
Abstract
The two efficient non-doped blue emitters with hybridized local and charge transfer state namely, NDBNPIN and DBTPIN have been synthesised and characterised. These materials are employed as a host for green and red phosphorescent OLEDs. The white device based on DBTPIN:Ir(MDQ)2(acac) (4%) exhibit maximum external quantum efficiency (ηex) −24.8%; current efficiency (ηc) −57.1 cdA−1; power efficiency (ηp) −64.8 lmW−1 with Commission Internationale de l’Eclairage (CIE:0.49, 0.40) than NDBNPIN:Ir(MDQ)2acac (4%) device [ηex − 23.1%; ηc −54.6 cd A−1; ηp− 60.0 lm W−1 with CIE (0.47, 0.42)].
Similar content being viewed by others

Introduction
Development of blue emitter is crucial in organic light emitting devices (OLEDs) to reduce power consumption effectively1. For an OLED with stable emission the current efficiency (CE) is proportional to external quantum efficiency (ƞex): power efficiency (PE) is determined by CE and operating voltage (V) [PE = π CE/V]2,3,4. Iridium and platinum based phosphorescent complexes and TADF (thermally activated delayed fluorescent materials) exhibit high ƞex, however, suffered with short lifetime and roll-off efficiency and also the production cost of phosphorescent materials are unfavourable for practical applications5. Therefore, low driving voltage with high brightness become the major issue to achieve efficient OLEDs6. In OLEDs, balanced hole: electron recombination leads to formation of CT exciton (charge-transfer) which undergo decay directly or relaxes to LE (local exciton), thus, utilization of both CT exciton and LE provides efficient EL (electroluminescence). From 4-(dicyanomethylene)-2-methyl-6-[4-(dimethylaminostyryl)-4H-pyran] with CT state maximum efficiency have been harvested7,8,9. Donor–acceptor (D–A) compounds with low % CT leads to RISC (reverse intersystem crossing) process which results high singlet utilisation efficiency(ƞs), however, colour-purity is still poor due to broadened PL (photoluminescence) and EL (electroluminescence) spectra10,11,12,13. D-A architecture with high % LE state leads to higher efficiency because of maximum orbital overlap whereas high % CT provides low efficiency due to partial hole and electron overlap. However, because of small energy splitting (ΔES-T ≈ 0) CT state undergo RISC process results in enhanced ηs11,12. These issues are overcome, by employing D–A configured emissive materials with HLCT emissive state: stabilised LE and CT states results in ηPL(photoluminance efficiency) and high ηs.. Thus, construction of D–A emitters with HLCT emissive state is novel strategy to design efficient blue emitters.
One strategy for constructing blue emitters is the integration of high-energy emissive moieties via twisted arrangement which reduce the conjugation. The twisted structure with high thermal properties promotes blue emission which need for non-doped blue OLEDs14,15. The development of efficient host having high ET (triplet-exited state energy) with good carrier transport properties is critical for efficient PhOLEDs16. The triplet energy of diphenylphenanthrimidazole based hosts is lower compared to individual imidazole molecule owing to enlarged π- conjugation. Therefore, tuning the molecular architecture to highly twisted molecular conformation may be an effective strategy to achieve hosts with high ET. In this communication, we report NDBNPIN and DBTPIN composed of phenanthrimidazole and phenyltriphenylamine with naphthyl and thienyl as spacer components used as blue emitters and host for green and red PHOLEDs.
Result and Discussion
The structure of the emissive materials NDBNPIN and DBTPIN are confirmed by CHN analysis, NMR and mass spectral studies. The glass transition temperature (Tg)/Td (decomposition temperature) of 115°/440° & 121°/501 °C were determined for NDBNPIN and DBTPIN, respectively (Fig. 1a). The high Td/Tg could enhance the life time and stability of OLEDs17. The high Td (NDBNPIN-440 °C and DBTPIN-501 °C) indicates the high resistance of fused aromatic ring on thermolysis and high Td could enhance the device lifetime17. These materials has the ability to form an amorphous glass with high glass-transition temperature (Tg) of 115 °C – NDBNPIN and 121 °C - DBTPIN which is beneficial for the formation of stable, homogeneous and amorphous film upon thermal evaporation. Absence of endothermic peak during the measuring process reveal that no phase separation of host–guest system occur when used as host material.

(a) TGA (inset: DSC) graph. (b) Normalized absorption and emission spectra; (c) cyclic voltamogram; (d) Life time spectra of NDBNPIN and DBTPIN and (e) normalized absorption and emission spectra of NDBNPIN and DBTPIN in film and solution (CHCl3).
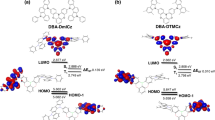
The onset oxidation potential of NDBNPIN and DBTPIN measured by cyclic voltammetry is 0.44 and 0.38 V (Fig. 1), respectively and their HOMO energies are 5.24 and 5.18 eV, respectively. Natural transition orbitals (HONTOs-hole- & LUNTOs-particle) of S1 state reveal spatial separation (CTstate), however, some orbital delocalised on entire molecule (LE) of their excited states shows presence of CT and LE components. i.e., HLCT emissive state. The energy of S1 (1.0376 eV - NDBNPIN and 1.7013 eV - DBTPIN) and T3 states (1.0274 eV - NDBNPIN and 1.6617 eV- DBTPIN) are almost same. A wider energy gap (Eg) (T3 − T2/T1) for NDBNPIN (0.42 eV) and DBTPIN (0.57 eV) is because of same acceptor (phenanthrimidazole) group and the energy gap of DBTPIN is larger when compared to NDBNPIN. In both NDBNPIN and DBTPIN, very small ΔEST ≈ 0 facilitates RISC (T3 → S1) with hot exciton due to HLCT state. Thus, DBTPIN show high ηPL and high ηS compared with NDBNPIN. The ηEQE (external quantum efficiency) of device with DBTPIN is increased due to high % LE. The HOMO as well as LUMO of NDBNPIN and DBTPIN exhibit partial separation which enhanced hole- and electron-transportation (bipolar nature) with electron/hole transfer integrals, NDBNPIN (0.24/0.47 eV) and DBTPIN (0.21/0.41 eV) and minimised the ΔEST (Fig. 2).

Frontier molecular orbitals and HOMO & LUMO at 40 and 90° of NDBNPIN and DBTPIN.
The optical characteristics of NDBNPIN and DBTPIN were studied in solution as well as in film (Fig. 1 & Fig. S1). Absorption (λabs) around 276 and 378 nm is attributed to π − π* and CT transitions, respectively and strong absorption is due to CT from triphenylamine (donor) to acceptor (naphthonitrilephenanthrimidazole)18. The suppressed π-π* stacking in film induced red-shifted λabs relative to solution19 and the larger red shift supports the CT in twisted DBTPIN or NDBNPIN. From the onset absorption in film, optical Eg (band gap) is calculated as NDBNPIN (2.98 eV) and DBTPIN (2.90 eV). The emitters NDBNPIN and DBTPIN show emission maxima at 440 and 428 nm, respectively (Fig. 1). As solvent polarity increased the emission spectra is red-shifted with broadened structure (Fig. S1) and exhibits larger variation in ground state dipole moment (μg) relative to excited state dipole moment (μe). By employing DFT and Lippert-Mataga plot (Fig. S1) μg/μe was calculated as NDBNPIN (9.02/27.8 D) and DBTPIN (8.11/26.1 D). Solvents with f ≥ 0.2, CT state is stabilised20,21,22 (strong interaction between solvent field and CT state, LE remains unchanged) whereas solvents with f ≤ 0.1 LE state is stabilised. Transformation in the slope observed between butyl ether (f = 0.10) and ethyl acetate (f = 0.20) reveal that the emitters show HLCT emissive state i.e., intercrossed excited state of LE and CT [ECT = ELE] (Fig. S1 and Table 1). The λemi of DBTPIN and NDBNPIN in film and ether is almost same due to HLCT emissive state.
The PES of NDBNPIN and DBTPIN reveal twisting of D-A linkage with 20–50° angle be the origin for intercross of CT and LE states. At 90° twist angle, frontier orbitals (HOMO and LUMO) on TPA and PPI are separated results in CT transition from HOMO (donor) → LUMO (acceptor). At 90°, twisted conformation of NDBNPIN and DBTPIN is less stable because of higher energy NDBNPIN (≈0.6 eV) and DBTPIN (≈0.04 eV) than at ≈40° (stable conformation)20,21,22. The HOMO and LUMO orbital map is displayed in Fig. 2. The high ɸ soln/film (quantum yield) of NDBNPIN (83/80%) and DBTPIN (92/90%) is due to co-emission from LE and CT which is essential for efficient blue OLEDs and the enhanced quantum yield is due to decreased non-radiative (knr) transition23 (Table 1). The oscillator strength(f) for λabs/λemi of NDBNPIN (gas phase-372 (f-1.5283)/383 (f-1.7692); CHCl3-368 (f-1.7982)/412 (f-2.0462) and DBTPIN [gas phase-380 (f-1.5846)/392 (f-1.8146); CHCl3-370 (f-1.8268)/422 (f-1.9432)] show that oscillator strength of these compounds in CHCl3 is high relative to gaseous phase due to higher luminance of HLCT state in CHCl3 (Fig. S1). To further investigate the excited state properties, transient PL decay was recorded using time-correlated single photon counting method. The single-exponential lifetime of 1.61 ns (NDBNPIN) and 1.20 ns (DBTPIN) indicates that the hybridization of LE and CT components into a single emissive HLCT state (Fig. S1)24,25,26. The lifetime measurements in nanosecond scale further confirmed that they are fluorescent materials22.
The triplet energies (ET) estimated as 2.62 eV (DBTPIN) and 2.74 eV (NDBNPIN) which are sufficient for exciting red as well as green phosphorescent emitters. Also small ΔEST is sufficient for energy-transfer from host27,28,29 triplet to green and red emitters. The charge transportation of DBTPIN and NDBNPIN, CBP:Ir(ppy)3, DBTPIN:Ir(ppy)3 and NDBNPIN:Ir(ppy)3 was examined by single carrier device fabrication (Fig. 3). Current-density difference of DBTPIN and NDBNPIN compared to CBP device reveal that these bipolar-materials transport electrons and holes effectively30.

(a) Hole-only and electron-only devices based on NDBNPIN and DBTPIN; (b) Energy level diagram of non- doped devices; (c) Schematic representation of (i) Carrier trapping at Ir(ppy)3, (ii) Carrier hopping through both Ir(ppy)3 and NDBNPIN and DBTPIN and (d) NTOs squint of NDBNPIN and DBTPIN.
The carrier-current decreases in control device CBP: Ir(ppy)3 due to trapping at HOMO of Ir(ppy)3 (Fig. 3c (i)) whereas carrier current increased in DBTPIN: Ir(ppy)3 or NDBNPIN:Ir(ppy)3 devices due to direct carrier- injection into HOMO of Ir(ppy)3 followed by hopping transport via DBTPIN/NDBNPIN sites (Fig. 3c (ii)). Hole-current density of DBTPIN < NDBNPIN because cyanonaphthyl limits carrier (hole) injection/transportation significantly12. For devices with DBTPIN and NDBNPIN, similar electron/hole current charges were measured by high/low electric-field, respectively which shows that these materials are potential emissive candidate at low V for efficient OLEDs.
The fabricated blue device, ITO/NPB (60 nm)/DBTPIN or NDBNPIN (30 nm)/LiF (1 nm)/Al (100 nm) with HOMO-LUMO energies are depicted in Fig. 3. Generally, flat-decay was shown by TADF materials because of slow TADF process in conversion of triplet state exciton → singlet state. The single-exponential decay of DBTPIN and NDBNPIN reveal that the radiative exciton are short-lived without TADF contribution and also supports single emissive state (HLCT) (Fig. 1). Therefore, high ηs of DBTPIN and NDBNPIN is not due to TTA or TADF process31. The similar PL and EL emission of DBTPIN and NDBNPIN shows that both PL and EL stems from same source with similar radiative route. The DBTPIN device exhibit superior performance (Fig. 4): (ηc − 7.0 cd A−1, ηp − 7.4 lm W−1, ηex − 6.5%,CIE (0.14, 0.13)) than NDBNPIN based device (ηex − 4.8%; ηc − 5.8 cd A−1; ηp − 6.1 lm W−1, CIE (0.14, 0.13)). Further, doped devices [ITO/NPB (40 nm)/TCTA(5 nm)/CBP:DBTPIN or CBP:NDBNPIN (20 nm)/TPBi (50 nm)/LiF (1 nm)/Al (100 nm): CBP-LUMO -2.7 eV: HOMO -6.1 eV] were constructed to examine the efficiencies and CBP:DBTPIN show maximum efficiencies [ηex −6.70%; ηc −7.40 cd A−1; ηp − 7.8 lm W−1), CIE: (0.14, 0.11] (Fig. 4). Higher ηex harvested from doped devices relative to non-doped one ascribed to doping concentration which reduced the exciton concentration quenching and minimised the intermolecular CT leads to bathochromic shift32.

Device efficiencies: Luminous efficiency [CE (cd/m2), PE (lm/W), EQE (%)] -Current density (a–d); Luminance –Voltage (e); and (f) EL spectra of NDBNPIN and DBTPIN, CBP: NDBNPIN, CBP:DBTPIN.
The color purity of DBTPIN and NDBNPIN based devices supports that these emitters are potential candidates for full-color display. The calculated theoretical maximum ηEQE is of 4.6% and 4.15% [ηEQE = ηout × ηrc × ηγ × ΦPL33, ɸPL: DBTPIN (92%) and NDBNPIN (83%), ηout - out-coupling efficiency (20%), ηrc - product of charge recombination efficiency (100%), ηγ - radiative exciton-production (25%)] and the experimental ηEQE is of 6.50% and 4.8%, respectively. Experimental ηEQE > Theoretical ηEQE because larger triplet exciton are converted to singlet exciton in EL process34,35,36. The ηr calculated for DBTPIN (31–38%) and NDBNPIN (25–32%) indicates γ ˂ 100% because of poor unbalanced carrier transportation in the emissive layer. Enhanced ηIQE 25.2% (NDBNPIN); 32.5% (DBTPIN) and maximum ηs 31.6% (NDBNPIN) and 38.2% (DBTPIN) [ηs = ηout × ηPL × ηres ÷ ηEL] is because of retained CT % due to CN group in D-Π-A compounds. Maximum ηs breaking 25% limit: 6.6% (NDBNPIN) and 13.2% (DBTPIN) of triplet exciton converted to singlet exciton by RISC and remaining follow non-radiative process leads to high efficiency blue OLEDs. The ηc and ηp of DBTPIN device (7.0 cd/A; 7.4 lm/W) and NDBNPIN device (5.8 cd/A; 6.10 lm/W) are larger relative to TPA-PA (1.16 cd/A; 0.65 lm/W), TPA-NzP (1.00 cd/A; 0.77 lm/W) and mTPA-PPI (0.84 cd/A; 0.48 lm/W) devices. The quantum yield of DBTPIN (92%) and NDBNPIN (83%) is larger when compared with (i) Cz-BzP (69.7%) and TPA-BzP (49.2%) (ii) CBI (21%) and MCB (24%) and (iii) PPI-pCNCz (54%). Thickness of LBPPI influences ηc (50 nm: 0.01 cd/A; 40 nm − 0.13 cd/A; 30 nm: 0.40 cd/A and 20 nm: 0.68 cd/A). The ηc harvested in the current study with 30 nm DBTPIN (7.0 cd/A) and 30 nm NDBNPIN (5.8 cd/A) is higher than reported ηc (Tables S1–S3). Thickness tuning of emissive layer enhanced DBTPIN and NDBNPIN efficiencies which also supports that these materials are the best fluorescent materials. These experimental results reveal that currently non-doped [DBTPIN and NDBNPIN] and doped [CBP:DBTPIN and CBP:NDBNPIN] devices are the efficient one. Additional triplet exciton is utilized in the OLEDs because of HLCT of DBTPIN and NDBNPIN as showing the accuracy for our molecular- design- strategy.
We have fabricated green and red PHOLEDs with configuration: ITO/NPB (40 nm)/TCTA (5 nm)/DBTPIN (30 nm): 5 wt % Ir(ppy)3 or NDBNPIN (30 nm): 5 wt % Ir(ppy)3/TPBi (50 nm)/LiF (1 nm)/Al (100 nm)]: ITO/NPB (40 nm)/TCTA (5 nm)/DBTPIN (30 nm): 8 wt% Ir(MDQ)2(acac)/NDBNPIN (30 nm): 8 wt% Ir(MDQ)2(acac)/TPBi (50 nm)/LiF (1 nm)/Al (100 nm), Ir(ppy)3-fac-tris(2-phenylpyridine) iridium(III) and Ir(MDQ)2(acac)-bis(2-methyldibenzo-[f,h] quinoxaline) acetylacetonate iridium(III) are emissive layers for green and red devices, respectively]. The device performances are presented in Fig. 5. Two emission peaks are observed at 4% Ir(ppy)3: part of the exciton transferred to Ir(ppy)3 triplet state and part of exciton transferred to ground state (So) to generate phosphorescence and fluorescence, respectively32. At 5% doping concentration all the exciton were transferred to Ir(ppy)3 to generate phosphorescent emission (Fig. 5).

Device efficiencies: Luminous efficiency [CE (cd/m2), PE (lm/W), EQE (%)] - Current density (a–c) and Luminance –Voltage (d); EL spectra of green devices based on NDBNPIN:Ir(ppy)3, DBTPIN:Ir(ppy)3 and red devices based on NDBNPIN: Ir(MDQ)2(acac), DBTPIN:Ir(MDQ)2(acac) and white devices based on NDBNPIN: Ir(MDQ)2(acac) (0.4%), DBTPIN:Ir(MDQ)2(acac) (0.4%).
The green device DBTPIN (30 nm): 5 wt% Ir(ppy)3 shows maximum efficiency: ηc of 29.0 cd A−1 and ηp of 35.4 lm W−1 at 2.6 V; ηex of DBTPIN: Ir(ppy)3 and NDBNPIN:Ir(ppy)3 are of 23.0 and 20.1%, respectively. Red device with DBTPIN: Ir(MDQ)2(acac) exhibits excellent efficiencies (ηex − 24.1%; ηc − 29.8 cd A−1; ηp − 36.0 lm W−1) with CIE (0.64, 0.36) on comparison with NDBNPIN based device (ηex − 21.3%; ηc − 26.0 cd A−1; ηp − 31.9 lm W−1) with CIE (0.64, 0.36). The enhanced efficiencies reveal that DBTPIN and NDBNPIN are the best host-materials for green and red PHOLEDs. At 0.4% doping concentration both devices based on DBTPIN: Ir(MDQ)2(acac) [448 nm (sh)&590 nm] or NDBNPIN: Ir(MDQ)2(acac) [450 nm (sh)&582 nm] show two peaks leads to white emission (Fig. 5): part of the exciton transferred to Ir(MDQ)2(acac) triplet state to excite phosphorescence and another part of the exciton transferred to ground state to produce fluorescence. Increasing the doping concentration, generated exciton were completely transferred to Ir(MDQ)2(acac) which results in phosphorescent emission. The white device based on DBTPIN:Ir(MDQ)2(acac) exhibit maximum ηex − 24.8%; ηc − 57.1 cd A−1; ηp − 64.8 lm W−1 with CIE (0.49, 0.40) than NDBNPIN: Ir(MDQ)2(acac) based device [ηex − 23.1%; ηc -54.6 cd A−1; ηp − 60.0 lm W−1 with CIE (0.47, 0.42)]. At doping 0.4% concentration, blue emission of the EL spectrum of NDBNPIN: Ir(MDQ)2(acac) device was broader than DBTPIN:Ir(MDQ)2(acac). More blue fluorescence contribution in device NDBNPIN: Ir(MDQ)2(acac) than device DBTPIN: Ir(MDQ)2(acac) leads to lower efficiencies of NDBNPIN:Ir(MDQ)2(acac) than NDBNPIN: Ir(MDQ)2(acac). Outstanding efficiencies indicates, DBTPIN and NDBNPIN are potential host materials and transfer of exciton is shown in Fig. 5. A part of host singlet exciton transferred to singlet blue fluorophore (BS1) exhibits deep-blue emission whereas another part of singlet exciton transferred to singlet green/red phosphorescent emitters (GS1/RS1) and then delivered to triplet green and red phosphorescent emitters (GT1/RT1) by intersystem-crossing process and shows red and green phosphorescent emission. Furthermore, host triplet exciton (HT1) is transferred to GT1/RT1 to enhance the exciton utilization (Fig. 6).

Energy level diagram of (a) green, (b) red white devices with molecular structures of functional materials and (c) Molecule energy levels and energy-transfer diagrams.
Conclusion
We have reported two deep blue emitting materials DBTPIN and NDBNPIN with dual charge transport properties and exhibit high EQE of 6.5% and 4.8% with CIE (0.14, 0.13). The triplet energies (ET) estimated as 2.62 eV (DBTPIN) and 2.74 eV (NDBNPIN) are sufficient for the excitation of green and red phosphorescent dopants. Efficient green and red PhOLEDs with EQE of 23.0%/20.1% and 24.1%/21.3% have been harvested based on DBTPIN/NDBNPIN doped OLEDs,, respectively. White device with DBTPIN:Ir(MDQ)2(acac) (0.4%) exhibit maximum ηex − 24.8%; ηc − 57.1 cdA−1; ηp − 64.8 lmW−1 with CIE (0.49, 0.40) than NDBNPIN:Ir(MDQ)2(acac) (0.4%) based device [ηex − 23.1%; ηc − 54.6 cd A−1; ηp − 60.0 lm W−1, CIE (0.47, 0.42)]. We have reported structure modification strategy to harvest efficient full-color OLEDs by employing phenanthroimidazoles in doped, non-doped devices and red, green and white PHOLEDs.
Experimental Section
Synthesis of NDBNPIN and DPTPIN
4′-bromo-N,N-diphenyl-[1,1′-biphenyl]-4-amine (TPAB-Br)
About 1.42 g of 1-bromo-4-iodobenzene (5 mmol), 2.89 g of (4-(diphenylamino) phenyl)boronic acid (10.0 mmol), 50 mg Pd(PPh3)4 (0.1 mmol) and 2 M K2CO3 (10 mL) was refluxed in toluene (30 mL) at 90 °C for 32 h (Scheme S1). The reaction mixture was treated with CH2Cl2 and dried, crude TPAB-Br was purified by column chromatography to afford white solid [CH2Cl2–petroleum ether (60–90 °C) (1:15)], yield: 84%. MS (EI): m/z 399.1, 401.3 (M+)37.
4-naphthylcarbaldehyde-N,N-diphenyl-[1,1′-biphenyl]-4-amine (NCDBA)
The TPAB-Br (2 g, 5 mmol), 4-formylnaphthalen-1-yl-1-boronic acid (0.75 g, 5 mmol) and Pd(PPh3)4 (50 mg, 0.1 mmol) and 2 M K2CO3 (15 mL) was refluxed in toluene (30 mL) at 105 °C for 3 days with N2 stream. The reaction mixture was treated with CH2Cl2 and dried (Scheme S1).Yield 86.1%. 1H NMR (400 MHz, CDCl3): δ 6.45-6.68 (m, 8 H), 7.02 (t, 4 H), 7.25 (d, J = 8.42 Hz, 2 H), 7.41 − 7.68 (m, 7 H), 7.72 (d, J = 8.42 Hz, 2 H), 9.12 (d, J = 8.2 Hz, 1 H), 9.80 (s, 1 H); 13C NMR (400 MHz, CDCl3): δ 122.71, 123.68, 126.98, 128.32, 128.59, 129.74, 131.50, 132.86, 133.43, 135.47, 135.95, 136.89, 139.51, 141.12, 143.53, 191.52. MS (EI): m/z 475.23 (M+).
4′-thienylcarbaldehyde-N,N-diphenyl-[1,1′-biphenyl]-4-amine (TCDBA)
The TPAB-Br (2 g, 5 mmol), 5-formylthiophen-2-yl-2-boronic acid (0.75 g, 5 mmol), Pd(PPh3)4 (50 mg, 0.1 mmol) and 2 M K2CO3 (15 mL) was refluxed in toluene (30 mL) (105 °C ;3 days;N2 stream). The reaction mixture was treated with CH2Cl2 and dried.Yield 85%. 1H NMR (400 MHz, CDCl3): δ 6.46-6.67 (m, 8 H), 7.15 (t, 5 H), 7.23 (d, J = 8.42 Hz, 2 H), 7.54 − 7.68 (m, 5 H), 9.68 (s, 1 H); 13C NMR (400 MHz, CDCl3): δ 122.68, 128.41, 129.54, 130.51, 132.74, 136.53, 138.72, 139.92, 141.09, 143.21, 148.32, 182.83. MS (EI): m/z 431.21 (M+).
4-(2-(4-(4′-(diphenylamino)-[1,1′-biphenyl]-4-yl)naphthalen-1-yl)-1H-phenanthro [9,10-d]imidazol-1-yl)-1-naphthonitrile (NDBNPIN)
The NCDBA (4.5 mmol), 9,10-phenanthernequinone (5 mmol), 4-aminonaphthalene-1-carbonitrile (6 mmol) and ammonium acetate (61 mmol) was refluxed in ethanol (12 h; N2 atmosphere). The solution was extracted with dichloromethane and dried. Anal. Calcd. for C60H38N4: C, 88.43; H, 4.70; N, 6.87. Found: C, 88.36; H, 4.62; N, 6.75. 1H NMR (400 MHz, CDCl3): δ 6.43-6.52 (m, 6 H), 6.61 (d, J = 8.0 Hz, 2 H), 7.01 (t, 4 H), 7.22 (d, J = 8.4 Hz, 2 H), 7.32 (t, 2 H), 7.44 (d, J = 8.8 Hz, 1 H), 7.54-7.61 (m, 10 H), 7.80–7.88 (m, 6 H), 8.12–8.20 (m, 3 H) 8.93 (d, J = 7.8 Hz, 2 H) (Fig. S2); 13C NMR (100 MHz, CDCl3): δ 109.88, 116.13, 122.12, 122.93, 123.11, 123.45, 123.97, 124.96, 126.13, 126.65, 127.53, 128.32, 128.47, 129.67, 130.15, 131.54, 133.21, 133.61, 133.89, 134.59, 135.47, 136.08, 139.99, 141.15, 149.51 (Fig. S3). MALDI-TOF MS: m/z 814.98, [M+]; calcd: 814. 31 (Fig. S6).
4-(2-(5-(4′-(diphenylamino)-[1,1′-biphenyl]-4-yl)thiophen-2-yl)-1H-phenanthro[9,10-d] imidazol-1-yl)-1-naphthonitrile (DBTPIN)
The TCDBA (4.5 mmol), 9,10-phenanthernequinone (5 mmol), 4-aminonaphthalene-1-carbonitrile (6 mmol) and ammonium acetate (61 mmol) was refluxed in ethanol (12 h; N2 streame). The solution was extracted with dichloromethane and dried. Anal. Calcd. for C54H34N4S: C, 84.13; H, 4.45; N, 7.27. Found: C, 84.05; H, 4.37; N, 7.18. 1H NMR (400 MHz, CDCl3): δ 6.46–6.52 (m, 6 H), 7.0 (t, 6 H), 7.24 (d, J = 8 Hz, 2 H), 7.43 (d, J = 8.4 Hz, 1 H), 7.54 − 7.56 (t, 6 H), 7.80–7.88 (m, 8 H), 8.12–8.20 (m, 3 H), 8.96 (d, J = 7.6 Hz, 2 H) (Fig. S4); 13C NMR (100 MHz, CDCl3): δ 109.87, 116.15, 121.69, 122.45, 122.68, 123.35, 125.64, 126.79, 128.84, 129.75, 130.54, 131.58, 132.98, 134.10, 136.51, 138.32, 139.51, 141.06, 141.75, 143.54 (Fig. S5). MALDI-TOF MS: m/z 771.01, [M+]; calcd: 770. 25 (Fig. S6).
Devices fabrication
ITO glass (resistance 20 Ω/sq) was cleaned with acetone, isopropanol and deionised water and dried (120 °C) followed by UV-zone treatment (20 min). Fabrication was made by deposition method in a vacuum (4 × 10−5 mbar). Organic materials and metal electrodes are evaporated at a rate of 0.4 Å s−1 for 1–4 Å s−1, respectively. Thickness of each layer was measured with quartz crystal thickness monitor. The EL spectra/CIE coordinates were measured with USB-650-VIS-NIR spectrometer (Ocean Opitics, Inc, USA). Current density-voltage-luminance (J-V-L) were measured by computer-controlled source meter (Keithley 2450) equipped with light intensity meter LS-110 under ambient atmosphere. The EQEs were determined from luminance, current density and EL spectrum.
Single carrier device
Hole-only:(I) ITO/HATCN (8 nm)/DBTPIN or NDBNPIN (50 nm)/HATCN (8 nm)/LiF (1 nm)/Al (100 nm);(II) ITO/HATCN (8 nm)/DBTPIN (30 nm): 5 wt % Ir(ppy)3 or NDBNPIN (30 nm):5wt % Ir(ppy)3/HATCN (8 nm)/LiF (1 nm)/Al (100 nm); (III) ITO/HATCN (8 nm)/ CBP (30 nm):5 wt % Ir(ppy)3 or CBP (30 nm): 5 wt % Ir(MDQ)2 (acac)/HATCN (8 nm)/LiF (1 nm)/Al(100 nm) and electron-only:(IV)ITO/TPBi (8 nm)/DBTPIN or NDBNPIN (50 nm)/TPBi (8 nm)/LiF(1 nm)/Al (100 nm);(V) ITO/TPBi (8 nm)/DBTPIN (30 nm): 5 wt % Ir(ppy)3 or NDBNPIN (30 nm):5 wt % Ir(ppy)3/TPBi (8 nm)/LiF (1 nm)/Al (100 nm);(VI) ITO/TPBi (8 nm)/CBP (30 nm): 5 wt % Ir(ppy)3 or CBP (30 nm): 5 wt % Ir(MDQ)2 (acac)/TPBi (8 nm)/LiF (1 nm)/Al (100 nm) were made (Fig. 6; Tables S1–S3)38,39,40,41,42,43,44,45,46,47,48,49,50,51,52,53,54,55,56,57,58,59,60,61,62,63,64,65,66,67.
Measurement
1H and 13C NMR and mass spectra were recorded at 298 K on Bruker 400 MHz spectrometer and Agilent (LCMS VL SD), respectively. Absorption (solution and film) were recorded on Perkin-Elmer Lambda 35 and Lambda 35 spectrophotometer with integrated sphere (RSA-PE-20), respectively. PerkinElmer LS55 fluorescence spectrometer and fluorescence spectrometer Model-F7100 with integrating sphere was employed to analyse PL and absolute quantum yield, respectively. Thermogravimetric analysis (TGA) and differential scanning calorimetric (DSC) were recorded with PerkinElmer thermal analysis system and NETZSCH-DSC-204, respectively (10 °C min−1; N2 flow rate of 100 mL min−1). Lifetime was estimated with time correlated single-photon counting (TCSPC) method on Horiba Fluorocube-01-NL lifetime system. Cyclic voltammetry was performed with potentiostate CHI 630 A electrochemical analyzer. The HOMO [EHOMO = −(Eox + 4.8 eV)] energies and LUMO [ELUMO = (Ered − 4.8 eV)] energies were calculated using oxidation and reduction potentials, respectively.
Data availability
The authors declare that data in our manuscript are available.
References
Gurin, N. T., Paksyutov, K. V., Terent, M. A. & Shirokov, A. V. (CaO · Al2O3 · SiO2): Eu phosphors for violet/ultraviolet-to-white radiation conversion. Tech. Phys. 57, 308–310, https://doi.org/10.1134/S1063784212020089 (2012).
Chen, D. C., Su, S. J. & Cao, Y. Nitrogen heterocycle-containing materials for highly efficient phosphorescent OLEDs with low operating voltage. J. Mater. Chem. C 2, 9565–9578, https://doi.org/10.1039/C4TC01941E (2014).
Li, C. et al. Non-doped luminescent material based organic light-emitting devices displaying high brightness under very low driving voltage. J. Mater. Chem. C 4, 7013–7019, https://doi.org/10.1039/c6tc01975g (2016).
Qiu, C., Chen, H. & Wong, M. Dependence of the current and power efficiencies of organic light-emitting diode on the thickness of the constituent organic layers. IEEE Transactions on Electron Devices 48, 2131–2137, https://doi.org/10.1109/16.944206 (2001).
Kim, K. H. et al. Crystal organic light-emitting diodes with perfectly oriented non-doped Pt-based emitting layer. Adv. Mater. 28, 2526–2532, https://doi.org/10.1002/adma.201504451 (2016).
Ye, H. et al. Pyridine-containing electron-transport materials for highly efficient blue phosphorescent OLEDs with ultralow operating voltage and reduced efficiency roll-off. Adv. Funct. Mater. 24, 3268–3275, https://doi.org/10.1002/adfm (2014).
Chen, C. H., Tang, C. W., Shi, J. & Klunek, K. P. Improved red dopants for organic electroluminescent devices. Macromol. Symp. 125, 49–58, https://doi.org/10.1002/masy.19981250103 (1997).
Ma, C. Q. et al. A novel n-type red luminescent material for organic light-emitting diodes. J. Mater. Chem. 12, 1671–1675, https://doi.org/10.1039/B110934K (2002).
Yao, Y. S., Xiao, J., Wang, X. S., Deng, Z. B. & Zhang, B. W. Starburst DCM‐type red‐light‐emitting materials for electroluminescence applications. Adv. Funct. Mater. 16, 709–718, https://doi.org/10.1002/adfm.200500558 (2006).
Ye, J. et al. Carbazole/sulfone hybrid D-π-A-structured bipolar fluorophores for high-efficiency blue-violet electroluminescence. Chem. Mater. 25, 2630–2637, https://doi.org/10.1021/cm400945h (2013).
Zhang, Q. et al. Efficient blue organic light-emitting diodes employing thermally activated delayed fluorescence. Nat. Photonics 8, 326–332, https://doi.org/10.1038/nphoton.2014.12 (2014).
Zhang, S. et al. Achieving a significantly increased efficiency in nondoped pure blue fluorescent OLED: A quasi-equivalent hybridized excited state. Adv. Funct. Mater. 25, 1755–1762, https://doi.org/10.1002/adfm.201404260 (2015).
Chen, X. et al. Ambipolar charge-transport property for the D–A complex with naphthalene diimide motif. J. Mater. Chem. C 2, 2869–2876, https://doi.org/10.1039/C3TC32377C (2014).
Li, C. L. et al. Efficient deep-blue OLEDs based on phenanthro[9,10-d]imidazole-containing emitters with AIE and bipolar transporting properties. J. Mater. Chem. C. 4, 10120–10129, https://doi.org/10.1039/C6TC03923E (2016).
He, D. et al. High performance near ultraviolet emitter based on phenanthroimidazole via substitutions at C6- and C9-positions. Dyes Pigm. 136, 347–353, https://doi.org/10.1016/j.dyepig.2016.08.066 (2017).
Wang, K. et al. New multifunctional phenanthroimidazole–phosphine oxide hybrids for high-performance red, green and blue electroluminescent devices. J. Mater. Chem. C. 2, 6817–6826, https://doi.org/10.1039/C4TC00749B (2014).
Jayabharathi, J., Ramanathan, P. & Thanikachalam, V. Synthesis and optical properties of phenanthromidazole derivatives for organic electroluminescent devices. New J. Chem. 39, 142–154, https://doi.org/10.1039/C4NJ01515K (2015).
Yan, Y. N., Pan, W. L. & Song, H. C. The synthesis and optical properties of novel 1,3,4-oxadiazole derivatives containing an imidazole unit. Dyes Pigm. 86, 249–258, https://doi.org/10.1016/j.dyepig.2010.01.011 (2010).
Jayabharathi, J., Panimozhi, S. & Thanikachalam, V. Hot exciton transition for organic light-emitting diodes: tailoring excited-state properties and electroluminescence performances of donor–spacer–acceptor molecules. RSC Adv. 8, 37324–37338, https://doi.org/10.1039/c8ra07891b (2018).
Grabowski, Z. R., Rotkiewicz, K. & Rettig, W. Structural changes accompanying intramolecular electron transfer: focus on twisted intramolecular charge-transfer states and structures. Chem. Rev. 103, 3899–4032, https://doi.org/10.1021/cr940745l (2003).
King, S. M. et al. Exploiting a dual-fluorescence process in fluorene–dibenzothiophene-s,s-dioxideco-polymers to give efficient single polymer LEDs with broadened emission. Adv. Funct. Mater. 19, 586–591, https://doi.org/10.1002/adfm.200801237 (2009).
Ouyang, X. et al. Novel “hot exciton” blue fluorophores for high performance fluorescent/phosphorescent hybrid white organic light-emitting diodes with super high phosphorescent dopant concentration and improved efficiency roll-off. ACS Appl. Mater. Interfaces. 7, 7869–7877, https://doi.org/10.1021/am5081106 (2015).
Li, Y. P. et al. Supramolecular network conducting the formation of uniaxially oriented molecular crystal of cyano substituted oligo(p-phenylene vinylene) and its amplified spontaneous emission (ASE) behavior. Chem. Mater. 20, 7312–7318, https://doi.org/10.1021/cm801427s (2008).
Konidena, R. K., Thomas, K. R. J., Dubey, D. K., Sahoob, S. & Jou, J. H. A new molecular design based on hybridized local and charge transfer fluorescence for highly efficient (46%) deep-blue organic light emitting diodes. Chem. Commun. 53, 11802–11805, https://doi.org/10.1039/C7CC07139F (2017).
Wang, Z. Y. et al. Bipolar deep-blue phenanthroimidazole derivatives: Structure, photophysical and electroluminescent properties. Org. Electron. 52, 89–97, https://doi.org/10.1016/j.orgel.2017.09.051 (2018).
Zhu, Z. L. et al. Removing shortcomings of linear molecules to develop high efficiencies deep-blue organic electroluminescent materials. Org. Electron. 38, 323–329, https://doi.org/10.1016/j.orgel.2016.09.004 (2016).
Chen, K. et al. Macrospirocyclic oligomer based on triphenylamine and diphenylphosphine oxide as a bipolar host for efficient blue electrophosphorescent organic light-emitting diodes (OLEDs). Org. lett. 17, 1413–1416, https://doi.org/10.1021/acs.orglett.5b00292 (2015).
Kawamura, Y., Yanagida, S. & Forrest, S. R. Energy transfer in polymer electrophosphorescent light emitting devices with single and multiple doped luminescent layers. J. Appl. Phys. 92, 87, https://doi.org/10.1063/1.1479751 (2002).
Shen, F. et al. Electroluminescence of poly(vinylcarbazole) doubly doped with two phosphorescence dyes: investigation of spectral change as a function of driving voltage. J. Phys. Chem. B. 108, 1014–1019, https://doi.org/10.1021/jp036111c (2004).
Liu, C. et al. Low turn-on voltage, high-power-efficiency, solution-processed deep-blue organic light-emitting diodes based on starburst oligofluorenes with diphenylamine end-capper to enhance the HOMO Level. Chem. Mater. 26, 3074–3083, https://doi.org/10.1021/cm4039522 (2014).
Du, X. et al. Multifunctional phenanthroimidazole derivatives to realize high-performance deep-blue and white organic light-emitting diodes. Adv. Optical Mater. 5, 1700498, https://doi.org/10.1002/adom.201700498 (2017).
Bahers, T. L., Adamo, C. & Ciofini, I. A qualitative index of spatial extent in charge-transfer excitations. J. Chem. Theory Comput. 7, 2498–2506, https://doi.org/10.1021/ct200308m (2011).
Cho, S. H. et al. Highly efficient phosphor-converted white organic light-emitting diodes with moderate microcavity and light-recycling filters. Opt. Express. 18, 1099–1104, https://doi.org/10.1364/OE.18.001099 (2010).
Zhu, M. & Yang, C. Blue fluorescent emitters: design tactics and applications in organic light-emitting diodes. Chem. Soc. Rev. 42, 4963–4976, https://doi.org/10.1039/C3CS35440G (2013).
Chen, W. C., Tong, Q. X. & Lee, C. S. Sci. The development of phenanthroimidazole derivatives in blue-emitting organic electroluminescence. Adv. Mater. 7, 2193–2205, https://doi.org/10.1166/sam.2015.2264 (2015).
Xu, Y. et al. Highly efficient blue fluorescent OLEDs based on upper level triplet–singlet intersystem crossing. Adv. Mater. 31, 1807388–8, https://doi.org/10.1002/adma.201807388 (2019).
Herbich, J. & Kapturkiewicz, A. Electronic structure and molecular conformation in the excited charge transfer singlet states of 9-acridyl and other aryl derivatives of aromatic amines. J. Am. Chem. Soc. 120, 1014–1029, https://doi.org/10.1021/ja972474c (1998).
Jayabharathi, J., Ramya, R., Thanikachalam, V., Jeeva, P. & Sarojpurani, E. Efficient full-colour organic light-emitting diodes based on donor–acceptor electroluminescent materials with a reduced singlet–triplet splitting energy gap. RSC adv. 9, 2948–2966, https://doi.org/10.1039/C8RA09486A (2019).
Liu, H. et al. Highly efficient near ultraviolet organic light-emitting diode based on a meta-linked donor–acceptor molecule. Chem. Sci. 6, 3797–3804, https://doi.org/10.1039/C5SC01131K (2015).
Wang, Z. et al. Dimeric phenanthroimidazole for blue electroluminescent materials: the effect of substituted position attached to biphenyl center. Phys. Chem. Chem. Phys. 16, 10837–10843, https://doi.org/10.1039/C4CP00209A (2014).
Gao, Z. et al. High-efficiency violet-light-emitting materials based on phenanthro[9,10-d]imidazole. Chem. Eur. J. 19, 2602–2605, https://doi.org/10.1002/chem.201203335 (2013).
Li, X. L. et al. Highly efficient blue and warm white organic light-emitting diodes with a simplified structure. Nanotechnology 27, 124001, https://doi.org/10.1088/0957-4484/27/12/124001 (2016).
Zhang, Y. et al. A pyrene-phenanthroimidazole derivative for non-doped blue organic light-emitting devices. Dyes and Pigments 98, 190–194, https://doi.org/10.1016/j.dyepig.2013.01.022 (2013).
Fisher, A. et al. Highly efficient multilayer organic pure blue light emitting diodes with substituted carbazoles compounds in the emitting layer. J. Phys. D: Appl. Phys. 39, 917, https://doi.org/10.1088/0022-3727/39/5/004 (2006).
Zhang, Y. et al. High efficiency nondoped deep-blue organic light emitting devices based on imidazole-π-triphenylamine derivatives. Chem. Master. 24, 61–70, https://doi.org/10.1021/cm201789u (2012).
Wu, F. I. et al. Novel distyrylcarbazole derivatives as hole-transporting blue emitters for electroluminescent devices. J. Mater. Chem. 15, 4753–4760, https://doi.org/10.1039/B510035F (2005).
Liao, Y. L., Lin, C. Y., Wong, K. T., Hou, T. H. & Hung, W. Y. A novel ambipolar spirobifluorene derivative that behaves as an efficient blue-light emitter in organic light-emitting diodes. Org. Lett. 9, 4511–4514, https://doi.org/10.1021/ol701994k (2007).
Yuan, Y. et al. Bipolar phenanthroimidazole derivatives containing bulky polyaromatic hydrocarbons for nondoped blue electroluminescence devices with high efficiency and low efficiency roll-off. Chem. Mater 25, 4957–4965, https://doi.org/10.1021/cm4030414 (2013).
Tang, X. et al. Efficient deep blue electroluminescence with an external quantum efficiency of 6.8% and CIEy <0.08 based on a phenanthroimidazole–sulfone hybrid donor–acceptor molecule. Chem. Mater. 27, 7050–7057, https://doi.org/10.1021/acs.chemmater.5b02685 (2015).
Liu, B. et al. Ambipolar D–A type bifunctional materials with hybridized local and charge-transfer excited state for high performance electroluminescence with EQE of 7.20% and CIEy ∼ 0.06. J. Mater. Chem. C. 5, 5402–5410, https://doi.org/10.1039/C7TC01133D (2017).
Chen, W. C. et al. Achieving efficient violet-blue electroluminescence with CIEy <0.06 and EQE>6% from naphthyl-linked phenanthroimidazole–carbazole hybrid fluorophores. Chem. Sci. 8, 3599–3608, https://doi.org/10.1039/C6SC05619A (2017).
Zhao, J. et al. EQE climbing over 6% at high brightness of 14350 cd/m2 in deep-blue OLEDs based on hybridized local and charge-transfer fluorescence. ACS Appl. Mater. Interfaces. 10, 9629–9637, https://doi.org/10.1021/acsami.7b19646 (2018).
Wang, Z. et al. Phenanthro[9,10-d]imidazole as a new building block for blue light emitting materials. J. Mater. Chem. 21, 5451–5456, https://doi.org/10.1039/C1JM10321K (2011).
Wang, Z. M. et al. Tuning of the electronic and optical properties of 4,4′-bis(1-phenyl-phenanthro[9,10-d]imidazol-2-yl)biphenyl via cyano substitution in un-conjugated phenyl. RSC Adv. 2, 9635–9644, https://doi.org/10.1039/C2RA21054A (2012).
Chen, S., Wu, Y., Zhao, Y. & Fang, D. Deep blue organic light-emitting devices enabled by bipolar phenanthro[9,10-d]imidazole derivatives. RSC Adv. 5, 72009–72018, https://doi.org/10.1039/C5RA13814K (2015).
Chen, S., Wu, Y., Hu, S., Zhao, Y. & Fang, D. Non-doped deep blue and doped white electroluminescence devices based on phenanthroimidazole derivative. J. Fluoresc. 27, 451–461, https://doi.org/10.1007/s10895-016-1970-5 (2017).
Gao, Z., Liu, F., Li, J., Cheng, G. & Lu, P. Solution-processed UV light emitting diode based on butyltriphenylsilane modified phenanthro[9,10-d]imidazole with high efficiency. RSC Adv. 6, 81744–81749, https://doi.org/10.1039/C6RA18755B (2016).
Huang, Z. et al. Highly twisted bipolar emitter for efficient nondoped deep-blue electroluminescence. Dyes and Pigments 140, 328–336, https://doi.org/10.1016/j.dyepig.2017.01.028 (2017).
Tang, X. et al. Efficient deep-blue electroluminescence based on phenanthroimidazole-dibenzothiophene derivatives with different oxidation states of the sulfur atom. Chem. Asian J. 12, 552–560, https://doi.org/10.1002/asia.201601626 (2017).
Shen, Z., Burrows, P. E., Bulovic, V., Forrest, S. R. & Thompson, M. E. Three-color, tunable, organic light-emitting devices. Science 276, 2009–2011, https://doi.org/10.1126/science.276.5321.2009 (1997).
Sakakibara, Y., Okutsu, S., Enokida, T. & Tani, T. Red electroluminescence and photoluminescence properties of a reduced porphyrin compound, tetraphenylchlorin. Thin Solid Films 363, 29–32, https://doi.org/10.1016/S0040-6090(99)00976-1 (2000).
Zhang, X. H. et al. Red electroluminescence and photoluminescence properties of new porphyrin compounds. Chem. Phys. Lett. 382, 561–566, https://doi.org/10.1016/j.cplett.2003.10.104 (2003).
Yu, J. & Shirota, Y. A. A new class of high-performance red-fluorescent dyes for organic electroluminescent devices, [7-diethylamino-3-(2-thienyl)chromen-2-ylidene]-2,2-dicyanovinylamine and {10-(2-Thienyl)-2,3,6,7-tetrahydro-1H,5H-chromeno[8,7,6-ij]quinolizin-11-ylidene}-2,2-dicyanovinylamine. Chem. Lett. 31, 984–985, https://doi.org/10.1246/cl.2002.984 (2002).
Wu, W. C., Yeh, H. C., Chan, L. H. & Chen, C. T. Red organic light-emitting diodes with a non-doping amorphous red emitter. Adv. Mater 14, 1072–1075, https://doi.org/10.1002/1521-4095(20020805) (2002).
Thomas, K. R. J., Lin, J. T., Velusamy, M., Tao, Y. T. & Chuen, C. H. Color tuning in benzo[1,2,5]thiadiazole-based small molecules by amino conjugation/deconjugation: bright red-light-emitting diodes. Adv. Funct. Mater. 14, 83–90, https://doi.org/10.1002/adfm.200304486 (2004).
Chiu, C. W., Chow, T. J., Chuen, C. H., Lin, H. M. & Tao, Y. T. Bisindolylmaleimides as red electroluminescence materials. Chem. Mater. 15, 4527–4532, https://doi.org/10.1021/cm0303890 (2003).
Mi, B. X. et al. New polycyclic aromatic hydrocarbon dopants for red organic electroluminescent devices. J. Mater. Chem. 12, 1307–1310, https://doi.org/10.1039/B110153F (2002).
Frisch, M. J. et al. Gaussian, Inc., Wallingford., CT. http://www.gaussian.com/g_tech/g_ur/m_citation.htm (2009).
Lu, T. & Chen, F. “Multiwfn: a multifunctional wavefunction analyzer. J. Comput. Chem. 33, 580–592, https://doi.org/10.1002/jcc.22885 (2012).
Acknowledgements
This research was supported by the DST (Department of Science and Technology - EMR/2014/000094, F.No. SR/S1/1C-73/2010, F. No. SR/S1/1C-07/2007), DRDO (Defence Research and Development Organization -213/MAT/10-11), CSIR (Council of Scientific and Industrial Research -No. 01/(2707)/13EMR-II), UGC (University Grant Commission -6-21/2008, F. No. 30-71/2004(SR)) and DST-Nano Mission (SR/NM/NS-1001/2016).
Author information
Authors and Affiliations
Contributions
J.J. and V.T. designed and analyzed the data and prepared the manuscript. S.P. conducted the experiments, performed OLEDs fabrication and characterization.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Jayabharathi, J., Panimozhi, S. & Thanikachalam, V. A novel hot exciton blue fluorophores and white organic light-emitting diodes with simplified configuration. Sci Rep 10, 5114 (2020). https://doi.org/10.1038/s41598-020-62029-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-62029-4
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
