
Abstract
Many driver pathways for cancer cell proliferation have been reported. Driver pathway activation is often evaluated based on a single hotspot mutation such as EGFR L858R. However, because of complex intratumoral networks, the impact of a driver pathway cannot be predicted based on only a single gene mutation. Here, we developed a novel diagnostic system named the “EGFR impact score” which is based on multiplex mRNA expression profiles, which can predict the impact of the EGFR pathway in lung cancer cells and the effect of EGFR-tyrosine kinase inhibitors on malignancy. The EGFR impact score indicated robust predictive power for the prognosis of early-stage lung cancer because this score can evaluate the impact of the EGFR pathway on the tumor and genomic instability. Additionally, the molecular features of the poor prognostic group resembled those of biomarkers associated with immune checkpoint inhibitors. The EGFR impact score is a novel prognostic and therapeutic indicator for lung adenocarcinoma.
Similar content being viewed by others

Introduction
EGFR mutations are frequently reported in lung adenocarcinoma1,2. The missense mutation L858R and exon 19 deletion is a well-known hotspot mutation in EGFR3,4. EGFR hotspot mutations activate the EGFR pathway and promote tumor cell proliferation5. Conversely, next-generation sequencing has revealed the presence of many variants of uncertain significance (VUSs) in the EGFR gene6. To date, a method for evaluating these VUSs has not been established.
In the clinical setting, EGFR hotspot mutations are considered surrogate markers of tyrosine kinase inhibitor (TKI) treatment. However, approximately 30% of EGFR-mutant cancers exhibit resistance to TKI treatment without well-known secondary EGFR mutations such as T790M7. Some papers reported that these TKI-resistant tumors depend on both EGFR signaling and multiple driver pathways for survival8,9. Contrarily, the presence of tumors that respond to TKIs despite not carrying EGFR structural mutations has been reported10,11. These results indicated the existence of EGFR pathway activation in the absence of EGFR structural mutations. These findings illustrated the limitation of using single gene mutations to predict the effects on EGFR network signaling. We hypothesized that mRNA expression profiles can predict the complicated EGFR network status more comprehensively than the DNA structural status because of central dogma. Based on this hypothesis, this study first aimed to develop a predictive model of responsiveness to TKI based on mRNA expression profiles.
It is controversial whether EGFR mutation is a prognostic factor in early-stage lung adenocarcinoma12,13,14,15,16. Therefore, we clarified whether accurate evaluation of EGFR networks can predict the prognosis of early-stage lung adenocarcinoma. In this study, we evaluated EGFR networks via multiplex gene expression profiling. Several prognostic prediction models based on expression profiles have been developed for lung cancer17,18,19,20,21. Some expression profiles are currently in clinical use and are commercially available22. However, these expression profiles do not influence treatment decision-making. Considering these situations, the second purpose of this study was to clarify whether EGFR network expression profiles can be used to predict the prognosis of early-stage lung adenocarcinoma. Furthermore, to develop new treatment strategies for the poor prognostic group identified using our new system, we clarified the molecular biological features of lung cancer using multi-omics data.
Results
Establishment of the EGFR impact score by analyzing a publicly available dataset
We identified 254 differently expressed genes (DEGs) in EGFR-mutant cancers in the GSE32863 dataset (Fig. 1a). We analyzed 58 patients in this dataset, 17 of whom had an EGFR mutation. Of the 254 DEGs, 42 were selected using the random forest algorithm. Of these selected DEGs, 23 were upregulated and 19 were downregulated in EGFR-mutant cancers. These gene sets were designated as the upregulated EGFR signature and downregulated EGFR signature, respectively. The ALK gene was added to the downregulated EGFR signature group (Fig. 1b). Using the expression profile of each EGFR signature, we calculated the EGFR impact score. We conducted receiver operating characteristic (ROC) analysis to determine the threshold of the EGFR impact score (Supplemental Fig. 1). Patients with an EGFR impact score higher than 2.2 comprised the EI-H-type group, and those with an EGFR impact score lower than 2.2 comprised the EI-L-type group (Fig. 1c).

(a) MA plot. Each dot denotes upregulated (red) and downregulated (blue) genes among patients with EGFR mutations in the GSE32863 dataset. (b) Heatmap of the EGFR signature in GSE32863. The rows of the heatmap represent gene symbols and Entrez IDs, and samples are presented in columns. The labels at the top denote the structural status of the EGFR and KRAS genes. (c) EGFR impact score in the GSE32863 dataset. The x-axis shows samples, and the y-axis shows the EGFR impact score. MT, mutant; WT, wild-type.
Prediction of responsiveness to TKI treatment based on the EGFR impact score
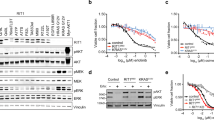
We analyzed GSE34228 data (n = 52) to clarify whether the EGFR impact score can predict responsiveness to TKIs. The GSE34228 dataset contains expression data from both gefitinib-sensitive and gefitinib-resistant cell lines. This dataset uses the PC-9 cell line, which carries the delE746-A750 mutation in EGFR23. The gefitinib-resistant cell line was created by exposing PC-9 cells to long-term culture with gefitinib. We calculated the EGFR impact score in these cell lines. The EGFR impact score of the gefitinib-resistant cell line was significantly lower than that of the gefitinib-sensitive cell line (Fig. 2a). Moreover, we analyzed CCLE (Cancer Cell Line Encyclopedia) and GDSC (Genomics of Drug Sensitivity in Cancer) data sets. CCLE and GDSC datasets have information of both IC50 of gefitinib and expression profile data in EGFR mutant cell lines. We circulated the EGFR impact score of each cell line. The EGFR impact score was clearly associated with IC50 of gefitinib in both CCLE and GDSC data sets (Supplementary Fig. 2a,b). Notably, In the GDSC data set, the EGFR impact score was clearly correlated with IC50 of gefitinib. Furthermore, we analyzed GSE37138 data to clarify whether the EGFR impact score can predict responsiveness to TKIs in clinical specimens. In GSE37138 data, patients with non-squamous non-small cell lung cancer (NSCLC) were treated with erlotinib and bevacizumab. The tumor shrinkage rate was assessed in all patients 12 weeks after treatment. Of these patients, four patients had EGFR mutations, and three patients had KRAS mutations. The dataset included patients with high EGFR impact scores and good responsiveness to treatment despite not carrying EGFR mutations (Fig. 2b). In the GSE37138 dataset, Although Fig. 2b necessitate the recruitment of more patients, EGFR impact score tends to predict responsiveness of TKI treatment in clinical samples.

(a) EGFR impact score in the GSE34228 dataset. The boxplot in yellow shows the EGFR impact score in gefitinib-resistant cell lines, and the boxplot in green shows gefitinib-sensitive cell lines. (b) Association between the EGFR impact score and tumor shrinkage rate in GSE37138. Each dot shows patients with mutant EGFR (red), mutant KRAS (blue), and other (grey).
Diagnosis of patients with VUSs using the EGFR impact score
We analyzed TCGA data to clarify the classification of patients with VUSs in the EGFR gene using the EGFR impact score. Patients with early-stage lung adenocarcinoma (n = 171) were selected, and of these patients, 24 had EGFR mutations. Six patients carried exon 19 deletions, six patients carried the L858R mutation, three patients had VUSs and the remaining patients had other mutations in EGFR. Sixty-one percent of patients with pathogenic mutations were diagnosed with EI-H-type cancer, and the remaining patients were diagnosed with EI-L-type cancer. All patients with VUSs were diagnosed with EI-L-type cancer (Fig. 3a).

(a) EGFR impact score in the TCGA dataset. The x-axis shows samples, and the y-axis shows the EGFR impact score. Larger dots denote patients with VUSs in EGFR. (b) EGFR impact score in the GSE31210 dataset. The x-axis shows samples, and the y-axis shows the EGFR impact score. Larger dots denote patients with recurrence. VUS, variant of unknown significance; WT, wild-type; MT, mutant.
Prediction of prognosis using the EGFR impact score
We analyzed the GSE31210 dataset to clarify whether the EGFR impact score can predict the prognosis of early-stage lung adenocarcinoma more accurately than EGFR structural mutations. In this dataset, patients who did not receive adjuvant therapy (n = 204) were selected, and of these patients, 116 had EGFR mutations. Thirty-one percent of patients with EGFR mutations were diagnosed with EI-H-type cancer, and the remaining patients were diagnosed with EI-L-type cancer (Fig. 3b). We compared recurrence-free survival (RFS) and overall survival (OS) between patients with wild-type EGFR and EGFR structural mutations. Patients with wild-type EGFR had poorer RFS and OS than patients carrying EGFR mutations (Fig. 4a,b). Similarly, we compared the prognosis of the EI-H-type and EI-L-type groups. The EI-L-type group also had poorer RFS and OS than the EI-H-type group (Fig. 4c,d). Among patients with EGFR-mutant cancer, the EI-L-type group had poorer prognosis than the EI-H-type group (Fig. 5a). Multivariate analysis using the Cox proportional hazard model indicated that the EGFR impact score can predict prognosis more accurately than the EGFR structural status (Fig. 5b). In addition to GSE31210, we analyzed GSE11969 data set which has early stage lung adenocarcinoma patients with EGFR mutation. In GSE11969, EI-L type tended to have poorer OS than EI-H type (Supplementary Fig. 3).

(a,b) Recurrence-free survival (RFS) and overall survival (OS) curves, respectively, according to the EGFR status in the GSE31210 dataset. The green line denotes patients with EGFR structural mutations, and the orange line denotes patients with wild-type EGFR. (c,d) RFS and OS curves, respectively, according to the EGFR impact status in the GSE31210 dataset. The red line denotes the EI-H-type group, and the blue line shows the EI-L-type group. MT, mutant; WT, wild-type.

(a) Recurrence-free survival (RFS) curves according to the EGFR structural status and EGFR impact status in the GSE31210 dataset. The different coloured lines denote the EGFR-mutant EI-H-type (red), EGFR-mutant EI-L-type (blue) and EGFR wild type group (green). (b) Multivariate analysis by the EGFR structural status and EGFR impact score regarding RFS in the GSE31210 dataset using the Cox hazard model. MT, mutant; WT, wild-type.
Biological features of the EI-H-type and EI-L-type groups
Using GSE31210 data, we compared biological molecular features between the EI-H-type and EI-L-type groups among patients with EGFR mutations via gene set enrichment analysis (GSEA). The GSEA results indicated that gene sets related to “EGFR signaling down,” “poor survival,” “the cell cycle,” and “immune response” were enriched in the EI-L-type group (Fig. 6a), and gene sets related to “good survival” were enriched in the EI-H-type group. We compared PD-L1 expression levels and somatic mutation burden between the EI-H-type and EI-L-type groups. In the GSE31210 and TCGA datasets, PD-L1 levels were significantly higher in the EI-L-type group than in the EI-H-type group (Fig. 6b,c). Restricting the analysis to only patients with EGFR mutations, PD-L1 expression was significantly higher in the EI-L-type group than in the EI-H-type group in the GSE31210 dataset (Fig. 6d). In the TCGA dataset, the EI-L-type group had a significantly higher total mutation burden than the EI-H type group (Fig. 6e). Among patients with EGFR mutations, the total mutation burden was significantly higher in the EI-L-type group than in the EI-H-type group (Fig. 6f).

(a) Results of gene set enrichment analysis using the GSE31210 dataset. These gene sets are enriched regarding patients with EI-L-type cancer and EGFR mutations. (b) Microarray data of PD-L1 expression in the GSE31210 dataset. (c) RNA sequence data of PD-L1 expression levels of PD-L1 expression levels in the EGFR-mutant EI-H-type (red), EGFR-mutant EI-L-type (navy), wild-type EGFR EI-H-type (blue), and wild-type EGFR EI-L-type groups (yellow). (d) Microarray data of PD-L1 expression in the EGFR-mutant EI-H-type (red), EGFR-mutant EI-L-type (blue), EGFR wild type groups (green) in the GSE31210 dataset.. (e) Total mutation burden of the exome sequence in the TCGA dataset. Red dots denote the mutation burden in the EI-H-type group, and blue dots denote the mutation burden in the EI-L-type group. (f) Total mutation burden of the exome sequence in the EGFR-mutant EI-H-type (red), EGFR-mutant EI-L-type (blue), EGFR wild type groups (green) in the TCGA dataset.
Discussion
We developed a novel diagnostic system to predict the EGFR network status comprehensively based on gene expression profiles, which we named the EGFR impact score. Using TCGA data, the EGFR impact score distinguished pathogenic mutations in the EGFR gene from VUSs. Whether these VUSs are pathogenic is unclear because we did not conduct forward genetic experiments using cell or animal models. However, the result that the EGFR impact score can distinguish hotspot mutations from VUSs in the EGFR gene means the strategy can potentially predict the functional abnormality of VUSs that have been unreported as pathogenic mutations. In the analysis of the cell line model, the EGFR impact score identified cells that are resistant to gefitinib despite carrying hotspot EGFR mutations. This result illustrates that the EGFR impact score can categories the same group classified by the EGFR structural status into more detailed groups based on differences in EGFR pathway dependency. Nakata et al., who established a gefitinib-resistant cell line, indeed reported activation of oncogenic pathways in addition to the EGFR pathway in the cell line24. In an analysis of clinical specimens, the EGFR impact score also predicted responsiveness to TKIs, although there is a limitation that patients were not treated with erlotinib alone. The EGFR impact score tends to predict the responsiveness to TKI treatment in clinical samples, indicating that this score can accurately predict EGFR pathway dependency irrespective of the presence of EGFR structural mutations. These results demonstrate that the EGFR impact score can predict responsiveness to TKIs by evaluating EGFR pathway dependency.
In addition, Although follow-up time is short, the EGFR impact score also predicted prognosis in early-stage lung adenocarcinoma. Yixin et al. reported that genomic instability is caused by defects in DNA damage checkpoints, DNA repair, and mitotic checkpoints25. Our results found that EI-L-type lung adenocarcinoma has a high mutation burden, suggesting that this type has genomic instability because of abnormality of DNA repair. Several studies reported that tumors with genomic instability have a poor prognosis26,27. Accordingly, the EGFR impact score is a novel diagnostic system that can evaluate genomic instability in addition to EGFR pathway dependency. The EGFR signature indeed includes genes associated with DNA repair. A prior report identified a subgroup of patients with EGFR-mutant lung adenocarcinoma featuring genomic instability28. Our results indicated that one reason why findings regarding the relationship of the EGFR structural status with the prognosis of early-stage lung adenocarcinoma differed in past reports is that genomic instability cannot be evaluated using the EGFR structural status alone. As the EGFR impact score can evaluate genomic instability, this method accurately identified a poor prognostic group that could not be classified using the EGRF structural status alone. In summary, the EI-H-type group has pure EGFR pathway dependency, whereas the EI-L-type group features genomic instability. An EGFR mutant tumor without pure EGFR dependency is a tumor with genomic instability, as a result of which the prognosis for this patient group is poor. The EGFR impact score is a new diagnostic system that can evaluate two major events in oncogenesis, namely driver gene and DNA repair defects.
The EI-L-type group exhibited a high mutation burden and high PD-L1 expression. These features are consistent with biomarkers associated with immune checkpoint inhibitors29,30. Patients with driver mutations such as EGFR mutations display a low mutation burden31, and the efficacy of immune checkpoint inhibitor in such patients is controversial32. The EGFR impact score identified patients with high PD-L1 expression and a high mutation burden. Tumors with high immunogenicity based on high genomic instability and a high mutation burden will likely respond to treatment with an immune checkpoint inhibitor. Accordingly, although EI-L-type lung adenocarcinoma is expected to exhibit resistance to TKIs and a poor prognosis, patients in this group can potentially respond to immune checkpoint inhibitors. Although the low EGFR impact group may respond to an immune checkpoint inhibitor, further study of the efficacy of immune checkpoint inhibitors is required.
We developed a novel diagnostic system that can evaluate EGFR networks. The EGFR impact score can predict the responsiveness to TKI treatment in patients with EGFR mutant lung adenocarcinoma. Additionally, this diagnostic system can predict the prognosis of early-stage lung adenocarcinoma because it reflects genomic instability. Furthermore, immune checkpoint inhibition could represent an alternative treatment strategy for patients predicted to have poor prognoses using the EGFR impact system. In summary, the EGFR impact score is a novel diagnostic system with the potential to change standard treatment strategies.
Methods
Publicly available datasets
The gene expression datasets GSE32863, GSE37138, GSE34228, GSE31210, and GSE11969 were downloaded from the National Center for Biotechnology Information Gene Expression Omnibus database24,33,34,35,36,37. The TCGA dataset (Lung Adenocarcinoma in TCGA Provisional) and CCLE (Cancer Cell Line Encyclopedia) were downloaded via cBioportal38,39. GDSC (Genomics of Drug Sensitivity in Cancer) was downloaded via GDSC homepage40,41. Among these datasets, the expression data of GSE31210 and GSE37138 were calculated using the RMA algorithm from CEL files using “R package Affy Ver. 1.58.0” and “R package oligo Ver. 1.46.0,” respectively42,43. Other downloaded expression data were normalized data. All expression profiles were transformed into the logarithmic scale for ssGSEA. In the GSE31210 dataset, the EGFR mutations are defined as exon 19 del and L858R. In the TCGA dataset, the pathogenicity of the EGFR mutation is defined by OncoKB44.
Determination of DEGs in EGFR-mutant lung adenocarcinoma
Using expression data from the GSE32863 dataset (n = 58, tumor samples), we identified DEGs between EGFR-mutant and wild-type samples. As the KRAS gene is a downstream molecule of the EGFR signaling pathway, we excluded tumor samples with KRAS mutations from analyses of DEGs, which were identified using “R package limma Ver. 3.36.5”45. DEGs were defined as follows: genes with p < 0.01 and fold change >1.2 or <1/1.2. The variable importance of DEGs was calculated via the random forest algorithm using “R package RandomForest Ver. 4.6.1446, and excluding genes with a variable importance of 0, genes with variable importance exceeding the third quartile were selected. We considered these DEGs and the ALK gene as EGFR signatures. Accordingly, EGFR signatures that were upregulated in samples with EGFR mutations were referred to as upregulated EGFR signatures, and the remaining DEGs were referred to as downregulated EGFR signatures.
Diagnostic algorithm of the EGFR score classification
We classified early-stage lung adenocarcinoma as EI-H-type or EI-L-type based on the EGFR impact score. The EGFR impact score was calculated on the basis of ssGSEA using the expression profile of the EGFR signature. The EGFR impact score was calculated as follows:
ssGSEA was performed using “R package GSVA Ver. 1.30.0”47. The threshold of the EGFR impact score was determined via ROC analysis, which was performed using “R package ROCR Ver. 1.0.7”48.
Evaluation of responsiveness to TKIs
The authors of the GSE34228 dataset (n = 52, untreated samples) examined the differences between gefitinib-sensitive and gefitinib-resistant cell lines and established a gefitinib-resistant cell line from gefitinib-sensitive PC9 cells via long-term exposure to the drug. We compared the EGFR impact score between gefitinib-sensitive and gefitinib-resistant cell lines. Welch’s t-test was used to evaluate the difference in the EGFR impact score. EGFR mutant lung adenocarcinoma cell lines were analyzed CCLE (n = 6) and GDSC (n = 8). Correlation between EGFR impact score and IC50 of gefitinib was evaluated using Pearson’s product moment correlation coefficient. Furthermore, we analyzed GSE37138 data. In these data, patients with non-squamous NSCLC were treated with erlotinib and bevacizumab. The patients whose tumor shrinkage rates were available (n = 28) were selected, and the EGFR impact score of these patients was calculated. The association between the EGFR impact score and the tumor shrinkage rate was evaluated using Pearson’s product moment correlation coefficient.
Evaluation of VUSs
We classified patients with early-stage lung adenocarcinoma in the TCGA dataset (n = 171) using the EGFR impact score.
Evaluation of RFS and OS
The log-rank test was used to evaluate the differences in RFS between the EGFR-mutant and wild-type groups and the differences in OS between the EI-H-type and EI-L-type groups in the GSE31210 dataset. Multivariate analysis using the Cox proportional hazard model was conducted to compare the prediction of recurrence based on the EGFR structural status and EGFR impact score. In addition to GSE31210, GSE11969 was analyzed. Only patients with EGFR mutant lung adenocarcinoma were used for evaluating differences in OS between the EI-H-type and EI-L-type groups (n = 32).
GSEA
GSEA was performed using the javaGSEA Desktop Application between the EI-H-type and EI-L-type groups among patients with EGFR-mutant cancer49,50. We used the gene sets of the predefined C2 and C5 categories51.
Evaluation of PD-L1 expression levels
The PD-L1 expression levels in the EI-H-type and EI-L-type groups were evaluated using Welch’s t-test with GSE31210 and TCGA data.
Evaluation of tumor mutation burden
Non-synonymous mutations were summed and defined as the somatic mutation burden in the TCGA dataset. Welch’s t-test was used to compare the mutation burden between the EI-H-type and EI-L-type groups.
Statistical analysis
All analyses were performed using the programming language R Ver. 3.5.0.
References
Kohno, T. et al. Beyond ALK-RET, ROS1 and other oncogene fusions in lung cancer. Transl. lung cancer research. 4, 156–164 (2015).
Kohno, T. et al. KIF5B-RET fusions in lung adenocarcinoma. Nat. Med. 18, 375–377 (2012).
Ohashi, K., Maruvka, Y. E., Michor, F. & Pao, W. Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitor–Resistant Disease. J. Clin. Oncol. 31, 1070–1080 (2013).
da Cunha Santos, C., Shepherd, F. A. & Tsao, M. S. EGFR Mutations and Lung Cancer. Annu. Rev. Pathology: Mechanisms Disease. 6, 49–69 (2011).
Cooper, W. A., Lam, D. C. L., O’Toole, S. A. & Minna, J. D. Molecular biology of lung cancer. J. Thorac. Dis. 5, S479–90 (2013).
Kohsaka, S. et al. A method of high-throughput functional evaluation of EGFR gene variants of unknown significance in cancer. Sci. Transl. medicine. 9, eaan6566 (2017).
Kobayashi, Y. & Mitsudomi, T. Not all epidermal growth factor receptor mutations in lung cancer are created equal: Perspectives for individualized treatment strategy. Cancer Sci. 107, 1179–1186 (2016).
Santoni-rugiu, E., Melchior, L. C., Urbanska, E. M. & Jakobsen, J. N. Intrinsic resistance to EGFR-tyrosine kinase inhibitors in EGFR-mutant non-small cell lung cancer: differences and similarities with acquired resistance. Cancers (Basel) 1–57 (2019).
Inhibitors, R. K. Mechanism of Resistance to Epidermal Growth Factor Receptor-Tyrosine Kinase Inhibitors and a Potential Treatment Strategy. Cells 1–16 (2018).
Kim, S. Y. et al. Factors that predict clinical benefit of EGFR TKI therapy in patients with EGFR wild-type lung adenocarcinoma. Tuberc. Respir. Dis. 3536, 62–70 (2019).
Laurie, S. A. & Goss, Role of Epidermal Growth Factor Receptor Inhibitors in Epidermal Growth Factor Receptor Wild-Type Non–Small-Cell Lung Cancer. J. Clin. Oncol. 31, 1061–1069 (2013).
Suda, K. & Mitsudomi, T. Role of EGFR mutations in lung cancers: prognosis and tumor chemosensitivity. Arch. Toxicol. 89, 1227–1240 (2015).
Okamoto, T., Kitahara, H. & Shimamatsu, S. Prognostic Impact of EGFR Driver Mutations on Postoperative Disease Recurrence in Lung Adenocarcinoma. Anticancer. Res. 3064, 3057–3063 (2016).
Hayasaka, K., Shiono, S. & Matsumura, Y. Epidermal Growth Factor Receptor Mutation as a Risk Factor for Recurrence in Lung Adenocarcinoma. Ann. Thorac. Surg. 105, 1648–1654 (2018).
Lin, C.-Y. et al. Prognostic implication of EGFR gene mutations and histological classification in patients with resected stage I lung adenocarcinoma. PLoS One. 12, e0186567 (2017).
Pang, W. L. L. Z. Q. Prognostic value of epidermal growth factor receptor mutations in resected lung adenocarcinomas. Med Oncol., https://doi.org/10.1007/s12032-013-0771-9 (2014).
Larsen, J. E. et al. Gene Expression Signature Predicts Recurrence in Lung Adenocarcinoma. Clin. Cancer Res. 13, 2946–2955 (2007).
He, R. & Zuo, S. A robust 8-gene prognostic signature for early-stage non-small cell lung cancer. Front. oncology. 9, 1–14 (2019).
Lau, S. K. et al. Three-Gene Prognostic Classifier for Early-Stage Non – Small-Cell Lung Cancer. J. Clin. Oncology. 25, 5562–5569 (2007).
Boutros, P. C. et al. Prognostic gene signatures for non-small-cell lung cancer. Proc. Natl Acad. Sciences. 106, 2824–2828 (2009).
Xie, Y., Xiao, G., Coombes, K. R., Behrens, C. & Solis, L. M. Robust gene expression signature from formalin-fixed paraffin-embedded samples predicts prognosis of non-small-cell lung cancer patients. Clin. Cancer Res. 17, 5705–5714 (2012).
Zheng, Y. & Bueno, R. models in early-stage lung cancer: a review of the tests Commercially available prognostic molecular models in early-stage lung cancer: a review of the Pervenio Lung RS and Myriad myPlan Lung Cancer tests. Expert Rev Mol Diagn. 7159 (2015).
Noro, R. et al. Gefitinib (IRESSA) sensitive lung cancer cell lines show phosphorylation of Akt without ligand stimulation. BMC Cancer 6, 1–12 (2006).
Nakata, A. et al. Elevated β-catenin pathway as a novel target for patients with resistance to EGF receptor targeting drugs. Sci. Rep. 5, 13076 (2015).
Yixin Yao, W. D. Genomic Instability and Cancer. J. Carcinog. Mutagen. 5, 1–3 (2014).
Yamaguchi, S. et al. Molecular and clinical features of the TP53 signature gene expression profile in early-stage breast cancer. Oncotarget. 9, 14193–14206 (2018).
Knijnenburg, T. A. et al. Genomic and Molecular Landscape of DNA Damage Repair Deficiency across The Cancer Genome Atlas. Cell Rep. 23, 239–254.e6 (2018).
Nahar, R. et al. Elucidating the genomic architecture of Asian EGFR-mutant lung adenocarcinoma through multi-region exome sequencing. Nat. Commun. 9, 216 (2018).
Savic Prince, S. & Bubendorf, L. Predictive potential and need for standardization of PD-L1 immunohistochemistry. Virchows Arch. (2018).
Greillier, L., Tomasini, P. & Barlesi, F. The clinical utility of tumor mutational burden in non-small cell lung cancer. Transl Lung Cancer Res. 1, 639–646
Nagahashi, M. et al. Common driver mutations and smoking history affect tumor mutation burden in lung adenocarcinoma. J. Surg. Res. 230, 181–185 (2018).
Kunimasa, K., Nishino, K. & Kumagai, T. Is an immune checkpoint inhibitor really a hopeless therapeutic choice for EGFR -mutant non-small cell lung cancer (NSCLC) patients? Annals of translational medicine. 7 (2019).
Selamat, S. A. et al. Genome-scale analysis of DNA methylation in lung adenocarcinoma and integration with mRNA expression. Genome research. 22, 1197–1211 (2012).
Cathomas, R. et al. EGFR Exon-Level Biomarkers of the Response to Bevacizumab / Erlotinib in Non-Small Cell Lung Cancer. PLoS One. 8 (2013).
Okayama, H. et al. Identification of Genes Upregulated in ALK -Positive and EGFR/KRAS/ALK -Negative Lung Adenocarcinomas. Cancer Res. 72, 100–112 (2012).
Yamauchi, M., Yamaguchi, R., Nakata, A., Kohno, T. & Nagasaki, M. Epidermal Growth Factor Receptor Tyrosine Kinase Defines Critical Prognostic Genes of Stage I Lung Adenocarcinoma. PLoS One. 7 (2012).
Takeuchi, T. et al. Expression Profile – Defined Classification of Lung Adenocarcinoma Shows Close Relationship With Underlying Major Genetic Changes and Clinicopathologic Behaviors. J Clin Oncol. 24 (2006).
Ghandi, M. et al. Next-generation characterization of the Cancer Cell Line Encyclopedia. Nature. 569, 503–508 (2019).
Cerami, E. et al. The cBio Cancer Genomics Portal: An Open Platform for Exploring Multidimensional Cancer Genomics Data. Cancer Discov. 2, 401–404 (2014).
Iorio, F. et al. A Landscape of Pharmacogenomic Interactions in Cancer. Cell. 740–754, https://doi.org/10.1016/j.cell.2016.06.017 (2016).
Yang, W. et al. Genomics of Drug Sensitivity in Cancer (GDSC): a resource for therapeutic biomarker discovery in cancer cells. Nucleic Acids Res. 41, 955–961 (2013).
Gautier, L., Cope, L., Bolstad, B. M. & Irizarry, R. A. affy–analysis of Affymetrix GeneChip data at the probe level. Bioinformatics 20, 307–315 (2004).
Carvalho, B. S. & Irizarry, R. A. A framework for oligonucleotide microarray preprocessing. Bioinformatics. 26, 2363–2367 (2010).
Chakravarty, D. et al. OncoKB: A Precision Oncology Knowledge Base. JCO Precis Oncol. 1–23 (2018).
Ritchie, M. E. et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47–e47 (2015).
Breiman, L. Random Forests. Mach. Learn. 45, 5–32 (2001).
Hänzelmann, S., Castelo, R. & Guinney, J. GSVA: Gene set variation analysis for microarray and RNA-Seq data. BMC Bioinformatics 14 (2013).
Sing, T., Sander, O., Beerenwinkel, N. & Lengauer, T. ROCR: Visualizing classifier performance in R. Bioinformatics 21, 3940–3941 (2005).
Subramanian, A. et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. (2005).
Mootha, V. K. et al. PGC-1α-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat. Genet. (2003).
Liberzon, A. et al. Molecular signatures database (MSigDB) 3. 0. Bioinformatics. 27, 1739–1740 (2011).
Acknowledgements
The authors would like to thank Enago (www.enago.jp) for the English language review.
Author information
Authors and Affiliations
Contributions
S.Y. designed this study and wrote the manuscript. K.T. analyzed datasets and wrote the manuscript. K.M. advised bioinformatics techniques. S.K. advised interpretation of all the results. M.H., T.F., M.H., A.Y., T.A., C.Y. and T.H. contributed to the correction and interpretation of data. All the authors have approved the submitted version and have agreed both to be personally accountable for the author’s contributions and to ensure that questions related to the accuracy or integrity of any part of the work.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Takakuwa, K., Mogushi, K., Han, M. et al. A novel diagnostic system to evaluate epidermal growth factor receptor impact as a prognostic and therapeutic indicator for lung adenocarcinoma. Sci Rep 10, 6214 (2020). https://doi.org/10.1038/s41598-020-63200-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-63200-7
This article is cited by
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.

{kind=link}
{kind=link}
{kind=link}