
Abstract
One-carbon metabolism fuels the high demand of cancer cells for nucleotides and other building blocks needed for increased proliferation. Although inhibitors of this pathway are widely used to treat many cancers, their global impact on anabolic and catabolic processes remains unclear. Using a combination of real-time bioenergetics assays and metabolomics approaches, we investigated the global effects of methotrexate on cellular metabolism. We show that methotrexate treatment increases the intracellular concentration of the metabolite AICAR, resulting in AMPK activation. Methotrexate-induced AMPK activation leads to decreased one-carbon metabolism gene expression and cellular proliferation as well as increased global bioenergetic capacity. The anti-proliferative and pro-respiratory effects of methotrexate are AMPK-dependent, as cells with reduced AMPK activity are less affected by methotrexate treatment. Conversely, the combination of methotrexate with the AMPK activator, phenformin, potentiates its anti-proliferative activity in cancer cells. These data highlight a reciprocal effect of methotrexate on anabolic and catabolic processes and implicate AMPK activation as a metabolic determinant of methotrexate response.
Similar content being viewed by others
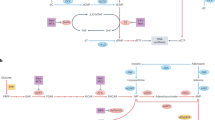


Introduction
Cell growth and proliferation require the production of numerous macromolecules. One-carbon metabolism covers a complex metabolic network important for the production of nucleotides, lipids, reducing power, and substrates for methylation reactions1,2. Indeed, many genetic and functional studies have highlighted the importance of this pathway, along with hyperactivation of serine/glycine biosynthesis, in driving tumourigenesis3,4. Enzymes of the mitochondrial folate pathway, such as methylenetetrahydrofolate dehydrogenase/cyclohydrolase 2 (MTHFD2), which are normally low or absent in normal adult tissues5,6, are highly upregulated in cancer and are negatively correlated with survival in breast cancer patients7. Also, enzymes implicated in serine biosynthesis, such as phosphoglycerate dehydrogenase (PHGDH), are upregulated in triple-negative breast cancer and melanoma8,9, while serine hydroxymethyltransferase 2 (SHMT2) expression and glycine consumption correlate with cancer cell proliferation and poor prognosis across several cancer cell types10,11,12. In support of this point, a recent report showed that patients with colorectal and lung adenocarcinomas that have high expression of mitochondrial one-carbon metabolism genes have shorter overall survival compared to those with low expression of these genes13.
The effectiveness of targeting one-carbon metabolism in cancer was initially recognized over 60 years ago. In 1948, Sydney Farber discovered that treatment of patients with a folic acid antagonist, aminopterin, produced temporary remission in children with acute lymphoblastic leukemia (ALL)14. This landmark paper led the way for the development of a class of drugs called anti-folates. Methotrexate (MTX), a member of this class of drugs, is one of the most commonly used anti-folates in chemotherapy treatment for various cancers, including acute lymphoblastic leukemia, breast cancer, bladder cancer and lymphomas15. Although MTX can be used as a single agent, such as to treat choriocarcinoma16, it is most commonly used in combination with cyclophosphamide and 5-fluorouracil as part of the CMF (cyclophosphamide, methotrexate, 5-fluorouracil) treatment for breast cancer17. MTX inhibits dihydrofolate reductase (DHFR), an enzyme required to produce tetrahydrofolate (THF) that is needed to build nucleotides18. MTX also inhibits 5-aminoimidazole-carboxyamide ribonucleotide formyltransferase (ATIC) and thymidylate synthetase (TYMS), which are enzymes of the purine and pyrimidine biosynthesis pathways respectively19. Despite its wide use as a cancer drug, MTX can have high toxicity by targeting metabolic enzymes found in both transformed and non-transformed cells, hence reducing its therapeutic index20,21. In this manuscript, we uncover that MTX promotes AMPK signaling in breast cancer, which results in stimulation of mitochondrial metabolism and inhibition of cellular proliferation. This knowledge will assist in refining cancer-specific therapeutic strategies involving MTX.
Results
Methotrexate induces AMPK signaling
Given that MTX inhibits purine biosynthesis, we first quantified the levels of metabolic intermediates in this pathway upon treatment. MTX caused a strong increase (~30 fold) in endogenous AICAR levels in breast cancer cells and mouse embryonic fibroblasts (MEFs) (Fig. 1A, Supplementary Fig. 1), with little effect on the downstream metabolites IMP and AMP, suggesting a blockage in de novo purine biosynthesis at the ATIC step. AICAR is used as an exogenous compound to activate AMPK in various cell models22, hence we assessed whether the increase in endogenous AICAR levels upon methotrexate treatment was sufficient to promote AMPK activation. MTX treatment increased the phosphorylation of Ser79 on acetyl-CoA carboxylase (pACC)23, and the phosphorylation of Thr172 on AMPK, indicating that AMPK is activated (Fig. 1B,C). PGC-1α signaling is a known downstream effector of AMPK activation in both non-transformed and transformed cells24,25,26. Accordingly, MTX treatment increased the expression of PPARGC1A and its partner ESRRA in BT-474 cells, indicating that MTX upregulates the PGC-1α/ERRα axis (Fig. 1D). In addition, MTX decreases the expression of MTHFD1L (Fig. 1D), a folate cycle gene that is repressed by AMPK/PGC-1α/ERRα signaling26. Collectively, these data show that MTX treatment promotes AMPK signaling.

Methotrexate activates AMPK signaling by increasing endogenous AICAR levels. (A) Analysis of purine metabolites (AICAR, IMP, AMP) following treatment with 0.1 µM MTX (blue) or control (black) for 72 hours in BT-474 cells, normalized to control treatment (dashed line) (n = 3). (B) Immunoblots of phosphorylated-ACC (Ser79), total ACC, phosphorylated-AMPKα (T172), total AMPK, or Actin in BT-474 cells treated with 0.1 µM MTX or control for 72 hours (n = 3). (C) Quantitation of immunoblots from (B) (n = 3). (D) Expression of PPARGC1A, ESRRA and MTHFD1L in BT-474 cells treated with 0.1 µM MTX (blue) or control for 72 hours, normalized to control treatment (dashed line) (n = 3). Full length blots are presented in Supplementary Fig. 3. All data are presented as means + SEM, *p < 0.05, Student’s t test.
Methotrexate promotes AMPK-dependent mitochondrial respiration
To test the biological implications of AMPK activation upon MTX treatment, we first performed respirometry experiments given that AMPK engages the PGC-1α/ERR axis, which is a central regulator of mitochondrial oxidative phosphorylation. In accordance with the role of AMPK in promoting catabolic reactions, MTX increased cellular respiration in breast cancer cells and non-transformed mammary cells, including the respiration linked to ATP synthesis (coupled respiration) and the respiration linked to proton leak (uncoupled respiration) (Fig. 2A, Supplementary Fig. 2A–F). We also formally quantified the impact of MTX on global cellular bioenergetics28. MTX treatment increased basal total cellular ATP production (J ATP total), which was largely due to an increase in oxidative phosphorylation (J ATP ox), with a small contribution from glycolysis (J ATP glyc) (Fig. 2B). MTX treatment also increased maximal total bioenergetic capacity (Fig. 2C,D) and the levels of aspartate, a metabolite linked to increased respiration in proliferating cells27 (Fig. 2E). In addition, MTX promoted mitochondrial metabolism in non-transformed MEFs. Indeed, MEFs treated with MTX displayed increased total, uncoupled and coupled respiration at baseline, similar to cancer cells (Fig. 2F,G–I blue bars). To determine if the MTX-induced increase in oxidative metabolism was AMPK-dependent, MEF cells deficient for AMPKα1/2 were treated with MTX. AMPK-null MEF cells showed no significant increase in oxidative metabolism upon MTX treatment (Fig. 2F,G–I purple bars). Taken together, these results demonstrate that MTX promotes mitochondrial respiration in an AMPK-dependent manner.

Methotrexate promotes cellular respiration and increases global bioenergetic capacity in an AMPK-dependent manner. (A) Respiration of BT-474 cells treated with 0.1 µM MTX or control for 72 hours. Size of pie chart indicates fold change of total respiration upon MTX treatment (Fold change of 1.98 of MTX-treated cells compared to control); % of coupled respiration (beige) and uncoupled respiration (green) are shown (n = 4). (B) Quantification of total ATP production (J ATP total) for BT-474 cells treated with 0.1 µM MTX or control for 72 hours under basal conditions (10 mM glucose). J ATP total is the sum of J ATP ox (oxidative phosphorylation, orange) and J ATP glyc (glycolysis, brown) (n = 3). (C) Quantification of total bioenergetic capacity in BT-474 cells treated with 0.1 µM MTX (blue) or control (black), compared to control treatment (dashed line) (n = 3). (D) Bioenergetic capacity of BT-474 cells treated with 0.1 µM MTX (blue box/data points) or control (black box/data points). Data points represent J ATP values as a function of J ATP glyc (x-axis) and J ATP ox (y-axis) after the following additions: basal (square), FCCP (triangle), monensin (circle). The size of the box represents the theoretical bioenergetic space that can be occupied by each condition (n = 3). (E) Aspartate levels of BT-474 cells treated with 0.1 μM MTX (blue) for 72 hours, normalized to control treatment (dashed line) (n = 3). (F) Respiration of WT and AMPK-null MEFs treated with 0.02 µM MTX or control for 72 hours. Size of pie chart indicates fold change of total respiration upon MTX treatment (Fold change of WT MTX: 2.13, AMPK-null: 1.25, AMPK-null MTX: 0.93, compared to WT); % of coupled respiration (beige) and uncoupled respiration (green) are shown (n = 4). (G–I) Total, uncoupled, and coupled respiration of WT MEF cells treated with MTX (blue) and AMPK-null MEF cells treated with MTX (purple) compared to each respective control (dashed line) (n = 4). All data are presented as means + SEM, *p < 0.05, Student’s t test for (A–C,E,G–I), and two-way ANOVA, Dunnett’s post hoc test for (F).
Methotrexate exerts AMPK-dependent anti-proliferative effects
It is well established that MTX acts as a chemotherapeutic agent by inhibiting cellular proliferation29. Given that AMPK-mediated metabolic reprogramming has previously been shown to impact proliferation and tumour growth30,31, we tested if the anti-proliferative effect of MTX is dependent on AMPK. We first examined this in eμ-Myc B cell lymphoma cells using an AMPKα1/α2 hairpin construct (shAMPK) targeting AMPK. Knockdown of AMPK in cancer cells decreased the anti-proliferative effects of MTX compared to controls (shCTRL) (Fig. 3A–D). Similarly, the proliferation of AMPK-null MEF cells was less inhibited by MTX treatment than WT controls (Fig. 3E–H). Together, these results indicate that the growth inhibitory effects of MTX are dependent on AMPK.

Methotrexate decreases cellular proliferation in an AMPK-dependent manner. (A) Immunoblots of phosphorylated-ACC (Ser79), total ACC, phosphorylated-AMPKα (T172), total AMPK, and RPLP0 in eμ-Myc B cell lymphoma cells transfected with shCTRL or shAMPK and treated with 2 nM MTX or control for 72 hours (n = 3). (B) Quantitation of immunoblots from (A) (n = 3). (C) Cell proliferation of eμ-Myc B cell lymphoma cells transfected with shCTRL or shAMPK treated with 2 nM MTX for 72 hours compared to shCTRL control (dashed line) (n = 4). (D) Cell proliferation of eμ-Myc B cell lymphoma cells transfected with shCTRL or shAMPK and treated with MTX compared to each respective control (dashed line) (n = 4). (E) Immunoblots of phosphorylated-ACC (Ser79), total ACC, phosphorylated-AMPKα (T172), total AMPK, and RPLP0 in WT and AMPK-null MEF cells treated with 0.05 μM MTX or control for 72 hours (n = 3). (F) Quantitation of immunoblots from (E) (n = 3). (G) Cell proliferation of WT and AMPK-null MEF cells treated with 0.05 μM MTX for 72 hours compared to WT control (dashed line) (n = 3). (H) Cell proliferation of WT and AMPK-null MEF cells treated with MTX compared to each respective control (dashed line) (n = 3). Full length blots are presented in Supplementary Figs. 4 and 5. All data are presented as means + SEM, *p < 0.05, Student’s t test for (B,D,F,H), and two-way ANOVA, Dunnett’s post hoc test for (C,G).
Formate supplementation does not rescue the AMPK-dependent effects of MTX treatment
Genetic defects in one-carbon metabolism have been rescued by formate supplementation32. To determine if the AMPK-dependent effects of MTX treatment can be rescued by formate supplementation, BT-474 cells were treated with a combination of MTX and sodium formate. Formate supplementation did not rescue the MTX-mediated induction of AICAR or ZMP, which resulted in unchanged AMPK signaling activation (Fig. 4A–D), and unaltered cellular bioenergetics (Fig. 4E,F). Furthermore, formate did not rescue the MTX-mediated decrease in cell count (Fig. 4G). Ultimately, the AMPK-dependent effects of MTX do not appear to arise due to lack of formate.

Formate does not rescue the AMPK-dependent metabolic effects of MTX. (A,B) Analysis of metabolites (AICAR and ZMP) following treatment with 0.1 µM MTX (red) or a combination of 0.1 µM MTX and 1 mM sodium formate (dark blue) in BT-474 cells for 72 hours, normalized to control treatment (n = 3). (C) Immunoblots of phosphorylated-ACC (Ser79), total ACC, phosphorylated-AMPKα (T172), total AMPK, or Actin in BT-474 cells treated with MTX, MTX and formate, or control for 72 hours (n = 3). (D) Quantitation of immunoblots from (C) (n = 3). (E-F) Quantification of basal and maximal oxidative ATP production (J ATP ox) and glycolytic ATP production (J ATP glyc) in BT-474 cells treated with MTX, MTX and formate, or control for 72 hours (n = 3). (G) Cell proliferation of BT-474 cells treated with MTX, MTX and formate, or control for 72 hours, compared to control (dashed line) (n = 5). Full length blots are presented in Supplementary Fig. 6. All data are presented as means + SEM, *p < 0.05, one-way ANOVA, Dunnett’s post hoc test (A,B,D-G).
Biguanides potentiate the anti-neoplastic effect of methotrexate
Cancer cells are dependent on one-carbon metabolism to build nucleotides de novo2. Recently, we have shown that one-carbon metabolism is inhibited through activation of AMPK/PGC-1α/ERRα signaling, resulting in increased sensitivity to MTX26. In addition, the AMPK/PGC-1α/ERRα axis is activated by treatment with biguanides24,26,33,34,35. To investigate whether co-treatment with biguanides can potentiate the anti-proliferative effects of MTX, cells were pre-treated with phenformin for 24 hours before treatment with MTX for 72 hours. Cells treated with both phenformin and methotrexate demonstrated lower cell counts than those treated with methotrexate or phenformin alone (Fig. 5A,B). AMPK-null MEF cells were also less sensitive to the combination of phenformin and MTX in terms of cell proliferation, viability, and cell cycle progression compared to WT cells (Fig. 5C–E), further highlighting the impact of AMPK on MTX response. These data show that activation of AMPK using phenformin can potentiate the anti-proliferative effect of MTX in cancer cells.

Phenformin treatment potentiates MTX response. (A) Cell proliferation of BT-474 cells treated with 0.02 mM phenformin or vehicle for 24 hours, then treated with 0.02 mM phenformin or vehicle and 0.1 µM MTX for 72 hours (n = 5). (B) Cell proliferation of sarcoma-180 (S-180) cells treated with 0.01 mM phenformin for 24 hours, then treated with 0.01 mM phenformin and 0.01 µM MTX for 72 hours (n = 7) (C) Cell proliferation, (D) cell viability, and (E) cell cycle analysis of WT and AMPK-null MEF cells treated with MTX (0.02 μM), phenformin (0.1 mM) or both for 72 hours (n = 3). (F) Schematic indicating that AMPK signaling plays a functional role in controlling the catabolic and anabolic procresses induced by methotrexate. All data are presented as means + SEM, *p < 0.05, ***p < 0.001, ****p < 0.0001, one-way ANOVA, Dunnett’s post hoc test for (A,B), and two-way ANOVA, Dunnett’s post hoc test for (C–E).
Discussion
In this study, we report that AMPK signaling plays a key role in mediating the metabolic and anti-proliferative effects of MTX (Fig. 5F). MTX treatment increases endogenous levels of AICAR, leading to AMPK activation. Increased AMPK signaling results in a decrease in one-carbon metabolism gene expression and cell proliferation. In addition, elevated AMPK signaling augments cellular global bioenergetic capacity, mainly by promoting oxidative phosphorylation. The anti-proliferative effects of MTX was potentiated by phenformin, a complex I inhibitor that activates AMPK, which suppresses one-carbon metabolism through PGC-1α/ERRα signaling26. These data highlight potential synergy between MTX and modulators of mitochondrial bioenergetics, and underscore the importance of understanding the metabolic impact of cancer drugs to improve their chemotherapeutic action.
MTX is a potent inhibitor of one-carbon metabolism and nucleotide biosynthesis. Previous work has shown that MTX treatment causes an accumulation of AICAR36 sufficient to activate AMPK in breast cancer cells37,38,39,40. MTX-induced AMPK signaling was shown to promote glucose uptake and lipid oxidation39, as well as inhibit protein synthesis40. The anti-folate drug pemetrexed also induces AICAR-mediated AMPK activation and inhibits anabolic processes in colon cancer cells and lymphoblastic leukemia cells41,42. Here, we show that MTX does not only inhibit anabolic processes, but also promotes catabolic metabolism by stimulating mitochondrial respiration.
Our group has previously shown that the AMPK/PGC-1α/ERRα axis can inhibit one-carbon metabolism and purine biosynthesis, resulting in decreased proliferation26. Given that the anti-proliferative effects of MTX are linked to AMPK activity, we show in this study that the AMPK activator phenformin can improve the anti-proliferative action of MTX. This is line with reports showing that pharmacological AMPK activation with AICAR also increases the antineoplastic effect of MTX in breast, skin, and prostate cancer models37. However, given that AICAR has poor bioavailability43, combining MTX with AMPK activators that have higher bioavailability, such as biguanides, may prove to be an effective option in the clinic.
There has been increased interest in repurposing biguanides for cancer treatment. Indeed, both phenformin and metformin display AMPK-dependent and –independent anti-neoplastic effects in several cancer subtypes including breast, prostate, and colorectal cancers44,45. Despite these successes, clinical trials regarding the usage of biguanides in cancer have been disappointing44. Given that biguanides and MTX both share a common effector in AMPK, the combination of both drugs may have strong clinical implications in improving AMPK-dependent anti-neoplastic response. Indeed, AMPK has been reported to have both anti-Warburg and anti-proliferative effects31. However, the overall outcome of AMPK activation in cancer is still unclear as AMPK can limit anabolic processes required for proliferation, yet promote metabolic adaptation of tumours to cope under energetic stress46.
Overall, our results highlight the importance of AMPK in mediating chemosensitivity. Our work will likely stimulate translational research aimed at testing whether biguanides could be used as neoadjuvant therapy to improve chemotherapeutic response to anti-folates.
Methods
Cell lines and reagents
BT-474, S-180, MCF10A, MCF7, and NMUMG cell lines were purchased from ATCC and cultured as previously described26,47. NT2196 is a stable cell line corresponding to immortalized NMuMG cells that were transformed with an oncogenic form of Neu (ErbB2), which was cultured as previously described48. AMPK-null MEFs and eμ-Myc B cell lymphoma shCTRL/shAMPK cell lines were generous gifts from Dr. Russell Jones’ laboratory, and were cultured as previously described31,45,49. The eμ-Myc B cell lymphoma cell line was engineered using the plasmid MSCV p2GM AMPK alpha2hp1 alpha1hp1 (Addgene plasmid # 89492; http://n2t.net/addgene:89492; RRID:Addgene_89492). Methotrexate (MTX) (dissolved in DMSO) and phenformin (dissolved in water) was purchased from Sigma. All LC/MS grade solvents and salts were purchased from Fisher (Ottawa, Ontario Canada: water, acetonitrile (ACN), Methanol (MeOH), formic acid, and ammonium acetate. The authentic metabolite standards were purchase from Sigma-Aldrich Co. (Oakville, Ontario, Canada).
Western Blotting
Protein extracts were prepared using lysis buffer (50 mmol/L Tris-HCl [pH 7.4], 1% Triton X-100, 0.25% sodium deoxycholate, 150 mmol/L NaCl, 1 mmol/L EDTA) supplemented with protease inhibitors (cOmplete Mini EDTA-free Protease Inhibitor Cocktail, cat# 4693159001) and phosphatase inhibitors (PhosphoStop cat# 4906837001). Immunoblots were incubated with the following primary antibodies from Cell Signaling: pAMPK (2531), AMPK (2532), pACC (3661), ACC (3662); from Santa Cruz: Actin (sc-1616); RPLP0 (11290-AP) The results were visualized with Western Lightning Plus-ECL (PerkinElmer) and analyzed with ImageJ software (NIH).
Quantitative RT-PCR
RNA was extracted and purified using the Aurum Total RNA Mini Kit (Bio-Rad) following the manufacturers’ protocols. Reverse transcriptase reactions were performed using iScript cDNA Synthesis Kit (Bio-Rad). Samples were then analyzed by qRT-PCR with SYBR-green-based qRT-PCR with a MyiQ2 Real-Time Detection System (Bio-Rad). Gene-specific primers are found in Supplementary Table 1. Values were normalized to PUM1.
Respirometry
Cellular respiration was determined using a Digital Model 10 Clark Electrode (Rank Brothers) as previously described50. 1 × 106 cells (BT-474, MCF10A, MCF7) or 1.5 × 106 (NMuMG, NT2196) cells were treated with methotrexate for 72 hours and used for respiration measurements. Oligomycin (2.5 μg/mL/1 × 106 cells) was added to inhibit the ATP synthase, which allows for the calculation of respiration coupled to ATP synthesis (coupled respiration) and respiration linked to proton leak (uncoupled respiration). Myxothiazol (0.5 μM/1 × 106 cells) was added to inhibit complex III, and is used to determine the contribution of non-mitochondrial respiration, which was not observed.
Seahorse bioenergetic analysis
Oxygen Consumption Rate (OCR) and Extracellular Acidification Rate (ECAR) were measured using the XFe24 Extracellular Flux Analyzer (Agilent Technologies, Santa Clara, CA, USA) according to the manufacturer’s protocol. BT-474 cells were treated for 72 hours with methotrexate, then trypisizinzed and 50,000 cells per well were plated overnight in Seahorse cell culture plates (Agilent Technologies, Santa Clara, CA, USA). The next day, cells were washed with XF media (Seahorse Bioscience) without glucose and incubated in a CO2-free incubator at 37 °C for 2 hours to establish equilibrium. Basal conditions include XF media with 10 mM glucose. Oligomycin (1 μM), FCCP (1.5 μM), rotenone and antimycin A (0.5 μM each), and monensin (20 μM) were used. Oligomycin is an inhibitor of ATP synthase. FCCP uncouples the inner mitochondrial membrane, thereby allowing for maximal oxygen consumption. The combination of rotenone (complex I inhibitor) and antimycin A (complex III inhibitor) is used to maximally perturb mitochondrial respiration. Monensin is used to calculate the maximum glycolytic capacity of cells, by driving increased ATP demand by the Na+/K+–ATPase. In addition, HCl (hydrochloric acid) was injected into wells containing media with no cells (four injections of 0.25 mM each) in order to calculate the buffer capacity of the XF media. OCR, ECAR, and PPR (proton production rate) measurements were taken before and after each injection, and were used to calculate ATP production (J ATP total) and bioenergetic capacity as previously described28. Briefly, bioenergetic capacity is the product of the maximal ATP from glycolysis (J ATP glyc) and ATP from oxidative phosphorylation (J ATP ox). Values were normalized to cell counts.
Cell proliferation
Cell counts were quantified using an automated TC10 cell counter (Bio-Rad) and viability was determined using trypan blue exclusion.
Metabolomics
Nucleotide detection and analysis was performed using LC-MS/MS at the Metabolomics Core Facility of the Goodman Cancer Research Centre. Cultured cells were treated with methotrexate for 72 hours. Cells were washed in ammonium formate three times, then quenched in cold 50% methanol (v/v) and acetonitrile. Cells were lysed following bead beating at 30 Hz for 2 minutes. Cellular extracts were partitioned into aqueous and organic layers following dichloromethane treatment and centrifugation. Aqueous supernatants were dried down using a refrigerated speed-vac. Dried samples were subsequently resuspended in 25 μl of water. A 10 μl volume of sample was injected onto an Agilent 6430 Triple Quadrupole (QQQ)-LC-MS/MS for targeted metabolite analysis of AICAR (5-aminoimidazole-4-carboxamide-1-β-D-ribofuranoside) and a 10-fold dilution was injected for nucleotides: ZMP (5-aminoimidazole-4-carboxamide ribonucleotide), IMP (inosine monophosphate), and AMP (adenosine monophosphate). The liquid chromatography separation was performed using a 1290 Infinity ultra-performance binary LC system (Agilent Technologies, Santa Clara, CA, USA). The chromatography run was conducted as follows: column temperature was maintained at 10 °C and the separation was realised by reverse phase separation using a flow rate of 0.4 mL/min with a Scherzo SM-C18 column 3 μm, 3.0 × 150 mm (Imtakt Corp, JAPAN) The gradient started at 100% mobile phase A (5 mM ammonium acetate in water) with a 5 min gradient to 100% B (200 mM ammonium acetate in 80% water/20% ACN) at a flow rate of 0.4 ml/min. This was followed by a 5 min hold time at 100% mobile phase B and a subsequent re-equilibration time (6 min) before next injection.
The mass spectrometer was equipped with an electrospray ionization (ESI) source and samples were analyzed in positive ionization mode. Multiple reaction monitoring (MRM) transitions were optimized on standards for each metabolite quantitated. Transitions for quantifier and qualifier ions were as follows: AICAR (259.1 → 127.0 and 110.0, 82.1, 55.1), ZMP (339.1 → 127.0 and 110.0), IMP (349.0 → 147 and 110.0), and AMP (348.0 → 146.1 and 348.0 → 118.9). Gas temperature and flow were set at 350 °C and 10 l/min respectively, nebulizer pressure was set at 40 psi and capillary voltage was set at 3500 V. Relative concentrations were determined from external calibration curves prepared in water and compared to sample area under the curve. Note that no corrections were made for ion suppression or enhancement. Data were analyzed using MassHunter Quant (Agilent Technologies, Santa Clara, CA, USA).
Flow cytometry analysis of cell cycle
WT and AMPK-null MEF cells were seeded in 12-well plates and grown for 24 h, after which they were treated with control, MTX, phenformin or both for 72 h. Cells were counted and 100,000 cells were washed with cold wash buffer (PBS + 5% FBS + 0.01 M NaN3), spun down, and resuspended in hypotonic buffer (0.1% sodium citrate, 0.1% Triton X-100 in water). Cells were stained with Propidium iodide (50 μg/ml) (cat #: P4170, Sigma) for 30 min at 37 °C in the dark. Samples were analyzed with a FACS Canto II (San Jose, CA). Fluorescence was detected by excitation at 488 nm and acquisition on the 585/42 PI-A channel.
Data availability
The datasets generated and/or analysed during the current study are available from the corresponding author on reasonable request.
References
Deberardinis, R. J., Sayed, N., Ditsworth, D. & Thompson, C. B. Brick by brick: metabolism and tumor cell growth. Curr. Opin. Genet. Dev. 18, 54–61, https://doi.org/10.1016/j.gde.2008.02.003 (2008).
Locasale, J. W. Serine, glycine and one-carbon units: cancer metabolism in full circle. Nat. Rev. Cancer 13, 572–583, https://doi.org/10.1038/nrc3557 (2013).
DeBerardinis, R. J. Serine metabolism: some tumors take the road less traveled. Cell Metab. 14, 285–286, https://doi.org/10.1016/j.cmet.2011.08.004 (2011).
Ducker, G. S. & Rabinowitz, J. D. One-Carbon Metabolism in Health and Disease. Cell Metab. 25, 27–42, https://doi.org/10.1016/j.cmet.2016.08.009 (2017).
Pike, S. T., Rajendra, R., Artzt, K. & Appling, D. R. Mitochondrial C1-tetrahydrofolate synthase (MTHFD1L) supports the flow of mitochondrial one-carbon units into the methyl cycle in embryos. J. Biol. Chem. 285, 4612–4620, https://doi.org/10.1074/jbc.M109.079855 (2010).
Di Pietro, E., Wang, X. L. & MacKenzie, R. E. The expression of mitochondrial methylenetetrahydrofolate dehydrogenase-cyclohydrolase supports a role in rapid cell growth. Biochim. Biophys. Acta 1674, 78–84, https://doi.org/10.1016/j.bbagen.2004.06.014 (2004).
Nilsson, R. et al. Metabolic enzyme expression highlights a key role for MTHFD2 and the mitochondrial folate pathway in cancer. Nat. Commun. 5, 3128, https://doi.org/10.1038/ncomms4128 (2014).
Locasale, J. W. et al. Phosphoglycerate dehydrogenase diverts glycolytic flux and contributes to oncogenesis. Nat. Genet. 43, 869–874, https://doi.org/10.1038/ng.890 (2011).
Possemato, R. et al. Functional genomics reveal that the serine synthesis pathway is essential in breast cancer. Nature 476, 346–350, https://doi.org/10.1038/nature10350 (2011).
Jain, M. et al. Metabolite profiling identifies a key role for glycine in rapid cancer cell proliferation. Science 336, 1040–1044, https://doi.org/10.1126/science.1218595 (2012).
Lee, G. Y. et al. Comparative oncogenomics identifies PSMB4 and SHMT2 as potential cancer driver genes. Cancer Res. 74, 3114–3126, https://doi.org/10.1158/0008-5472.CAN-13-2683 (2014).
Kim, D. et al. SHMT2 drives glioma cell survival in ischaemia but imposes a dependence on glycine clearance. Nature 520, 363–367, https://doi.org/10.1038/nature14363 (2015).
Koseki, J. et al. Enzymes of the one-carbon folate metabolism as anticancer targets predicted by survival rate analysis. Sci. Rep. 8, 303, https://doi.org/10.1038/s41598-017-18456-x (2018).
Farber, S. & Diamond, L. K. Temporary remissions in acute leukemia in children produced by folic acid antagonist, 4-aminopteroyl-glutamic acid. N. Engl. J. Med. 238, 787–793, https://doi.org/10.1056/NEJM194806032382301 (1948).
Chabner, B. A. & Roberts, T. G. Jr. Timeline: Chemotherapy and the war on cancer. Nat. Rev. Cancer 5, 65–72, https://doi.org/10.1038/nrc1529 (2005).
Hertz, R., Li, M. C. & Spencer, D. B. Effect of methotrexate therapy upon choriocarcinoma and chorioadenoma. Proc. Soc. Exp. Biol. Med. 93, 361–366 (1956).
Munzone, E., Curigliano, G., Burstein, H. J., Winer, E. P. & Goldhirsch, A. CMF revisited in the 21st century. Ann. Oncol. 23, 305–311, https://doi.org/10.1093/annonc/mdr309 (2012).
Hitchings, G. H. & Burchall, J. J. Inhibition of folate biosynthesis and function as a basis for chemotherapy. Adv. Enzymol. Relat. Areas Mol. Biol. 27, 417–468 (1965).
Chabner, B. A. et al. Polyglutamation of methotrexate. Is methotrexate a prodrug? J. Clin. Invest. 76, 907–912, https://doi.org/10.1172/JCI112088 (1985).
Howard, S. C., McCormick, J., Pui, C. H., Buddington, R. K. & Harvey, R. D. Preventing and Managing Toxicities of High-Dose Methotrexate. Oncologist 21, 1471–1482, https://doi.org/10.1634/theoncologist.2015-0164 (2016).
Tennant, D. A., Duran, R. V. & Gottlieb, E. Targeting metabolic transformation for cancer therapy. Nat. Rev. Cancer 10, 267–277, https://doi.org/10.1038/nrc2817 (2010).
Merrill, G. F., Kurth, E. J., Hardie, D. G. & Winder, W. W. AICA riboside increases AMP-activated protein kinase, fatty acid oxidation, and glucose uptake in rat muscle. Am. J. Physiol. 273, E1107–1112 (1997).
Hardie, D. G. AMPK: a target for drugs and natural products with effects on both diabetes and cancer. Diabetes 62, 2164–2172, https://doi.org/10.2337/db13-0368 (2013).
Jager, S., Handschin, C., St-Pierre, J. & Spiegelman, B. M. AMP-activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC-1alpha. Proc. Natl Acad. Sci. USA 104, 12017–12022, https://doi.org/10.1073/pnas.0705070104 (2007).
Chaube, B. et al. AMPK maintains energy homeostasis and survival in cancer cells via regulating p38/PGC-1alpha-mediated mitochondrial biogenesis. Cell Death Discov. 1, 15063, https://doi.org/10.1038/cddiscovery.2015.63 (2015).
Audet-Walsh, É. et al. The PGC-1α/ERRα Axis Represses One-Carbon Metabolism and Promotes Sensitivity to Anti-folate Therapy in Breast Cancer. Cell Rep. 14, 920–931, https://doi.org/10.1016/j.celrep.2015.12.086 (2016).
Sullivan, L. B. et al. Supporting Aspartate Biosynthesis Is an Essential Function of Respiration in Proliferating Cells. Cell 162, 552–563, https://doi.org/10.1016/j.cell.2015.07.017 (2015).
Mookerjee, S. A., Gerencser, A. A., Nicholls, D. G. & Brand, M. D. Quantifying intracellular rates of glycolytic and oxidative ATP production and consumption using extracellular flux measurements. J. Biol. Chem. 292, 7189–7207, https://doi.org/10.1074/jbc.M116.774471 (2017).
Shuvalov, O. et al. One-carbon metabolism and nucleotide biosynthesis as attractive targets for anticancer therapy. Oncotarget 8, 23955–23977, https://doi.org/10.18632/oncotarget.15053 (2017).
Faubert, B., Vincent, E. E., Poffenberger, M. C. & Jones, R. G. The AMP-activated protein kinase (AMPK) and cancer: Many faces of a metabolic regulator. Cancer Lett. 356, 165–170, https://doi.org/10.1016/j.canlet.2014.01.018 (2015).
Faubert, B. et al. AMPK is a negative regulator of the Warburg effect and suppresses tumor growth in vivo. Cell Metab. 17, 113–124, https://doi.org/10.1016/j.cmet.2012.12.001 (2013).
Ducker, G. S. et al. Reversal of Cytosolic One-Carbon Flux Compensates for Loss of the Mitochondrial Folate Pathway. Cell Metab. 23, 1140–1153, https://doi.org/10.1016/j.cmet.2016.04.016 (2016).
Zakikhani, M., Dowling, R., Fantus, I. G., Sonenberg, N. & Pollak, M. Metformin is an AMP kinase-dependent growth inhibitor for breast cancer cells. Cancer Res. 66, 10269–10273, https://doi.org/10.1158/0008-5472.CAN-06-1500 (2006).
Zakikhani, M., Dowling, R. J., Sonenberg, N. & Pollak, M. N. The effects of adiponectin and metformin on prostate and colon neoplasia involve activation of AMP-activated protein kinase. Cancer Prev. Res. 1, 369–375, https://doi.org/10.1158/1940-6207.CAPR-08-0081 (2008).
Corominas-Faja, B. et al. Metabolomic fingerprint reveals that metformin impairs one-carbon metabolism in a manner similar to the antifolate class of chemotherapy drugs. Aging 4, 480–498, https://doi.org/10.18632/aging.100472 (2012).
Cronstein, B. N., Naime, D. & Ostad, E. The antiinflammatory mechanism of methotrexate. Increased adenosine release at inflamed sites diminishes leukocyte accumulation in an in vivo model of inflammation. J. Clin. Invest. 92, 2675–2682, https://doi.org/10.1172/JCI116884 (1993).
Beckers, A. et al. Methotrexate enhances the antianabolic and antiproliferative effects of 5-aminoimidazole-4-carboxamide riboside. Mol. Cancer Ther. 5, 2211–2217, https://doi.org/10.1158/1535-7163.MCT-06-0001 (2006).
Fodor, T. et al. Combined Treatment of MCF-7 Cells with AICAR and Methotrexate, Arrests Cell Cycle and Reverses Warburg Metabolism through AMP-Activated Protein Kinase (AMPK) and FOXO1. PLoS One 11, e0150232, https://doi.org/10.1371/journal.pone.0150232 (2016).
Pirkmajer, S. et al. Methotrexate promotes glucose uptake and lipid oxidation in skeletal muscle via AMPK activation. Diabetes 64, 360–369, https://doi.org/10.2337/db14-0508 (2015).
Tedeschi, P. M. et al. Quantification of folate metabolism using transient metabolic flux analysis. Cancer Metab. 3, 6, https://doi.org/10.1186/s40170-015-0132-6 (2015).
Rothbart, S. B., Racanelli, A. C. & Moran, R. G. Pemetrexed indirectly activates the metabolic kinase AMPK in human carcinomas. Cancer Res. 70, 10299–10309, https://doi.org/10.1158/0008-5472.can-10-1873 (2010).
Racanelli, A. C., Rothbart, S. B., Heyer, C. L. & Moran, R. G. Therapeutics by cytotoxic metabolite accumulation: pemetrexed causes ZMP accumulation, AMPK activation, and mammalian target of rapamycin inhibition. Cancer Res. 69, 5467–5474, https://doi.org/10.1158/0008-5472.can-08-4979 (2009).
Dixon, R. et al. AICA-riboside: safety, tolerance, and pharmacokinetics of a novel adenosine-regulating agent. J. Clin. Pharmacol. 31, 342–347 (1991).
Mallik, R. & Chowdhury, T. A. Metformin in cancer. Diabetes Res Clin Pract, https://doi.org/10.1016/j.diabres.2018.05.023 (2018).
Griss, T. et al. Metformin Antagonizes Cancer Cell Proliferation by Suppressing Mitochondrial-Dependent Biosynthesis. PLoS Biol. 13, e1002309, https://doi.org/10.1371/journal.pbio.1002309 (2015).
Hardie, D. G. Molecular Pathways: Is AMPK a Friend or a Foe in Cancer? Clin. Cancer Res. 21, 3836–3840, https://doi.org/10.1158/1078-0432.CCR-14-3300 (2015).
Andrzejewski, S., Gravel, S.-P., Pollak, M. & St-Pierre, J. Metformin directly acts on mitochondria to alter cellular bioenergetics. Cancer Metab. 2, 12–12, https://doi.org/10.1186/2049-3002-2-12 (2014).
Ursini-Siegel, J. et al. Elevated expression of DecR1 impairs ErbB2/Neu-induced mammary tumor development. Mol. Cell Biol. 27, 6361–6371, https://doi.org/10.1128/mcb.00686-07 (2007).
Yan, M. et al. The tumor suppressor folliculin regulates AMPK-dependent metabolic transformation. J. Clin. Invest. 124, 2640–2650, https://doi.org/10.1172/jci71749 (2014).
Fantin, V. R., St-Pierre, J. & Leder, P. Attenuation of LDH-A expression uncovers a link between glycolysis, mitochondrial physiology, and tumor maintenance. Cancer Cell 9, 425–434, https://doi.org/10.1016/j.ccr.2006.04.023 (2006).
Acknowledgements
We would like to thank staff scientists at the Goodman Cancer Research Centre (GCRC) Metabolomics Core Facility (McGill University) for guidance with metabolomics studies. We would like to thank Oro Uchenunu and Christian Young at the Lady Davis Institute (McGill University) for assistance with flow cytometry. The Metabolomics Core Facility is supported by Terry Fox Research Institute (#1048 in partnership with Fondation du cancer du sein du Quebec), the Fraser Fund and McGill University. This work was supported by grants from Terry Fox Research Institute and Québec Breast Cancer Foundation (TFF-242122 to J.St-P. and R.G.J.). We acknowledge salary support from Fonds de Recherche du Québec-Santé (FRQS) and the McGill Integrated Cancer Research Training Program (MICRTP) to D.J.P. J.St-P. is recipient of a Canada Research Chair in Cancer Metabolism.
Author information
Authors and Affiliations
Contributions
Conceptualization and design: D.J.P., M.R., G.B., D.A., R.G.J., J.St-P.; Development of methodology: D.J.P., M.R., G.B., D.A.; Acquisition of data: D.J.P., E.M., D.R., M.R., G.B., D.A.; Analysis and interpretation of data: D.J.P., M.R., G.B., D.A., J.St-P. Writing of the manuscript: D.J.P., M.R., D.A., J.St-P. Study supervision: J.St-P.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Papadopoli, D.J., Ma, E.H., Roy, D. et al. Methotrexate elicits pro-respiratory and anti-growth effects by promoting AMPK signaling. Sci Rep 10, 7838 (2020). https://doi.org/10.1038/s41598-020-64460-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-64460-z
This article is cited by
-
Metformin sensitises hepatocarcinoma cells to methotrexate by targeting dihydrofolate reductase
Cell Death & Disease (2021)
-
Folate metabolism: a re-emerging therapeutic target in haematological cancers
Leukemia (2021)
-
Juvenile Dermatomyositis: New Clues to Diagnosis and Therapy
Current Treatment Options in Rheumatology (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
