including some Saudi Arabian varieties and species. The Arabian Peninsula and Saudi Arabia are home to Z. qatarense26. According to different authors27,29,33,38,39,40,41,42,43,44,45,46,47,48,49,50,51, Z. mandavillei, Z. hamiense and Z. qatarense are three different species, whereas others (Chaudhary31) believed that the other two are varieties of Z. hamiense because of similar fruit and leaflet morphology. Sheahan and Chase36 studied the phylogenetic relationship of Zygophyllaceae based on anatomy, morphology, and rbcl gene sequence and found that Fagonia is a sister to the Zygophyllaceae family. Interestingly, Z. fabago is sister to Augea genus while Z. simplex might not belong to Zygophyllum but at the same time is sister to the Tetraena genus. Zygophyllum simplex L. is a salt-tolerant plant that belongs to Zygophyllaceae locally (in Saudi Arabia)38,52. Later, Sheahan and Chase36 selected thirty-six Zygophyllaceae taxa, including fifteen Zygophyllum species of regions like Australia, Africa, and Southeast Asia based on plastome genes rbcL and trn-F (non-coding), investigated the phylogenetic relationship. It was found that Zygophyllum is polyphyletic and eventually classified into five clades with high bootstrap values. Furthermore, according to molecular investigations, Tetraena is also located within the large, paraphyletic Zygophyllum36. Later on, these species, with other 33 species, were transferred from Zygophyllum to Tetraena by Beier et al.34 and are currently known as T. hamiensis (Schweinf.) Beier & Thulin, 2003; T. mandavillei (Hadidi) Beier & Thulin, 2003; and T. hamiensis var. qatarensis (Hadidi)34. According to Ghazanfar and Osborne53, Tetraena species found in the Arabian Peninsula and Saudi Arabia include T. qatarensis, T. decumbens, T. coccinea, T. alba, T. propinqua, T. hamiensis, T. dumosa, T. mandavillei, and T. simplex.
Compared to the phylogenetic tree based on individual barcodes, the full plastomes tree has a greater supporting rate and lesser discrimination54. Li et al.18 suggested that the complete plastome be used as a super-barcode to identify closely related species accurately. This study sequenced the full plastomes of Tetraena qatarensis and Tetraena simplex and compared them to previously published complete plastomes of Zygophyllum and Tetraena species. We analyzed the full plastomes of the Tetraena and Zygophyllum species, which are difficult to distinguish based on appearance and taxonomy alone. Our study had three objectives: (i) understand whether the plastome can be used as a super-barcode to identify closely related species, (ii) sequence new plastomes from Tetraena species and explore polymorphic regions within plastomes, and (iii) assess the discrimination power of plastomes in the genus Tetraena. This study's findings may assist in investigating the possibility of using the plastome to differentiate between different Tetraena species and serve as a barcode to distinguish between closely related species.
Results
General features of T. hamiensis var. qatarensis and T. simplex of plastomes and their comparison to related species plastomes
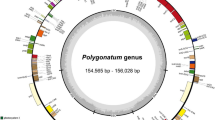
The complete sequenced plastomes of Tetraena species, T. hamiensis var. qatarensis (OM809718) and T. simplex (OL943588) have typical circular quadripartite structures like other angiosperm plastomes. However, plastome sizes of T. hamiensis var. qatarensis and T. simplex are 106,720 bp and 106,446 bp, respectively, which are smaller than typical angiosperms plastomes; each circular genome is divided into large single-copy (LSC) region (80,964 bp, 80,554 bp), small single-copy (SSC) region (17,416 bp, 17,280 bp) and two inverted repeats (IR) regions (4170 bp, 4306 bp) (Figs. 1, S1 and Table 1). The plastomes were analyzed and compared with eleven other PCD (protein coding region) Zygophyllaceae (three Tetraena, five Zygophyllum, one Larrea, one Tribulus, and one Guaiacum) species. In all these plastomes, the most significant size was noted for Tr. terrestris (158,184 bp) followed by L. tridentate (136,194 bp) and G. angustifolium (130,809 bp), while the smallest genome size was noted for Z. fabago2 (104,590 bp). Similarly, the highest LSC and SSC regions were found in Tr. terrestris, 88,878 bp and 17,622 bp respectively and the smallest LSC in Z. fabago2 (79,170 bp), while the smallest SSC was identified in Z. xanthoxylon2 (15,674 bp). Furthermore, the IR region in T. hamiensis var. qatarensis and T. simplex are 4170 bp and 4306 bp respectively; among the other species of Tetraene and Zygophyllum, the IR regions were found in a similar range between 4288 bp (Z. xanthoxylon1) to 4669 bp (Z. fabago1). Interestingly, the smallest IR region was found in T. hamiensis var. qatarensis. The GC content in both sequenced plastomes of T. hamiensis var. qatarensis and T. simplex were 33.6% and 33.9% respectively. The highest GC content (35.8%) was determined in Tr. terrestris plastome.

Circos view of complete plastome of T. hamiensis var. qatarensis with related species. Genes shown on the outside outer circle are transcribed counterclockwise and, on the inside, clockwise. (Track 1) the pink color track show guanine-cytosine (GC) skew. The heatmap track from outside to inside is nucleotide diversity (Pi) of T. hamiensis var. qatarensis with three Tetraena species (track 2), five Zygophyllum species (track 3), and twelve species plastomes from the family Zygophyllaceae used in this study (track 4). The inner three heatmap tracks show SNP and InDels of T. hamiensis var. qatarensis with three Tetraena species (track 5), five Zygophyllum species (track 6), and twelve species plastomes from the family Zygophyllaceae (track 7). Dark green color links show the inverted repeats (IR) region of T. hamiensis var. qatarensis. The palindromic, forward, and reverse repeats are linked by lines in different colors: forward repeats (green), reverse repeats (light blue), and palindromic repeats (red) color. The heatmap was generated using CIRCOS (http://circos.ca/software/).
Gene content and gene loss in Tetraena plastomes
The gene content of the Zygophyllaceae plastomes varied considerably. These plastomes contained 67–84 protein-coding genes, 4–8 rRNA genes, and 32–37 tRNA genes (Fig. 1; Table 1). The number of genes annotated in a plastome ranged from 104 (T. mongolica1) to 130 (Tr. terrestris). The plastomes T. hamiensis var. qatarensis and T. simplex sequenced in this study had 106 and 105 genes including four rRNA genes, 34 and 33 tRNA genes, and 69 and 68 protein-coding genes, respectively (Table S1). In both plastomes, the protein-coding region, tRNA, and rRNA lengths were 46,598–47,091 bp, 2461–2561 bp, and 4523–4524 bp, respectively (Table 1). In most higher plants the rRNA genes are located in the IRs region however, these are present in the SSC region Tetraena and Zygophyllum species plastomes. Subsequently, the copy number of rRNA genes changes from 2 to 1. We compared the five Tetraena, five Zygophyllum, Tribulus, Larrea, and Guaiacum plastomes. We found that all ndh genes encoding subunits of NADH oxidoreductase were lost in all Tetraena and Zygophyllum species except Z. fabago1 and Z. xanthozylon2 where one copy of ndhI retain (Fig. S2). Similarly, one copy of ndhE gene was also detected in the Z. xanthozylon2 plastome. These genes are usually located in SSC and IR regions. Moreover, rpl12, ycf2, ycf1, and infA, common chloroplast genes, were lost in all Tetraena and Zygophyllum plastomes. Furthermore, the ycf15 gene was absent in T. simplex, T. mongolica1, L. tridentata, Tr. terrestris and G. angustifolium plastome. Notably, the accD was absent in L. tridentata and G. angustifolium plastomes. Two copies of rps19, rpl2 and rpl23 genes were found in all Tetraena and Zygophyllum species while one copy of ndhB, rrn4.5, rrn16, rrn5 and rrn24 were lost with the contraction of inverted repeat regions (Fig. S2).
In both T. hamiensis var. qatarensis and T. simplex about 13.40% and 14.7% of the functional genes contain introns, including 5 and 7 tRNA, 9 and 10 protein-coding genes, respectively and only one gene ycf3 contains two introns (Table S2). Interestingly, in both T. hamiensis var. qatarensis and T. simplex and other Zygophyllum and Tetraena plastomes, we observed the absence of clpP and rps12 three end introns. Observation of loss of both introns represents the first documented loss within the Zygophyllaceae plastomes. Our study revealed that the loss of the clpP and rps12 introns are limited to the loss of large IR regions in these plastomes with only one exception where rps13 both introns were detected in Z. fabago3 (MK341052) plastome. Furthermore, the length of these introns ranges from 458 bp (trnL-UAA) to 2579 bp (trnK-UUU). The trnA-UGC gene was found with one intron in T. simplex; however, in T. hamiensis var. qatarensis, it was found to be an intron-less gene (Table S2).
Functional repeats analysis
In functional repeat analysis, various repetitive sequences in plastomes of T. hamiensis var. qatarensis and T. simplex and other related species were determined (Figs. 1, 3). Total of 19 palindromic, 26 forward, five reverse and 61 tandem repeat sequences were identified in T. hamiensis var. qatarensis plastome. The T. simplex plastome contained 20 palindromic, 17 forward, 13 reverse and 47 tandem repeat sequences (Fig. 2E). The L. tridentata plastome contained the highest number of palindromic repeats (27), while the Z. fabago2 plastome had the lowest (15) palindromic repeat sequences. The Z. xanthoxylon plastomes had maximum forward repeat sequences (33) and minimum reverse repeats (1). The highest number of reverse repeats was identified in plastome of T. mongolica2 (15). The minimum number of forwarding repeats (12) was found in Z. fabago3. Similarly, in Z. xanthoxylon2 plastome, 65 tandem repeats were determined, which is the highest in all the plastomes included in the present study (Fig. 2D).

Repetitive sequences in T. hamiensis var. qatarensis, T. simplex, and related species plastomes. (A) Lengthwise frequency of palindromic repeats in plastomes, (B) Lengthwise frequency of forward repeats, (C) Lengthwise frequency of reverse repeats, (D) Lengthwise frequency of tandem repeats, and (E) Total number of repetitive sequences.
Furthermore, maximum number of palindromic, forward and reverse repeat sequences in all plastomes identified were 21–30 bp in length except in plastomes of Z. fabago1, Z. fabago2 and Z. xanthoxylon1 in which the highest number of mentioned repetitive sequences were 11–20 bp in length (Fig. 2). Among all plastomes the forward repeat sequences of > 50 bp length were most in Z. Fabago1 (31) and Z. xanthoxylon2 (29) (Fig. 2A). The highest frequency of tandem repeat sequences was identified in 1–30 bp length, with highest number (28) in T. hamiensis var. qatarensis (Fig. 2D). As noted for Z. Fabago1 (31) and Z. xanthoxylon2 (29), in forward repeat sequences, the highest number of tandem repeats were also maximum in > 45 bp length.
Simple sequence repeats (SSRs) analysis
The SSRs were analyzed in all plastomes included in the study (Fig. 3). Like other functional repeat sequences, SSR numbers differed in all these plastomes. Total number of SSRs ranged from 162 (T. mongolica3) to 225 (Z. fabago1). The total SSRs in sequenced plastomes of T. hamiensis var. qatarensis and T. simplex were 166 and 170 respectively (Fig. 3K). Moreover, T. hamiensis var. qatarensis have 162 mono, one di, one tri and two penta-nucleotides SSRs while T. simplex has 164 mono, two di, one tri and three penta-nucleotides SSRs. Surprisingly, no tetra-nucleotides SSRs were identified in all plastomes included in this study. Unlike the other plastomes, the SSR numbers were not correlated with the size of plastomes. The highest sized plastome (Tr. terrestris) has a similar number of SSRs as the smaller-sized plastome (T. hamiensis var. qatarensis). Interestingly, the highest number of total SSRs were determined in the plastome of Z. fabago2 which has the smallest size compared to plastomes.

Simple sequence repeats (SSRs) in T. hamiensis var. qatarensis, T. simplex, and related species plastomes. (A) Mono-nucleotides SSRs (B) Di-nucleotides SSRs, (C) tri-nucleotides SSRs, (D) Penta-nucleotides SSRs, (E) hexa-nucleotides SSRs, (F) number of SSRs in protein-coding region, (G) number of SSRs in inverted repeats region, (H) number of SSRs in inverted repeats region small single-copy region, (I) number of SSRs in the large single-copy region, (J) SSR motif frequency in plastomes, (K) total number of SSRs in plastomes.
As for the complete plastomes the SSRs numbers were also determined in various parts of the plastomes like LSC, SSC, IR and protein coding regions. LSC regions of all plastomes had the highest number of SSRs (Fig. 3I). The plastomes of T. hamiensis var. qatarensis and T. simplex have 140 (84% of total SSRs) and 142 (83% of total SSRs) in LSC regions respectively, in which 137, 137 mono, 1,2 di and 2, 3 pentanucleotides were identified respectively. The sequenced plastomes contained 18 (17 mono and one tri nucleotides SSRs each) in the SSC region (Fig. 3H). Furthermore, the IR regions of the sequenced plastomes of T. hamiensis var. qatarensis and T. simplex have 4 and 5 (only mono-nucleotides) SSRs. interestingly the plastomes of L. tridentate and Tr. terrestris have 11 and 10 SSRs in IR regions (Fig. 3G). The difference among the SSR numbers in IR regions is attributed to the IR sizes in these plastomes. The highest number of SSRs in protein-coding region was detected in Tr. terrestris (41), L. tridentate (33) followed by T. hamiensis var. qatarensis (33). At the same time, lowest was determined in T. mongolica2 T. mongolica3 Z. fabago1 and Z. fabago2 (26 each) (Fig. 3F).
Comparative analysis and divergence
Comparison of complete plastomes of T. hamiensis var. qatarensis, T. simplex and 11 other closely related species were performed. The 13 complete plastomes and 58 shared protein coding genes were aligned and compared to determine the average pairwise distance among these species using T. hamiensis var. qatarensis as a reference (Table S3). Larrea tridentata (0.177384) showed the highest average pairwise distance, followed by Tribulus terrestris (0.163060). The minimum average pairwise distance was exhibited by T. mongolica1 (0.023) and T. simplex (0.027). Among the 58 shared protein coding genes, the atpF, clpP psbH, rpl20, rpl22, and rps2 were most divergent (Table S4). The highest average pairwise sequence divergence was determined for clpP gene across all compared species. Still, the highest divergence was recorded in Tribulus terrestris (0.642) and (0.505) followed by Z. fabago3 (0.177) and Z. xanthoxylon2 (0.172) (Table S4).
Furthermore, the values of nucleotide diversity (Pi) were determined in plastomes T. hamiensis var. qatarensis, T. simplex and other related species (Figs. 1, S3). The genomes were aligned in three different groups: (i) Two currently sequenced and three Tetraena plastomes, (ii) Two sequenced and five Zygophyllum plastomes, (iii) all plastomes included in the study to better evaluate and understand the nucleotide diversity (Pi). The nucleotide diversity (Pi) values within 200 bp window size and 100 bp step size across these plastomes vary from 0 to 0.2 (Fig. S3A), 0 to 0.3 (Fig. S3B) and 0 to 0.4 (Fig. S3C). Only 2 variable loci (psbE-PetL, clpP) were found with Pi > 0.1 in Tetraena plastomes while in Tetraena-Zygophyllum 4 variable loci (atpF, clpP, rps15-trnL, ycf2) were found with Pi > 0.2. In overall comparison of all 13 plastomes, several variable loci were identified having Pi value > 0.2, in which the highest Pi value of 0.323 was noted for (rps12) followed by (rpoC1) having Pi 0.285 (Fig. S3C).
Similarly, the codon usage frequency T. hamiensis var. qatarensis and T. simplex protein-coding genes were determined. The protein-coding genes in T. hamiensis var. qatarensis, and T. simplex were composed of 20,122 and 27,231 codons respectively (Table S5). The highest codon used in T. hamiensis var. qatarensis plastome was ATT (884) which codes for isoleucine. In T. simplex plastome, the maximum used codons were AAA (1142) which codes for lysine and AAG (1142) which codes for isoleucine (Table S5). Furthermore, Relative Synonymous Codon Usage (RSCU) was also analyzed for all 13 plastomes (Fig. 4). Surprisingly, almost all of these codons had half-synonymous codons that ended in A or T and had high RSCU values. The other half had low RSCU ended with C and G. The third codon position showed biased codon usage toward A and U, which is consistent with the pattern seen in the majority of angiosperm plastomes55.

Heatmap plot of codon distribution of all shared protein-coding genes in T. hamiensis var. qatarensis, T. simplex, and related plastomes. Color key: red indicates lower, yellow means moderate, while black indicates higher RSCU values.
Genome divergence in all 13 plastomes was characterized by mVISTA using T. hamiensis var. qatarensis as a reference genome (Fig. S4). The divergence in LSC region was higher than SSC and IRs. In all LSC, SSC, and IRs, the non-coding region diverges more than the coding region. A significant divergence was identified among the protein-coding regions in matK, atpA, atpF, rpoC1, rpoC2, rpoB, rps11, petB, petD and accD genes in LSC region. The divergence in these genes was lower in Tetraena plastomes compared to Zygophyllum and other related plastomes. The LSC region non-coding regions trnQ-trnR, trnC-trnT, trnM, trnT-trnV had higher divergence in all plastomes than T. hamiensis var. qatarensis (Fig. S4). Most of the SSC region's divergence was found in non-coding regions like trnV-trnI and trnL-trnI. Interestingly, in IR regions the divergences in rps19 and rpl2 genes were only detected in Tr. terrestris and L. tridentata.
The SNPs (single nucleotide polymorphism) and InDels were evaluated in the complete plastome of T. hamiensis var. qatarensis to that of all other plastomes (Fig. 1, Table 2). 5506 SNPs and 1600 InDels were detected in plastome T. hamiensis var. qatarensis to T. simplex. The minimum number of SNPs and InDels were detected in T. hamiensis var. qatarensis to T. mongolica3 (4812 SNPs) and T. hamiensis var. qatarensis to T. mongolica2 (1781 InDels) respectively. In Zygophyllum species the highest SNPs and InDel were identified in plastome T. hamiensis var. qatarensis to Z. fabago1 13,144 and 13,739 respectively. Overall, the highest number of SNPs were identified in plastome T. hamiensis var. qatarensis to L. tridentata (28,348) The highest InDels were 49,513 in T. hamiensis var. qatarensis to Tr. terrestris. Similarly, the SNPs and InDels were also evaluated in different parts of plastomes. In plastome T. hamiensis var. qatarensis to T. simplex the number of SNPs and InDels were 4,274 and 1455 (LSC), 792 and 168 (SSC), 132 and 8 (IR) and 3076 and 911 (protein coding region) respectively (Table 2). IRs had a minimum number of SNPs and InDels in all plastomes in all parts of the plastomes.
Unusual contraction of IR region detected in Tetraena plastomes
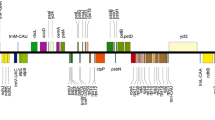
The IR regions in a plastome are considered the most preserved portions.. The IR regions contraction and expansion determine the variability in plastome sizes and are common evolutionary events. Herein, four IR junctions JLB (LSC/IRb), JSB (SSC/IRb), JSA (SSC/IRa), and JLA (LSC/IRa), boundary comparison analysis was performed, to determine the contraction and expansion events (Figs. 5, S5). The sub-familial members (Tetraena and Zygophyllum belonging to Zygophylloideae) showed high contraction in IR regions compared to the other family members L. tridentata, G. angustifolium and other family members Tr. terrestris, resulting in gene loss, gene order changes and rearrangements in the plastomes. The IR lengths in L. tridentata, G. angustifolium and Tr. terrestris, are 19,350, 17,744 bp and 25,842 bp respectively while the IR lengths in the sequenced plastomes T. hamiensis var. qatarensis, T. simplex is 4170 bp and 4306 bp, respectively. Furthermore, the location of genes at these junctions was also determined. The contraction of IR regions in Tetraena and Zygophyllum species including the two sequenced plastomes, resulted in altered gene locations. At JLB junction, all plastomes have noted variation in rpl22 (LSC) and rps19 (IRb) locations. The rpl22 gene is located 126 bp and 27 bp away from JLB in LSC region in plastomes T. hamiensis var. qatarensis and T. simplex respectively. Similarly, the rpl19 gene is 178 bp and 265 bp away from JLB in the IRb region in plastomes T. hamiensis var. qatarensis and T. simplex, respectively (Fig. 5). Moreover, in T. hamiensis var. qatarensis at the JSB ycf15 (842 bp away in IRb) and ccsA (909 bp away in SSC) are located while in T. simplex and T. mongolica1, the ycf15 gene is lost so rpl23 takes this position into IRb (1886 bp away) at JSB. Similarly, in Z. xanthoxylone2, the position of the rps19 gene is different than in other plastomes (Figs. 5, S5). In Tr. terrestris and L. tridentata plastomes, the location of genes has changed at JLB; the rps19 gene shifted from IRb to LSC in both plastomes with slight variation in position while in G. angustifolium it was like Tetraena and Zygophyllum plastomes (Fig. 5). Similarly, variations have been observed at JSB among Tetraena, Zygophyllum and other species. In Z. xanthoxylon1 the rpl32 gene is present in IRb unlike other plastomes at JSB. Furthermore, due to full lengths of IRs in Tr. terrestris and L. tridentata and G. angustifolium the genes arrangements at JSB and JSA are different from Tetraena and Zygophyllum plastomes. In these three plastomes the ycf1 genes are in IRb and the ndhF gene is located in SSC at JLB (Fig. 5).

Distances between adjacent genes and junctions of the small single-copy (SSC), large single-copy (LSC), and two inverted repeats (IR) regions among T. hamiensis var. qatarensis, T. simplex, and related plastomes. Boxes above and below the primary line indicate the adjacent border genes. The figure is not scaled regarding sequence length and only shows relative changes at or near the IR/SC borders.
Phylogenetic analysis and divergence time
We conducted phylogenetic analyses using ML, NJ, MP, and BI based on five distinct data sets, such as complete plastomes, to examine evolutionary relationships between Tetraena and Zygophyllum species and preliminarily assess species boundaries (Fig. S6), 58 concatenated protein-coding genes (Fig. S7), matK gene (Fig. S8), rbcL gene (Fig. S9) and cssA gene (Fig. S10) independently. Protein-coding genes, matK, rbcL and cssA genes showed almost similar topologies in all the trees resulted from complete plastomes.
The reconstructed phylogenies from the complete plastome (Figs. 6, S6) and shared protein-coding genes (Figs. 7, S7) datasets showed that Zygophyllum and Tetraena clustered into two main clades and Zygophyllum species were divided into two further clustered. The first Zygophyllum cluster comprised Z. kaschgaricum and Z. xanthoxylon with high bootstrap values. The second cluster was divided further into various clades. According to both trees (complete plastomes and shared protein-coding genes) Z. rosowii cluster with Z. jaxarticum and Z. pterocarpum. However, Z. fabago showed different results and Z. fabago (MK341052.1) and Z. fabago (NC_052768.1) clustered with Z. mucronatum, Z. gobicum and Z. kansuense in same clades (i). In contrast, the other Fabago species Z. fabago (MW417250.1), Z. fabago (MW551564.1) and Z. fabago (MW417249.1) are clustered with Z. macropdum in another clade (III) based on concatenated shared genes and complete plastomes. Similar results were obtained from trees based on matK, rbcL and cssA genes data sets. The Tetraena species were clustered into one clade sister to the other Zygophyllum species. Within the Tetraena species, the T. mongolica formed a monophyletic group and T. hamiensis var. qatarensis and T. simplex were clustered into one group based on complete plastomes, proteins coding genes and matK datasets. However, based on rbcL and ccsA genes T. simplex and T. hamiensis var. qatarensis were not clustered into one clade. In cssA based tree T. hamiensis var. qatarensis was clustered with T. mongolica (NC039985) while T. simplex was found in a separate clade. The L. tridentata species clustered with G. angustifolium species, and Tr. terrestris was found sister clade. The two outgroups from Krameria species had a far phylogenetic relationship to Zygophyllum. Overall, the phylogenetic trees based on the above data sets show almost the same result except for the rbcL and cssA genes, which show variation in some species.

The maximum credible molecular chronogram (time tree) of Tetraena from BEAST is based on whole plastomes, with branch lengths proportional to time and lognormal fossil-based calibrations. The GTR + G substitution model was used with four rate categories and a Yule tree speciation model was applied with a lognormal relaxed clock model in BEAST. Different color branches represent subgroupings in Zygophyllaceae. The 95% highest posterior density credibility intervals for the node ages in black circles (mya). Numbers indicate date estimates for different nodes. A geological time scale is shown at the bottom of the figure.

Divergence time estimates of Zygophyllaceae based on 58 protein-coding shared genes from T. hamiensis var. qatarensis, T. simplex and related species. The GTR + G substitution model was used with four rate categories and a Yule tree speciation model was applied with a lognormal relaxed clock model in BEAST. Different color branches represent subgroupings in Zygophyllaceae. The 95% highest posterior density credibility intervals for the node ages in black circles (mya). Numbers indicate date estimates for different nodes. A geological time scale is shown at the bottom of the figure.
The backbone topology in Zygophyllaceae was largely congruent among ML, NJ, MP, and the BEAST2 analyses, receiving moderate to high bootstrap support and posterior probabilities. The final alignment for complete plastome and 58 shared concatenated genes had 77,526 bp and 42,229 bp, respectively. Phylogenetic trees based on complete plastomes (Fig. 6) and protein-coding genes (Fig. 7) showed similar trees and divergence time estimations with few differences. Based on complete plastomes, we estimated the stem node age of Zygophyllaceae and Krameriaceae at 77.7 Ma (95% HPD: 77.2–78.0), i.e., Cretaceous. The stem age of Zygophylloideae was found at 64.2 Ma (95% HPD: 57.2–72.6), while the crown age was estimated at 36.6 Ma (95% HPD: 32.9–43.8). Furthermore, Tetraena species diverged from Zygophyllum species in Eocene at 36.6 Ma (95% HPD: 32.9–43.8). On the other hand, tree divergence time based on concatenated protein-coding genes showed similar results in the case of stem node age of Zygophyllaceae and Krameriaceae was estimated at 77.7 Ma (95% HPD: 77.1–78.6), while the crown age of Zygophylloideae was very different from whole plastome tree and estimated 63.2 Ma (95% HPD: 53.8–70.6). Similarly, the Tetraena species were estimated at 34.4 Ma (95% HPD: 28.2–47.7) from the Zygophyllum species. Furthermore, in both trees based on complete plastomes and protein-coding genes, T. hamiensis var. qatarensis and T. simplex diverged from other T. mongolica at 31.7 and 18.2 Ma.
Discussion
T. hamiensis var. qatarensis and T. simplex plastomes are almost similar in size to previously reported Tetraena and Zygophyllum plastomes56 and significantly shorter than other angiosperms57,58,59. Both Tetraena plastomes had a quadripartite structure, standard in vascular plants60, and the GC content was similar to that of many other angiosperm species59,61. Plastomes of most angiosperms are 120–160 kb long, but the plastomes of T. hamiensis var. qatarensis and T. simplex are between 106,720 and 106,446 bp long. The LSC sections of most angiosperms are around 80–90 kb long, whereas the SSC regions are about 16–27 kb long, and the size of two IRs is approximately 20–28 kb long. The sizes of T. hamiensis var. qatarensis and T. simplex LSC and SSC do not differ significantly from those of most angiosperms, with the most noticeable difference being in two decreased IRs by approximately 16–24 kb in size. As previously reported in Tetraena and Zygophyllum species plastomes, the shrinking of IRs is primarily responsible for the lower sizes of plastomes in these two Tetraena species56. Although even smaller plastomes have been reported in non-photosynthetic, parasitic plants, such as Rhizanthella gardneri (Orchidaceae) with just 59,190 bp, Zygophyllum and Tetraena mongolica contain genomes on the small end of the range56,62.
Several plastomes are substantially smaller than most other plants, even though the chloroplast genome is a very conservative63. The most common reports of small plastomes come from investigations of parasitic plants, such as Taxillus chinensis and Taxillus sutchuenensis in the Loranthaceae family of Santalales64 Epifagus virginiana in Orobanchaceae family65, Cuscuta chinensis and C. japonica in Convolvulaceae family66. Some gymnosperms, such as Welwitschia mirabilis in the Welwitschiaceae of the Welwitschiales and Gnetum ula in the Gnetaceae of the Gnetales67,68, have smaller plastomes. Except for Astragalus membranaceus, which has a plastome of roughly 124 kb owing to the lack of an IR, non-parasitic angiosperms rarely have a plastome less than 130 kb69. The reduction in the size of SSCs was linked to the contraction of plastomes in other plant species70,71. LSC and SSC sizes drop slightly in both Tetraena plastomes, whereas IR lengths decrease considerably, as found before in other Zygophyllaceae taxa. As a result, these two new plastomes and others from the Zygophyllaceae family might be used as new models to study plastome structure and size evolution.
Aside from genome size, the number of genes in T. hamiensis var. qatarensis and T. simplex, as well as other related Tetraena and Zygophyllum plastomes, ranges from 104 to 106, which is lower than other Zygophyllaceae members such as L. tridentata and Tr. terrestris (125–130 respectively) (Table 1) and other land plants72,73. When five Tetraena and five Zygophyllum plastomes are compared to three other Zygophyllaceae species, four rRNA genes were found in the SSC region in these Tetraena and Zygophyllum plastomes, resulting in the reduction of copy number of rRNA genes (Table 1). Moreover, the contraction and expansion in IR regions forced the transferring of some genes to the LSC region and as result the IR regions became the single copies. Most of the SSC region's genes assigned to the IR regions duplicated, changing the number of genes, growing the LSC region's size, and shrinking the SSC region70. Furthermore, as previously observed, some genes within the NADH dehydrogenase complex (i.e., ndh genes) generally found in the SSC and IRs region encoding NADH oxidoreductase subunits are missing plastomes74. Furthermore, both T. hamiensis var. qatarensis and T. simplex plastomes lack rps16, rpl12, ycf2, and infA, found in most angiosperm plastomes. All of the factors above might contribute to the IRs area's shrinkage. This is uncommon in non-parasitic plants75. However, it has been found in Najas76, several orchid species77, Pinaceae67, and gametophytes67,78. The NADH dehydrogenase complex has role in photosynthesis against the environmental stress in plant plastids. Although infrequent, ndh gene deletion or pseudogenization is expected in the plastomes of photoautotrophic seed plants of many lineages79. The phenomena described in which plant plastid ndh genes were specifically deleted, and nuclear-encoded NDH subunits were expressed in Pinaceae67, Orchidaceae80 gametophytes78 and Geraniales16,81. Plants that thrive in drought (arid and semi-arid) conditions must adapt to their surroundings more than others. The current finding reveals the loss of 11 ndh genes in the plastomes of T. hamiensis var. qatarensis and T. simplex; it is unclear whether plastid-encoded ndh genes have been lost entirely or functionally moved to the cell nucleus for these two Zygophylloideae species. Similar results were seen in the plastid genome of the highly drought-resistant saguaro cactus (Carnegiea gigantea), where all ndh genes were absent or non-functional82.
The current study assessed the SNP and InDel numbers in all plastomes compared to T. hamiensis var. qatarensis in whole genomes and different regions, i.e., LSC, SSC IRs, and protein coding region (Table 2). Minimum SNPs were identified in T. qatarensis–T. mongolica3 (4812), while minimum InDels were found in T. qatarensis–T. simplex. Plastome size is directly proportional to the number of SNPs and InDels. The large plastome had the most significant number of SNPs, InDels and vice versa. Similarly, in the same genus, the SNP and InDels were lower compared to subfamilies members. The genus and subfamily-based relation of SNPs and InDels was reported previously83. Mutational events occur in plastomes, including SNP, InDels, SSR repeats, and tandem inversions84,85,86. The association between SNP and InDels was previously reported83,87,88.
The IR regions of both Tetraena plastomes also lacked typically duplicated genes and the genes mentioned above. These genes, on the other hand, appeared in the SC regions. Four rRNA genes, for example, that are typically duplicated in plant plastomes, were found only in the SSC of Tetraena and Zygophyllum species plastomes. Similar results have previously been described in parasitic plants62. Unlike Tetraena and Zygophyllum, the plastomes of L. tridentata, G. angustifolium, and Tr. terrestris were far more extensive and included the often duplicated rRNA genes. The rps19, rps12, ndhB, rps7, and rps8 genes were duplicated in L. tridentata, G. angustifolium, and Tr. terrestris; we found only one copy in all Tetraena and Zygophyllum species (Figs. 1, S1). As a result, the smaller number of genes in Tetraena and Zygophyllum's IRs may have contributed to the genus's small plastome size. Recent studies of the number and distribution of repetitive sequences in fully sequenced rearranged plastid genomes such as legumes have revealed considerably more scattered repeats than in non-rearranged genomes89. Intron losses for the rps12 and clpP genes have been found in the plastomes of T. hamiensis var. qatarensis and T. simplex and other angiosperm lineages. For example, both introns of the clpP gene have been deleted in Oleaceae, Onagraceae, Poaceae, and Pinus90.
In contrast, the intron in the 3′-end of rps12 has been lost twice in the monocot order Asparagales91. However, the loss of introns from both clpP and rps12 genes in Teteraena and Zygophyllum species is the first reported example of intron loss from both genes in the same plastid genome. We found that gene positions in IR boundaries have changed due to IR contraction in T. hamiensis var. qatarensis and T. simplex and related species, based on IR boundary studies. There is a slight variance near the IR boundary in both T. hamiensis var. qatarensis and T. simplex plastomes. The borders between the IRs and the SSC/LSC were significantly varied between Tetraena and other sampled species (Fig. 5). The shrinkage of the IR region in both Tetraena and Zygophyllum species was attributed to all these variations. Similar findings have already been observed in the Zygophyllaceae family56,71.
We used sliding window analysis and MISA to find highly variable regions and SSRs within the T. hamiensis var. qatarensis and T. simplex plastomes and compared them to 11 related plastomes. The sliding windows study revealed that, like in other taxa, the average mutation rate of the intergenic areas inside the SC regions was substantially more significant than that of the IR regions92. Due to lower selection pressures, higher mutation rates often result in more variety within intergenic spacers93. Like other functional repeat sequences, SSRs numbers differed in all these plastomes. The number of SSRs found varied from 161 (T. mongolica3) to 225 (Z. fabago1). There were 166 and 170 SSRs in the sequenced plastomes of T. hamiensis var. qatarensis and T. simplex, respectively (Fig. 3K) PolyA and polyT were the most prevalent mononucleotide repeats in Tetraena and Zygophyllum. Lagerstroemia94, Primula95, Fritillaria96, and Allium96 have all been shown to have polyA and polyT SSRs97.
The majority of Saudi Arabian Zygophyllum species were shifted to Tetraena, according to Beier et al.34’s most current taxonomic proposal of Tetraena Maxim. and Zygophyllum. Previously, researchers utilized several barcodes to accurately identify Tetraena and Zygophyllum species, including rbcL, trnL-F, and matK.36,98,99. However, to differentiate Tetraena and Zygophyllum species, we used chloroplast barcodes such as matK, rbcL, and cssA genes combined with 66 shared protein-coding genes. To know the evolutionary position of T. hamiensis var. qatarensis and T. simplex, phylogenetic analyses were performed using ML, NJ, MP, and BI based techniques. The trees generated by analyzing the whole plastomes (Figs. 6, S6), shared protein-coding genes (Figs. 7, S7), matK (Fig. S8), rbcL (Fig. S9), and cssA (Fig. S10) genes exhibited very identical topologies, indicating that T. hamiensis var. qatarensis and T. simplex are sister to the genus Tetraena and may not belong to Zygophyllum. Our findings support previous phylogenetic reports36,100 that Z. simplex is a sister to the genus Tetraena, not Zygophyllum, which is based on the sequence of rbcl morphological, anatomical attributes. They supported our recent findings that Zygophyllum is polyphyletic and may be subdivided into clusters36,100. Furthermore, the Tetraena species in our research were grouped into a single clade that was sister to the rest of the Zygophyllum species. T. mongolica formed a monophyletic group within the Tetraena genus. In contrast, T. hamiensis var. qatarensis was grouped with T. simplex into a single group based on complete genomes, proteins coding genes, and matK datasets. Similar findings have been reported in the others36. Phylogenetic relationships of 44 specimens representing seven species of Saudi Arabian Tetraena Maxim. and Zygophyllum L. were investigated using individual and combined chloroplast DNA data of rbcL and trnL-F by Alzahrani and Albokhari101 used individual and combined chloroplast DNA data of rbcL and trnL-F to study phylogenetic relationships of 44 specimens representing seven species of Saudi Arabian Tetraena Maxim. and Zygophyllum L. This is the first research to use super barcodes to investigate the evolutionary relationships of Tetraena and Zygophyllum species. Taxonomists have already conducted preliminary investigations to assess the possibility of plastomes in plant groups of closely related species. Bayly et al.102 provided a phylogenetic study on three taxa (Eucalyptus, Corymbia, and Angophora) and showed that the plastome might be used in lower-level genetic research. The plastome identifies species as organelle-scale "barcodes," according to Yang et al.103. The plastome, according to Zhang et al.104, might be considered a super-barcode for the closely related species. The ability of super-barcode was investigated in Chrysanthemum and Ligularia, respectively, by Xia et al.105 and Chen et al.106.
According to phylogenetic analysis and time divergence using beast2 the time divergence between Zygophyllaceae and Krameriaceae was calculated at 77.7 Ma (95 percent HPD: 77.2–78.0), i.e., Cretaceous, based on phylogenetic analysis and time divergence using beast2. The stem age of Zygophylloideae was 64.2 Ma (95% HPD: 57.2–72.6), whereas the crown age was 36.6 Ma (95% HPD: 32.9–43.8). Bell et al.107 published a similar conclusion, stating that the separation between Zygophyllaceae and Krameriaceae occurred about 70 Ma (49–88 Ma). In another study, the stem age of Zygophyllaceae at 60.9 Ma (34–90 Ma) by Magallón et al.108. However, our results are similar to a recent study where the stem age of Zygophyllaceae was set at 70 Ma (49–88 Ma)107 to infer the ages of Zygophylloideae and Asian Zygophyllum. Another study estimated the crown-group age of Zygophyllaceae to be 59.89 Ma (95% HPD: 38.14–80.95 Ma), which agrees with ages calculated using rbcL data having two secondary calibrations and ITS data having one secondary calibration109. Similarly, based on entire plastome (Fig. 6) and protein-coding genes (Fig. 7) datasets, the time divergence of Zygophyllum and Tetraena was 36.6 Ma and 34.4 Ma, respectively. Zygophyllum stem ages were 32.7 and 29.4 Ma, respectively, whereas Tetraena stem ages were 31.7 and 18.2 Ma based on full plastome and protein-coding genes. The stem age of Asian Zygophyllum was estimated to be 30.39 Ma (95% HPD: 21.53–39.81 Ma) in a previous study110. Both data based on the overall plastome dataset and protein-coding genes exhibited different conclusions in the instance of T. simplex and T. hamiensis var. qatarensis time divergence. T. hamiensis var. qatarensis and T. simplex separated from T. mongolica around 31.7 Ma based on the full plastome dataset, and 18.2 Ma based on protein-coding genes. T. simplex split 3.7 Ma from T. hamiensis var. qatarensis based on protein-coding genes data, whereas the divergence period was calculated to be 18.7 Ma based on entire plastome data. Wu et al.110 found similar results using the rbcL dataset, with a T. simplex divergence timing of 3.04 Ma. Our findings are consistent with previous findings, suggesting that Zygophyllum and Tetraena evolved separately, according to Bellstedt et al.35 and Wu et al.110.
Conclusion
In the current study we sequenced and analyzed the plastomes of T. hamiensis var. qatarensis and T. simplex, revealing shorter length, shorter IR regions, and 16 deleted genes in SSC and IRs regions. Comparative analysis with other species showed genetic variations. Phylogenetic analysis supported current understanding of Tetraena's status, with divergence times estimated at 36.6 Ma and 34.4 Ma for Zygophyllum and Tetraena, respectively. This dataset will provide a genetic resource for future research on Tetraena's evolution, population genetics, and other biological functions.
Methodology
The fresh juvenile leaves were collected from T. hamiensis var. qatarensis and T. simplex growing in their natural habitat Nizwa (22°46′22.7″N 57°27′56.8″E) were collected and transported in liquid nitrogen to the − 80 °C facility. The specimens were submitted to the University of Nizwa herbarium center under the voucher numbers UoN-TQ1 (T. hamiensis var. qatarensis) and UoN-TS1 (T. simplex). Saif Al-Hathmi one of the leading taxonomists at the Oman Botanical Garden, Muscat, Oman, identified the plants. The plant samples were collected and processed per the national guidelines and legislation. Hence, a permission permits (6210/10/73) was obtained from the Director-General of Nature Conservation, Ministry of Environment and Climate Affairs, Sultanate of Oman.
DNA extraction and sequencing
The chloroplast (cp) DNA was isolated111. The genomic libraries were constructed according to the manufacturer’s protocol (Life Technologies USA, Eugene, OR, USA). Ion Shear™ Plus Reagents kit and Ion Xpress™ Plus, gDNA Fragment Library kit, were used to prepare (enzymatically) 400 bp fragments of the cp DNA. The prepared libraries were quantified using a Qubit 3.0 fluorometer followed by bioanalyzer (Agilent 2100 Bioanalyzer system, Life Technologies USA). The template was amplified using Ion OneTouch™ 2 and enriched using Ion OneTouch™ ES enrichment system Ion 530 & 520 OT2 reagents. Sequencing was performed using an Ion s5 sequencer (Life Technologies USA, Eugene, OR, USA). Samples were loaded on S5 530 chip according to the manufacturer’s protocol.
Genome assembly and annotation
215,240,872 and 109,991,167 raw reads were obtained for T. hamiensis var. qatarensis and T. simplex plastomes, respectively. To eliminate the low-quality sequences, we screened the reads for a Phred score < 30. The plastomes were assembled using two methods to ensure the accuracy of plastomes assembly. GetOrganelle v 1.7.5 pipeline112 with SPAdes version 3.10.1 (http://bioinf.spbau.ru/spades) as assemblers were employed to de novo assemble the plastomes T. hamiensis var. qatarensis and T. simplex. Annotations were performed using CpGAVAS2113 and DOGMA (http://dogma.ccbb.utexas.edu/, China)114, whereas tRNAs can-SE (v.1.21)115 was used to detect the tRNA genes. Similarly, the start-stop codons, intron boundaries and manual alterations were performed by comparing the plastomes to reference genomes using Geneious Pro v.10.2.3.116 and tRNAs can-SE (v.1.21)115. The plastomes' structural features were illustrated using the chloroplot117 and circos118. Moreover, the genomic divergence was determined by mVISTA119 in shuffle-LAGAN mode using plastomes T. hamiensis var. qatarensis as reference.
Repeat identification
We determined different functional repetitive sequences in T. hamiensis var. qatarensis and T. simplex in plastomes. Palindromic, forward, and reverse repeat sequences were determined using the REPuter120 online tool with 8 bp minimum repeat size conditions and 50 maximum computed repeats. Similarly, simple sequence repeats (SSRs) were calculated using MISA software121 using the conditions of ≥ 8 repeat units for one bp repeats; ≥ 6 repeat units for two bp repeats; ≥ 4 repeat units for 3 and 4 bp repeats and ≥ 3 repeat units for 5 and 6 bp repeats. Furthermore, tandem repeats were calculated using the online tool Tandem Repeats Finder v.4.09122.
Genome divergence
The divergence in shared protein-coding genes and complete plastomes T. hamiensis var. qatarensis, T. simplex, and related species were determined. Comparative analysis was performed through multiple sequence alignment, and the gene order was compared and analyzed to refine the missing and dubious gene annotations. MAFFT version 7.222123 employed for plastomes annotations with default parameters. The pairwise sequence divergence was determined with Kimura’s two-parameter model (K2P)123. In DnaSP software v 6.13.03124 the sliding window analysis (200 bp window size and 100 bp step size) was used to calculate the relative synonyms codon usage (RSCU) value, variable sites and nucleotide variations (Pi).
Phylogenetic analyses and divergence time
To determine the phylogenetic position of T. hamiensis var. qatarensis and T. simplex within the family Zygophyllaceae, 30 published plastome sequences of Zygophyllum species, three plastomes sequences of Tetraena species, Larrea, Guaiacum and Tribulus plastomes were downloaded from the NCBI database for phylogenetic analysis. Two Krameria species plastomes sequences were used as outgroup. Based on conserved gene order and structure, several alignments of whole plastomes were created125. The phylogenetic trees were generated using the four techniques listed below: MrBayes 3.1.2 was used for Bayesian inference; MEGA 6126 was used for maximum likelihood (ML) and neighbor-joining (NJ); and PAUP127,128 was used for maximum parsimony (MP). As per Akaike information criterion (AIC) for Bayesian posterior probabilities (PP) in BI analyses, the optimal substitution model GTR + G was examined using jModelTest version v2.1.02129. Starting with random trees and sampling 1 out of every 100 generations, the Markov Chain Monte Carlo (MCMC) technique was used to simulate 1,000,000 generations utilizing four incrementally heated chains. The values of the first 25% of trees were removed as burn-in for estimating the posterior probability. The maximum parsimony run was based on a heuristic search using the tree-bisection-reconnection (TBR) branch-swapping tree search criterion and 1000 random additions of sequence repetitions. Similarly, using a BIONJ tree130 as the starting tree and 1000 bootstrap replicates, the parameters for ML analysis were adjusted using the Kimura 2-parameter model with invariant sites and gamma-distributed rate heterogeneity.
A set of 58 shared genes, rbcL gene, matK gene, and cssA gene from the plastomes of the above species were aligned using MAFFT version 7.222131 under default parameters and by making various manual adjustments to preserve and improve reading frames in the second, third, fourth, and fifth tiers of phylogenies. As previously indicated and proposed by Asaf et al.24, the ML approach generated trees utilizing 58 shared genes, matK gene, rbcL gene, and cssA gene.
To conclude the divergence time of Tetraena with those of 30 Zygophyllum species, we used both entire plastomes and the concatenated data matrix. In BEAST132, a general time reversible (GTR + G) substitution model with four rate categories and a Yule tree speciation model with a lognormal relaxed clock model was utilized. We employed a fossil-based method to calibrate the molecular divergence by calculating an average substitution rate of 3.0 × 10−9 substitutions per site per year (s/s/y). The fossil record within Zygophyllaceae family is very sparse (reviewed by Bellstedt et al.35), no member of the present genera can be ascribed to the few reported fossils. Therefore, we employed a supplementary calibration strategy. We examined the data using concatenated protein-coding genes and entire plastomes to assess the robustness of our method. The Zygophyllum and Tetraena clades were specified as monophyletic, and we applied age constraints on four nodes with normal prior distributions107,110. To root the calibration time, we included two available outgroups species, Krameria bicolor and Krameria lanceolata from family Krameriaceae (the sister group to Zygophyllaceae). These outgroups were selected because they are all closely related to our research model species and have fossil records dating back to before the Zygophyllum genus110. Following the results of Bell et al.107 and Wu et al.110, the split between Zygophyllaceae and Krameriaceae was set at 70 Ma (49–88 Ma). Three separate MCMC runs of 50 million generations were used in the dating studies. The tree files from all three runs were combined with LOGCOMBINER. TRACER 1.5133 was used to test convergence and adequate sample sizes. We burned off 25% of the trees in each analysis. Finally, TREEANNOTATOR134 was used to construct the tree, and FIGTREE1.4 was used to display the tree with a 95% greatest posterior density (HPD).
Data availability
All data generated or analyzed during this study are included in this published article. T. hamiensis var. qatarensis and T. simplex plastomes were submitted to NCBI with accession numbers (OM809718) and (OL943588) respectively.
References
Liu, J. et al. Integrating a comprehensive DNA barcode reference library with a global map of yews (Taxus L.) for forensic identification. Mol. Ecol. Resour. 18, 1115–1131 (2018).
Parveen, I., Singh, H. K., Raghuvanshi, S., Pradhan, U. C. & Babbar, S. B. DNA barcoding of endangered Indian Paphiopedilum species. Mol. Ecol. Resour. 12, 82–90 (2012).
Li, D.-Z. et al. Comparative analysis of a large dataset indicates that internal transcribed spacer (ITS) should be incorporated into the core barcode for seed plants. Proc. Natl. Acad. Sci. 108, 19641–19646 (2011).
Gueuning, M. et al. Evaluating next-generation sequencing (NGS) methods for routine monitoring of wild bees: Metabarcoding, mitogenomics or NGS barcoding. Mol. Ecol. Resour. 19, 847–862 (2019).
Gonzalez, M. A. et al. Identification of Amazonian trees with DNA barcodes. PLoS ONE 4, e7483 (2009).
Chase, M. W., Reveal, J. L. & Fay, M. F. A subfamilial classification for the expanded asparagalean families Amaryllidaceae, Asparagaceae and Xanthorrhoeaceae. Bot. J. Linn. Soc. 161, 132–136 (2009).
Hebert, P. D., Cywinska, A., Ball, S. L. & DeWaard, J. R. Biological identifications through DNA barcodes. Proc. R. Soc. Lond. Ser. B Biol. Sci. 270, 313–321 (2003).
Hollingsworth, M. L. et al. Selecting barcoding loci for plants: Evaluation of seven candidate loci with species-level sampling in three divergent groups of land plants. Mol. Ecol. Resour. 9, 439–457 (2009).
Chase, M. W. et al. Land plants and DNA barcodes: Short-term and long-term goals. Philos. Trans. R. Soc. B Biol. Sci. 360, 1889–1895 (2005).
Chen, Q., Hu, H. & Zhang, D. DNA barcoding and phylogenomic analysis of the genus Fritillaria in China based on complete chloroplast genomes. Front. Plant Sci. 13, 764255–764255 (2022).
Fu, C.-N. et al. Prevalence of isomeric plastomes and effectiveness of plastome super-barcodes in yews (Taxus) worldwide. Sci. Rep. 9, 2773. https://doi.org/10.1038/s41598-019-39161-x (2019).
Ji, Y. et al. Testing and using complete plastomes and ribosomal DNA sequences as the next generation DNA barcodes in Panax (Araliaceae). Mol. Ecol. Resour. 19, 1333–1345 (2019).
Gitzendanner, M. A., Soltis, P. S., Yi, T.-S., Li, D.-Z. & Soltis, D. E. Advances in Botanical Research Vol. 85, 293–313 (Elsevier, 2018).
Li, H.-T. et al. Origin of angiosperms and the puzzle of the Jurassic gap. Nat. Plants 5, 461–470. https://doi.org/10.1038/s41477-019-0421-0 (2019).
Nie, Y. et al. Accounting for uncertainty in the evolutionary timescale of green plants through clock-partitioning and fossil calibration strategies. Syst. Biol. 69, 1–16. https://doi.org/10.1093/sysbio/syz032 (2019).
Asaf, S. et al. The dynamic history of gymnosperm plastomes: Insights from structural characterization, comparative analysis, phylogenomics, and time divergence. Plant Genome 14, e20130 (2021).
Chen, Q., Wu, X. & Zhang, D. Phylogenetic analysis of Fritillaria cirrhosa D. Don and its closely related species based on complete chloroplast genomes. PeerJ 7, e7480 (2019).
Li, X. et al. Plant DNA barcoding: From gene to genome. Biol. Rev. 90, 157–166 (2015).
Coissac, E., Hollingsworth, P. M., Lavergne, S. & Taberlet, P. (Wiley Online Library, 2016).
Huang, Y. et al. psbE-psbL and ndhA Intron, the promising plastid DNA barcode of fagopyrum. Int. J. Mol. Sci. 20, 3455 (2019).
Li, X. Study on conservation biology of Fritillaria cirrhosa (Chinese Academy of Medical Sciences and Peking Union Medical College, 2009).
Huang, H., Shi, C., Liu, Y., Mao, S.-Y. & Gao, L.-Z. Thirteen Camelliachloroplast genome sequences determined by high-throughput sequencing: Genome structure and phylogenetic relationships. BMC Evol. Biol. 14, 1–17 (2014).
Guo, H. et al. Complete chloroplast genome sequences of Schisandra chinensis: Genome structure, comparative analysis, and phylogenetic relationship of basal angiosperms. Sci. China Life Sci 60, 1286–1290 (2017).
Asaf, S. et al. Chloroplast genomes of Arabidopsis halleri ssp. gemmifera and Arabidopsis lyrata ssp. petraea: Structures and comparative analysis. Sci. Rep. 7, 1–15 (2017).
Khan, A. et al. Complete chloroplast genomes of medicinally important Teucrium species and comparative analyses with related species from Lamiaceae. PeerJ 7, e7260 (2019).
El Hadidi, M. N. Adumbratio Florae Aethiopicae. 30: Zygophyllaceae. Webbia 33, 45–101 (1978).
Migahid, A. & Hammouda, M. Flora of Saudi Arabia, vol. I–III (University Libraries, 1996).
Migahid, A. M. Flora of Saudi Arabia. (1978).
Hosny, A. Genus Zygophyllum L. Arabia. Taeckholmia 11, 19–32 (1988).
Mandaville, J. P. Flora of Eastern Saudi Arabia. (Routledge, 2013).
Chaudhary, S. Flora of the Kingdom of Saudi Arabia (Vascular Plants). (National Agriculture and Water Research Center, National Herbarium, Ministry of Agriculture and Water, 2001).
Soliman, M., El-Tarras, A. & El-Awady, M. Seed exomorphic characters of some taxa from Saudi Arabia. J. Am. Sci. 6, 906–910 (2010).
Waly, N. M., Al-Ghamdi, F. A. & Al-Shamrani, R. I. Developing methods for anatomical identification of the genus Zygophyllum L. (Zygophyllaceae) in Saudi Arabia. Life Sci. J. 3, 8 (2011).
Beier, B.-A., Chase, M. & Thulin, M. Phylogenetic relationships and taxonomy of subfamily Zygophylloideae (Zygophyllaceae) based on molecular and morphological data. Plant Syst. Evol. 240, 11–39 (2003).
Bellstedt, D. et al. Phylogenetic relationships, character evolution and biogeography of southern African members of Zygophyllum (Zygophyllaceae) based on three plastid regions. Mol. Phylogenet. Evol. 47, 932–949 (2008).
Sheahan, M. C. & Chase, M. W. Phylogenetic relationships within Zygophyllaceae based on DNA sequences of three plastid regions, with special emphasis on Zygophylloideae. Syst. Bot. 25, 371–384 (2000).
Shaltout, K., El-Halawany, E. & El-Garawany, M. Coastal lowland vegetation of eastern Saudi Arabia. Biodivers. Conserv. 6, 1027–1040 (1997).
Batanouny, K. H. Ecology and Flora of Qatar. Ecology and Flora of Qatar. (1981).
Collenette, S. Illustrated guide to the flowers of Saudi Arabia. (Scorpion, 1985).
Collenette, S. The ceropegias of Saudi Arabia. British Cactus Succulent J. 17, 181–187 (1999).
Mandaville, J. P. Plant life in the Rub’al-Khali (the Empty Quarter), south-central Arabia. Proc. R. Soc. Edinb. Sect. B. Biol. Sci. 89, 147–157 (1986).
Cornes, M. & Cornes, C. D. wild flowering plants of Bahrain (Immel, 1989).
Samuelsson, G. et al. Inventory of plants used in traditional medicine in Somalia. IV. Plants of the families Passifloraceae-Zygophyllaceae. J. Ethnopharmacol. 38, 1–29 (1993).
Wood, J. R. I. & Haig-Thomas, H. Handbook of the Yemen flora (Royal Botanic Gardens, 1997).
Western, A. R. The Flora of the United Arab Emirates: An Introduction (United Arab Emirates University, 1989).
Hosny, A. I. Revision of Genus Zygophyllum L., Sections Bipartia and Mediterranea in Egypt and Arabia. (Unpublished, 1978).
Böer, B. & Sargeant, D. Desert perennials as plant and soil indicators in Eastern Arabia. Plant Soil 199, 261–266 (1998).
Barth, H.-J. Desertification in the eastern province of Saudi Arabia. J. Arid Environ. 43, 399–410 (1999).
Sayed, O. H. Adaptational responses of Zygophyllum qatarense Hadidi to stress conditions in a desert environment. J. Arid Environ. 32, 445–452. https://doi.org/10.1006/jare.1996.0037 (1996).
Karīm, F. M. Wild Flowering Plants of the United Arab Emirates (Environmental Research and Wildlife Development Agency, 2002).
Kisksi, T., Guenaoui, C. & Fawzi, N. Early growth stages of the rare Acridocarpus orientalis in the UAE-A First step towards conservation. Nat. Resour. 3, 1–5 (2012).
Laurent-Täckholm, V. & Drar, M. Students' flora of Egypt. (1956).
Ghazanfar, S. & Osborne, J. Typification of Zygophyllum propinquum Decne. and Z. coccineum L. (Zygophyllaceae) and a key to Tetraena in SW Asia. Kew Bull. 70, 1–9 (2015).
Zeng, C.-X., Zhang, Y.-X., Triplett, J. K., Yang, J.-B. & Li, D.-Z. Large multi-locus plastid phylogeny of the tribe Arundinarieae (Poaceae: Bambusoideae) reveals ten major lineages and low rate of molecular divergence. Mol. Phylogenet. Evol. 56, 821–839 (2010).
Shapiro, J. A. & von Sternberg, R. Why repetitive DNA is essential to genome function. Biol. Rev. 80, 227–250 (2005).
Zhang, L. et al. Comparative chloroplast genomics and phylogenetic analysis of Zygophyllum (Zygophyllaceae) of China. Front. Plant Sci. 12, 723622 (2021).
Asaf, S., Ahmad, W., Al-Harrasi, A. & Khan, A. L. Uncovering the first complete plastome genomics, comparative analyses, and phylogenetic dispositions of endemic medicinal plant Ziziphus hajarensis (Rhamnaceae). BMC Genomics 23, 1–16 (2022).
Lubna, et al. The plastome sequences of Triticum sphaerococcum (ABD) and Triticum turgidum subsp. durum (AB) exhibit evolutionary changes, structural characterization, comparative analysis, phylogenomics and time divergence. Int. J. Mol. Sci. 23, 2783 (2022).
Mower, J. P. & Vickrey, T. L. Structural diversity among plastid genomes of land plants. Adv. Bot. Res. 85, 263–292 (2018).
Tonti‐Filippini, J., Nevill, P. G., Dixon, K. & Small, I. Vol. 90, 808–818 (Wiley Online Library, 2017).
Gruenstaeudl, M., Nauheimer, L. & Borsch, T. Plastid genome structure and phylogenomics of Nymphaeales: Conserved gene order and new insights into relationships. Plant Syst. Evol. 303, 1251–1270 (2017).
Delannoy, E., Fujii, S., Colas des Francs-Small, C., Brundrett, M. & Small, I. Rampant gene loss in the underground orchid Rhizanthella gardneri highlights evolutionary constraints on plastid genomes. Mol. Biol. Evol. 28, 2077–2086 (2011).
Dobrogojski, J., Adamiec, M. & Luciński, R. The chloroplast genome: A review. Acta Physiol. Plant. 42, 1–13 (2020).
Li, Y. et al. Gene losses and partial deletion of small single-copy regions of the chloroplast genomes of two hemiparasitic Taxillus species. Sci. Rep. 7, 1–12 (2017).
Wolfe, K. H., Morden, C. W. & Palmer, J. D. Function and evolution of a minimal plastid genome from a nonphotosynthetic parasitic plant. Proc. Natl. Acad. Sci. USA 89, 10648–10652. https://doi.org/10.1073/pnas.89.22.10648 (1992).
Park, I. et al. Cuscuta species identification based on the morphology of reproductive organs and complete chloroplast genome sequences. Int. J. Mol. Sci. 20, 2726 (2019).
Braukmann, T. W., Kuzmina, M. & Stefanović, S. Loss of all plastid ndh genes in Gnetales and conifers: Extent and evolutionary significance for the seed plant phylogeny. Curr. Genet. 55, 323–337. https://doi.org/10.1007/s00294-009-0249-7 (2009).
Lubna, et al. The dynamic history of gymnosperm plastomes: Insights from structural characterization, comparative analysis, phylogenomics, and time divergence. Plant Genome 14, e20130. https://doi.org/10.1002/tpg2.20130 (2021).
Lei, W. et al. Intraspecific and heteroplasmic variations, gene losses and inversions in the chloroplast genome of Astragalus membranaceus. Sci. Rep. 6, 21669. https://doi.org/10.1038/srep21669 (2016).
Henriquez, C. L. et al. Complete chloroplast genomes of Anthurium huixtlense and Pothos scandens (Pothoideae, Araceae): Unique inverted repeat expansion and contraction affect rate of evolution. J. Mol. Evol. 88, 562–574 (2020).
Wang, X., Dorjee, T., Chen, Y., Gao, F. & Zhou, Y. The complete chloroplast genome sequencing analysis revealed an unusual IRs reduction in three species of subfamily Zygophylloideae. PLoS ONE 17, e0263253 (2022).
Zhang, Y. et al. The complete chloroplast genome sequences of five Epimedium species: Lights into phylogenetic and taxonomic analyses. Front. Plant Sci. https://doi.org/10.3389/fpls.2016.00306 (2016).
He, L. et al. Complete chloroplast genome of medicinal plant Lonicera japonica: Genome rearrangement, intron gain and loss, and implications for phylogenetic studies. Molecules 22, 249 (2017).
Omelchenko, D. O. et al. Complete plastome sequencing of Allium paradoxum reveals unusual rearrangements and the loss of the ndh genes as compared to Allium ursinum and other onions. Gene 726, 144154 (2020).
Yu, J., Wang, C. & Gong, X. Degeneration of photosynthetic capacity in mixotrophic plants, Chimaphila japonica and Pyrola decorata (Ericaceae). Plant Divers. 39, 80–88. https://doi.org/10.1016/j.pld.2016.11.005 (2017).
Peredo, E. L., King, U. M. & Les, D. H. The plastid genome of Najas flexilis: Adaptation to submersed environments is accompanied by the complete loss of the NDH complex in an aquatic angiosperm. PLoS ONE 8, e68591 (2013).
Kim, H. T. et al. Seven new complete plastome sequences reveal rampant independent loss of the ndh gene family across orchids and associated instability of the inverted repeat/small single-copy region boundaries. PLoS ONE 10, e0142215 (2015).
Wu, F.-H. et al. Complete nucleotide sequence of Dendrocalamus latiflorus and Bambusa oldhamii chloroplast genomes. Tree Physiol. 29, 847–856 (2009).
Ruhlman, T. A. et al. NDH expression marks major transitions in plant evolution and reveals coordinate intracellular gene loss. BMC Plant Biol. 15, 100. https://doi.org/10.1186/s12870-015-0484-7 (2015).
Dong, W. L. et al. Molecular evolution of chloroplast genomes of orchid species: Insights into phylogenetic relationship and adaptive evolution. Int. J. Mol. Sci. https://doi.org/10.3390/ijms19030716 (2018).
Chris Blazier, J., Guisinger, M. M. & Jansen, R. K. Recent loss of plastid-encoded ndh genes within Erodium (Geraniaceae). Plant Mol. Biol. 76, 263–272. https://doi.org/10.1007/s11103-011-9753-5 (2011).
Sanderson, M. J. et al. Exceptional reduction of the plastid genome of saguaro cactus (Carnegiea gigantea): Loss of the ndh gene suite and inverted repeat. Am. J. Bot. 102, 1115–1127 (2015).
Abdullah, Henriquez, C. L., Croat, T. B., Poczai, P. & Ahmed, I. Mutational dynamics of aroid chloroplast genomes II. Front. Genet. 11, 610838. https://doi.org/10.3389/fgene.2020.610838 (2021).
Poczai, P. & Hyvönen, J. Identification and characterization of plastid trnF (GAA) pseudogenes in four species of Solanum (Solanaceae). Biotech. Lett. 33, 2317–2323 (2011).
Jheng, C.-F. et al. The comparative chloroplast genomic analysis of photosynthetic orchids and developing DNA markers to distinguish Phalaenopsis orchids. Plant Sci. 190, 62–73 (2012).
Shahzadi, I. et al. Comparative analyses of chloroplast genomes among three Firmiana species: Identification of mutational hotspots and phylogenetic relationship with other species of Malvaceae. Plant Gene 19, 100199 (2019).
Li, J., Su, Y. & Wang, T. The repeat sequences and elevated substitution rates of the chloroplast accD gene in cupressophytes. Front. Plant Sci. 9, 533 (2018).
Mes, T. H. et al. Hairpins involving both inverted and direct repeats are associated with homoplasious indels in non-coding chloroplast DNA of Taraxacum (Lactuceae: Asteraceae). Genome 43, 634–641 (2000).
Saski, C. et al. Complete chloroplast genome sequence of Glycine max and comparative analyses with other legume genomes. Plant Mol. Biol. 59, 309–322 (2005).
Jansen, R. K. et al. Analysis of 81 genes from 64 plastid genomes resolves relationships in angiosperms and identifies genome-scale evolutionary patterns. Proc. Natl. Acad. Sci. 104, 19369–19374. https://doi.org/10.1073/pnas.0709121104 (2007).
McPherson, M. A., Fay, M. F., Chase, M. W. & Graham, S. W. Parallel loss of a slowly evolving intron from two closely related families in Asparagales. Syst. Bot. 29, 296–307 (2004).
Perry, A. S. & Wolfe, K. H. Nucleotide substitution rates in legume chloroplast DNA depend on the presence of the inverted repeat. J. Mol. Evol. 55, 501–508 (2002).
Wang, A., Yang, M. & Liu, J. Molecular phylogeny, recent radiation and evolution of gross morphology of the rhubarb genus Rheum (Polygonaceae) inferred from chloroplast DNA trn LF sequences. Ann. Bot. 96, 489–498 (2005).
Zheng, G. et al. Comparative analyses of chloroplast genomes from 13 Lagerstroemia (Lythraceae) species: Identification of highly divergent regions and inference of phylogenetic relationships. Plant Mol. Biol. 102, 659–676 (2020).
Ren, T., Yang, Y., Zhou, T. & Liu, Z.-L. Comparative plastid genomes of Primula species: Sequence divergence and phylogenetic relationships. Int. J. Mol. Sci. 19, 1050 (2018).
Bi, Y. et al. Chloroplast genomic resources for phylogeny and DNA barcoding: A case study on Fritillaria. Sci. Rep. 8, 1–12 (2018).
Xie, D.-F. et al. Phylogeny of Chinese Allium species in section Daghestanica and adaptive evolution of Allium (Amaryllidaceae, Allioideae) species revealed by the chloroplast complete genome. Front. Plant Sci. 10, 460 (2019).
Bafeel, S. O. et al. Comparative evaluation of PCR success with universal primers of maturase K (matK) and ribulose-1, 5-bisphosphate carboxylase oxygenase large subunit (rbcL) for barcoding of some arid plants. Plant Omics 4, 195–198 (2011).
Maloukh, L. et al. Discriminatory power of rbcL barcode locus for authentication of some of United Arab Emirates (UAE) native plants. 3 Biotech 7, 1–7 (2017).
Sheahan, M. C. & Chase, M. W. A phylogenetic analysis of Zygophyllaceae R. Br. based on morphological, anatomical and rbc L DNA sequence data. Bot. J. Linn. Soc. 122, 279–300 (1996).
Alzahrani, D. A. & Albokhari, E. J. Molecular phylogeny of Saudi Arabian Tetraena Maxim. and Zygophyllum L. (Zygophyllaceae) based on plastid DNA sequences. Bangladesh J. Plant Taxon. 24, 155–164 (2017).
Bayly, M. J. et al. Chloroplast genome analysis of Australian eucalypts—Eucalyptus, Corymbia, Angophora, Allosyncarpia and Stockwellia (Myrtaceae). Mol. Phylogenet. Evol. 69, 704–716 (2013).
Yang, J.-B., Tang, M., Li, H.-T., Zhang, Z.-R. & Li, D.-Z. Complete chloroplast genome of the genus Cymbidium: Lights into the species identification, phylogenetic implications and population genetic analyses. BMC Evol. Biol. 13, 1–12 (2013).
Zhang, Z., Zhang, Y., Song, M., Guan, Y. & Ma, X. Species identification of Dracaena using the complete chloroplast genome as a super-barcode. Front. Pharmacol. 10, 1441 (2019).
Xia, Y. et al. The complete chloroplast genome sequence of Chrysanthemum indicum. Mitochondrial DNA Part A 27, 4668–4669 (2016).
Chen, X. et al. Identification of Ligularia herbs using the complete chloroplast genome as a super-barcode. Front. Pharmacol. 9, 695 (2018).
Bell, C. D., Soltis, D. E. & Soltis, P. S. The age and diversification of the angiosperms re-revisited. Am. J. Bot. 97, 1296–1303. https://doi.org/10.3732/ajb.0900346 (2010).
Magallón, S., Gómez-Acevedo, S., Sánchez-Reyes, L. L. & Hernández-Hernández, T. A metacalibrated time-tree documents the early rise of flowering plant phylogenetic diversity. New Phytol. 207, 437–453. https://doi.org/10.1111/nph.13264 (2015).
Yu, Y., Harris, A. J., Blair, C. & He, X. RASP (Reconstruct Ancestral State in Phylogenies): A tool for historical biogeography. Mol. Phylogenet. Evol. 87, 46–49. https://doi.org/10.1016/j.ympev.2015.03.008 (2015).
Wu, S.-D. et al. Evolution of Asian interior arid-zone biota: Evidence from the diversification of Asian Zygophyllum (Zygophyllaceae). PLoS ONE 10, e0138697 (2015).
Shi, C. et al. An improved chloroplast DNA extraction procedure for whole plastid genome sequencing. PLoS ONE 7, e31468 (2012).
Jin, J.-J. et al. GetOrganelle: A simple and fast pipeline for de novo assembly of a complete circular chloroplast genome using genome skimming data. BioRxiv 4, 256479 (2018).
Shi, L. et al. CPGAVAS2, an integrated plastome sequence annotator and analyzer. Nucleic Acids Res. 47, W65–W73 (2019).
Wyman, S. K., Jansen, R. K. & Boore, J. L. Automatic annotation of organellar genomes with DOGMA. Bioinformatics 20, 3252–3255 (2004).
Schattner, P., Brooks, A. N. & Lowe, T. M. The tRNAscan-SE, snoscan and snoGPS web servers for the detection of tRNAs and snoRNAs. Nucleic Acids Res. 33, W686–W689 (2005).
Kearse, M. et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28, 1647–1649 (2012).
Zheng, S., Poczai, P., Hyvönen, J., Tang, J. & Amiryousefi, A. Chloroplot: An online program for the versatile plotting of organelle genomes. Front. Genet. 11, 1123 (2020).
Krzywinski, M. et al. Circos: An information aesthetic for comparative genomics. Genome Res. 19, 1639–1645 (2009).
Frazer, K. A., Pachter, L., Poliakov, A., Rubin, E. M. & Dubchak, I. VISTA: Computational tools for comparative genomics. Nucleic Acids Res. 32, W273–W279 (2004).
Kurtz, S. et al. REPuter: The manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 29, 4633–4642 (2001).
Beier, S., Thiel, T., Münch, T., Scholz, U. & Mascher, M. MISA-web: A web server for microsatellite prediction. Bioinformatics 33, 2583–2585 (2017).
Benson, G. Tandem repeats finder: A program to analyze DNA sequences. Nucleic Acids Res. 27, 573–580 (1999).
Katoh, K. & Toh, H. Parallelization of the MAFFT multiple sequence alignment program. Bioinformatics 26, 1899–1900 (2010).
Librado, P. & Rozas, J. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25, 1451–1452 (2009).
Wicke, S., Schneeweiss, G. M., Depamphilis, C. W., Müller, K. F. & Quandt, D. The evolution of the plastid chromosome in land plants: Gene content, gene order, gene function. Plant Mol. Biol. 76, 273–297 (2011).
Kumar, S., Nei, M., Dudley, J. & Tamura, K. MEGA: A biologist-centric software for evolutionary analysis of DNA and protein sequences. Brief. Bioinform. 9, 299–306 (2008).
Asaf, S. et al. Complete chloroplast genome of Nicotiana otophora and its comparison with related species. Front. Plant Sci. 7, 843 (2016).
Wu, Z., Tembrock, L. R. & Ge, S. Are differences in genomic data sets due to true biological variants or errors in genome assembly: An example from two chloroplast genomes. PLoS ONE 10, e0118019 (2015).
Posada, D. jModelTest: Phylogenetic model averaging. Mol. Biol. Evol. 25, 1253–1256 (2008).
Gascuel, O. BIONJ: An improved version of the NJ algorithm based on a simple model of sequence data. Mol. Biol. Evol. 14, 685–695 (1997).
Katoh, K. & Standley, D. M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 30, 772–780 (2013).
Suchard, M. A. et al. Bayesian phylogenetic and phylodynamic data integration using BEAST 1.10. Virus Evol. 4, vey016 (2018).
Rambaut, A., Drummond, A. J., Xie, D., Baele, G. & Suchard, M. A. Posterior summarization in Bayesian phylogenetics using Tracer 1.7. Syst. Biol. 67, 901–904 (2018).
Helfrich, P., Rieb, E., Abrami, G., Lücking, A. & Mehler, A. In Proceedings of the Eleventh International Conference on Language Resources and Evaluation (LREC 2018).
Funding
The corresponding author (A.L.K.) wishes to thank the financial support of the University of Houston (NURF-R0507404) and the University of Nizwa.
Author information
Authors and Affiliations
Contributions
‘A.L.K.’, ‘and ‘S.A.’ performed experiments; ‘A.L.K.’, ‘S.A.’ and ‘W.A.’ wrote the original draft and Bioinformatics analysis: A.L.K. and A.H. supervision arranging resources. All authors have read and approved the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ahmad, W., Asaf, S., Al-Rawahi, A. et al. Comparative plastome genomics, taxonomic delimitation and evolutionary divergences of Tetraena hamiensis var. qatarensis and Tetraena simplex (Zygophyllaceae). Sci Rep 13, 7436 (2023). https://doi.org/10.1038/s41598-023-34477-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-34477-1
This article is cited by
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
