
Abstract
Monocytes are a critical component of the cellular innate immune system, and can be subdivided into classical, intermediate and non-classical subsets on the basis of surface CD14 and CD16 expression. Classical monocytes play the canonical role of phagocytosis, and account for the majority of circulating cells. Intermediate and non-classical cells are known to exhibit varying levels of phagocytosis and cytokine secretion, and are differentially expanded in certain pathological states. Characterisation of cell surface proteins expressed by each subset is informative not only to improve understanding of phenotype, but may also provide biological insights into function. Here we use highly multiplexed Tandem-Mass-Tag (TMT)-based mass spectrometry with selective cell surface biotinylation to characterise the classical monocyte surface proteome, then interrogate the phenotypic differences between each monocyte subset to identify novel protein markers.
Similar content being viewed by others


Introduction
Monocytes play critical roles in the response to infection and inflammation and antigen presentation. Key effector functions are mediated by differentiated cells, including macrophages and myeloid dendritic cells. Monocytes can be divided into subsets based on the presence of the cell surface lipopolysaccharide (LPS) co-receptor CD14, and Fc-Gamma Receptor III (CD16). CD14++ CD16− classical monocytes account for ~85% of circulating monocytes, intermediate cells (CD14++ CD16+) for ~5% and non-classical cells (CD14+ CD16++) for ~10% of circulating monocytes1,2. It has become increasingly well established that each subset plays divergent roles in different diseases, as well as differing in the ability to secrete cytokines and respond to pathogen associated molecular patterns (PAMPs). Classical monocytes are phagocytic and readily secrete inflammatory cytokines. Conversely, the CD16 positive non-classical cells are poorly phagocytic and are suggested to secrete TNFα in response to some stimuli, but less of other pro-inflammatory molecules2,3,4. Intermediate monocytes are increased in diseases such as severe asthma, rheumatoid arthritis and sarcoidosis, and there is some evidence for expansion of classical monocytes in atherosclerosis5,6,7,8. It is still unclear whether intermediate monocytes represent a truly distinct monocyte subset, or merely a transitional stage between classical and non-classical cells9.
Cells of the innate and adaptive immune systems can be categorised on the basis of microscopic appearance and expression of plasma membrane (PM) proteins, enabling separation by fluorescence activated cell sorting (FACS). As such, systematic evaluation of the entire cell surface proteome expressed by a given immune cell population is a powerful tool to characterise cellular function and distinguish cell types. Previous studies of monocytes subsets have examined transcriptional differences and offer varying depths of information about subset markers, suitability of individual proteins for discriminating subsets by cell surface flow cytometry and the importance of each protein in different pathological states2,8,10,11. Usage of multiple complementary grouping systems has the benefit of improving cellular assignment, in addition to enabling discovery of new cellular phenotypes8,12.
Here we use selective surface protein biotinylation with multiplexed tandem mass tag (TMT)-based mass spectrometry to directly measure the first comprehensive surface proteome of primary human classical monocytes from three donors. 373 classical monocyte cell surface proteins were quantified, and the relative abundance of each protein estimated. The surface proteome of classical, intermediate and non-classical cells was then compared to identify unique markers of each subset. Amongst these, Integrin alpha subunit 5 (ITGA5), complement receptor 1 (CR1/CD35) and Leukotriene B4 receptor (LTB4R) were defined as markers of classical monocytes, and Sialic Acid Binding Ig Like Lectin 10 (SIGLEC10) as a marker of non-classical cells.
Results
Establishing a definitive classical monocyte surface proteome
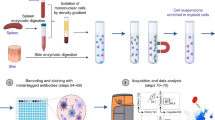
To establish a surface proteome map, monocytes were enriched from three independent peripheral blood mononuclear cell (PBMC) donations from healthy donors by negative selection with magnetic beads. Classical monocytes were then enriched by FACS after staining for CD86, CD14 and CD16 (Supplementary Fig. S1). Selective surface oxidation and aminoxybiotinylation was used to label cell surface glycoproteins, which were enriched from cellular lysates using streptavidin beads, then digested into peptides using trypsin. Peptides were labelled with TMT, samples combined and then quantified by MS3 mass spectrometry13,14 (Fig. 1a).

Overview of experimental strategy and protein enrichment scores. (a) A schematic overview of the experimental approach. (b) Enrichment of pathways within all 373 classical monocyte surface proteins in comparison to all human proteins as background, using DAVID software. Benjamini-Hochberg adjusted p-values are shown for each pathway.
437 proteins were identified from the three samples, of which 373 were annotated ‘cell surface’, ‘plasma membrane’, ‘extracellular’ or with a short Gene Ontology term as previously described13,14 (Supplementary Table S1d). Application of DAVID software to determine which pathways were enriched amongst these proteins indicated the presence of multiple components of integrin complexes and cell-cell junctions in addition to ‘glycosylation’ and ‘disulphide bond’, serving to validate our selective labelling approach (Fig. 1b).
Quantification of the classical monocyte cell surface proteome
We used a method derived from identity based absolute quantitation (iBAQ) to compare the contribution of each protein to the classical monocyte cell surface proteome. The summed MS1 maximum precursor intensity for each protein across all matching peptides was divided by the theoretical number of tryptic peptides 7–30 amino acids in length. Values thus effectively represent an average across three donors, offering the opportunity to provide precise information on the overall abundance of each PM protein, independent of individual genetic variation. Abundance values ranging over approximately five orders of magnitude were found, with 21 proteins collectively contributing 68.6% of the cell surface proteome, whilst individually contributing >1% (Fig. 2). The five most abundant surface proteins, CD44, SPN, ICAM3, ITGB2 and BSG accounted for ~25% of the surface proteome, with CD14 representing ~3.5% (Fig. 2). The summed abundance of class I Major Histocompatability antigen (MHC) accounted for 2.0% of surface proteins, and class II MHC 0.7%.

Pie chart showing the relative contribution of individual proteins to the classical monocyte surface proteome. Proteins contributing <1% are included in the ‘other’ category. The summed abundances of all MHC class I molecules and MHC class II molecules were considered in order to account for different alleles expressed by individual donors.
By using multiplexed TMT-based proteomics, this study offered the opportunity to directly measure the variability in expression of whole surface proteomes across different donors. 76.1% of proteins exhibited a <30% coefficient of variation (%CV). There was also no systematic inverse relationship between abundance and variability, i.e. less abundant proteins were not systematically more poorly quantified (Supplementary Fig. S2).
Comparison of the cell surface proteome of different monocyte subsets
Although the majority of circulating monocytes belong to the classical subset, the non-classical and intermediate subsets play an increasingly appreciated role in different diseases. Selective cell surface biotinylation with MS3-based mass spectrometry was used to investigate phenotypic differences between each subset in biological triplicate. From this analysis, 313 proteins annotated ‘cell surface’, ‘plasma membrane’, ‘extracellular’ or with a short GO term were quantified (Supplementary Table S1e). Principle component analysis (PCA) suggested that classical monocytes had a more distinct cell surface proteome in comparison to non-classical and intermediate cells (Fig. 3a). K-means clustering with 1–20 classes suggested there were at least eight distinct patterns of protein expression between monocyte subsets (Fig. 3b,c). Examples of proteins selectively enriched on classical monocytes (Cluster A) included CD99 antigen and Sialic Acid-Binding Ig-Like Lectin 3 (CD33), validating previous transcriptomic studies2,10. Clustering also highlighted a number of proteins enriched in non-classical cells (Cluster C), including CD16a (FCGRIIIA), the previously reported non-classical monocyte marker SIGLEC102 and Tetraspanin 14 (TSPAN14), which had not previously been reported. Distinguishing intermediate monocytes from other subsets was more challenging, however several candidate cell surface markers were identified including Solute Carrier Family 6 Member 6 (SLC6A6) (Cluster B). We then calculated the Benjamini-Hochberg corrected p-values for each monocyte subset comparison. This confirmed that the ‘classical’ and ‘intermediate’ clusters were enriched in proteins showing significantly differential abundance between the subsets (Supplementary Table S1e). The enrichment for the third ‘non-classical’ cluster was poor, in keeping with the appearance of the k-means analysis.

Analysis of the surface proteome of monocyte subsets. (a) Principal component analysis of all 313 proteins annotated ‘cell surface’, ‘plasma membrane’, ‘extracellular’ or with a short GO term. The grouping of the samples suggested that subset type was the major driver of variability as opposed to donor-specific differences. (b) The number of distinct classes of protein expression between monocyte subsets plotted against within-class variance. Proteins were clustered using a k-means approach with 1–20 classes, and the summed distance of each protein from its cluster centroid was determined. This summed distance becomes smaller as more clusters are added, but the rate of decline decreases with each iteration, eventually reaching a fairly steady rate of decline (orange line) that is reflective of overfitting. Clusters added before this point reflect the underlying structure in the protein data, whereas clusters subsequently added through overfitting add no additional useful information. For these data the point of inflexion was between eight and nine classes, suggesting that there are at least eight distinct surface protein profiles. (c) K-means based hierarchical cluster analysis of the 313 proteins identified at the surface of the different classes of monocyte. Right panels – enlargements of clusters particularly enriched in proteins of one class (the adjacent bar colour indicates where each enlargement matches the original cluster plot). C – Classical monocyte sample, I – Intermediate monocyte sample, NC – Non-classical monocyte sample.
Comparison to RNA microarray data
Previous analyses of monocyte subsets have mostly been performed at the level of RNA expression2,8,10,11. Hierarchical clustering was used to assess the complementarity between one of the most comprehensive previous transcriptomic studies2 and this proteomic analysis. For each protein, each data type was normalised to a maximum of 1 after averaging signal:noise (proteomics) or median fluorescent intensity values (microarray) across donors (Supplementary Fig. S3). RNA and protein data for the classical subset clustered separately from non-classical and intermediate subsets. However, intermediate protein-RNA did not neatly separate from the non-classical pair suggesting that these cells may be more phenotypically similar to one another than classical monocytes, in keeping with the PCA (Fig. 3a). Another recently published transcriptomic analysis15 used single-cell RNAseq to analyse Lin-HLA-DR+ index sorted cells. These included dendritic cells, monocytes and a population of contaminating NK cells. A phenograph-clustering algorithm aimed to identify within each cluster (i) cell types and (ii) differentially-expressed genes (DEGs). Eight clusters were identified, and clusters 1 and 3 were related most closely to classical monocytes (CD14hi CD16−, Fig. 3B from Duterte et al.15), with cluster 7 most closely related to mixed intermediate and non-classical monocytes (CD16+, Cluster 7 in the same figure). Comparison of DEGs to our cell surface protein-level data largely confirmed that these markers were differentially expressed at both the level of protein and RNA within classical and non-classical monocytes (Supplementary Fig. S4).
Validation of cell surface subset markers
Flow cytometry was used to validate a group of monocyte subset markers identified by proteomics, using independent samples from three different healthy donors of European heritage. SIGLEC10 validated as a marker most abundantly expressed by non-classical monocytes (Fig. 4a,b)2. ITGA5, CR1 and LTB4R also validated as markers of classical monocytes identified by proteomic data (Fig. 4a,b), in line with previous high throughput analyses for ITGA5 and LTB4R2.

Validation of proteomics data by flow cytometry. (a) Fresh human monocytes from three different donors were analysed by flow cytometry with antibodies specific to the indicated protein, in addition to anti-CD86, anti-CD14 and anti-CD16 to distinguish each monocyte subset. Corresponding profiles as determined by MS are shown in the lower panel. For both types of analyses, data were normalised to a maximum of one then averaged across the three replicates. Data are shown as mean +/− SEM. For proteomic data, a Benjamini-Hochberg-corrected two-tailed t-test was used to estimate p-values. For flow cytometry data, a two-tailed t-test was used to estimate p-values. *p < 0.05; **p < 0.005. (b) Representative contour plots from a single donor of flow cytometry data for each illustrated marker coloured by cell subset. FSC-A (Forward Scatter Area.) was used as an intrinsic description of cell size (which would not be expected to change between subsets) to separate the points on a second axis. This representation was advantageous compared to the alternative of histograms as it more clearly visually represents the distribution of data points with less confounding by absolute cell number. The relative proportion of each monocyte subset is different in peripheral blood, with classical monocytes predominating.
Discussion
Here we use cell surface specific protein enrichment in combination with isobaric tagging and MS3 mass spectrometry to define at high resolution the cell surface proteomes of monocyte subsets. This has not only identified novel markers of each subset but also provided a valuable resource for the future study of these crucial immune cells. This represents the first cell surface proteomic study in primary monocyte subsets. Up to now, other cell surface proteomic analyses in primary cells have been limited to CD4+ T-cells16, erythrocytes17,18, NK cells, adipocytes and a subset of tumour cells19. We quantify 373 cell surface proteins from classical monocytes, and further quantify 313 proteins across all monocyte subsets. Based on our data we predict that the majority of the classical monocyte cell surface proteome is composed of a relatively small number of different proteins. Limitations to this study include the number of donors. To ensure all samples could be directly compared in a single TMT analysis, three subpopulations of monocytes from three donors were analysed, since only 10 different TMT tags were available. Furthermore, demographic information for each donor was not available, meaning that characteristics such as gender might bias our analysis. Nevertheless, we believe this study provides important cell surface protein level data to complement the rapidly growing array of RNA studies of different leucocyte subsets.
Here we show CR1, LTB4R and ITGA5 are markers of classical monocytes, and SIGLEC10 is a marker of non-classical monocytes. CR1 (Complement Receptor type 1/CD35), acts as a cell surface receptor for particles opsonised by complement, facilitating their phagocytosis20. The specific elevation of CR1 on classical monocytes is consistent with their predominant phagocytic role.
LTB4R (also known as BLT1) is a cell surface G-protein coupled receptor for the proinflammatory leukotriene B4 (LTB4). It was originally functionally identified by subtractive cloning of cDNAs from cells differentially able to bind LTB4, and was observed to enable chemotactic response to LTB4 even in non-specialised non-immune cells21. Furthermore, LTB4R deletion has previously been reported to confer resistance to histopathological changes in response to a rodent model of inflammatory arthritis22. Given the previous link of certain monocytic subsets to inflammatory arthrititis6 it may be of interest to reassess the role of LTB4R positive classical monocytes in this pathology.
Integrin Alpha 5 (ITGA5, CD49e), is one of 18 mammalian alpha integrin chains which form cell surface heterodimers with integrin beta subunits23. Integrins are classically involved in extracellular adhesion and invasion, and also play roles in bidirectional transmembrane signalling24. SIGLEC10 was initially identified as a sialic acid binding protein expressed across a variety of tissues, but found at a particularly high level in organs rich in haematopoietic cells, and is notably expressed on dendritic cells and CD16+ cells10,25,26. SIGLEC10 acts as a ligand for vascular adhesion protein 1 (VAP1), and plays a role in lymphocyte adhesion to endothelial surfaces27. The differential expression of both of these adhesion molecules between classical and non-classical monocytes may thus contribute to independent mobility or effector function, although future studies will be required to assess the biological function of SIGLEC10 on these cell types.
The abundance of SLC6A6 was elevated at the surface of intermediate monocytes. SLC6A6 is a known taurine and beta-alanine transporter with proposed roles in regulation of apoptosis28,29. Further studies will be required to investigate whether this molecule modulates the survival of intermediate monocytes relative to the other subsets. Similarly, our data suggested that TSPAN14 was relatively enriched on non-classical monocytes. TSPAN14 is a member of a family of tetraspanins known to regulate the subcellular localisation of metalloproteinase ADAM1030; this might in part explain differences in ADAM10 expression we observed between different monocyte subsets.
To determine if any particular functional class of protein was enriched in each monocyte subpopulation, we used DAVID analysis to compare proteins enriched in different subsets from our k-means analysis (Fig. 3c). Unfortunately, at least partly due to the relatively small background of proteins (313 proteins in total), only a single term was significantly enriched (p < 0.05) within these groupings. It is therefore difficult to make confident, global statements about differences in the function of each monocyte subset. Manual inspection of the proteins enriched in each subset highlighted the presence of different cell adhesion and interaction molecules: classical – CD33, CD93, CD99; intermediate – ALCAM; non-classical – ICAM2, CD97, SELPLG (Supplementary Table S3). We speculate that the divergent role of the monocyte subsets in varying disease states could partially be explained by their differential interactions consequent on differential expression of these molecules. For example, CD33 polymorphisms have been implicated in genetic susceptibility to Alzheimer’s disease, with risk alleles correlating with increased CD33 expression and altered phagocytic activity31.
As standard in our cell surface proteomic analyses we included proteins annotated with the Gene Ontology Cellular Compartment terms ‘plasma membrane’ (267/313 proteins), ‘extracellular’ (110/313 proteins), ‘cell surface’ (95/313 proteins) and a short term that indicates that the protein is membrane-integral, however that the subcellular localisation is presently unknown (7/313 proteins) (shown in Supplementary Table 1E)13,32. This strategy facilitates a comprehensive coverage of the cell surface proteome. From the 313 proteins, 31 were annotated ‘extracellular’ but not ‘plasma membrane’ or ‘cell surface’. These included Lysozyme and Haptoglobin. Certain extracellular proteins can clearly bind to proteins or receptors at the plasma membrane. For example, Haptoglobin is known to bind CD16333, which we also detected in our analysis.
Although two previous reports have quantified the whole cell proteome of monocyte subsets34,35, ours represents the first selective study of the cell surface proteome of these primary cell types. Furthermore, one of the previous studies34 pooled non-classical and intermediate phenotype cells prior to analysis, and the other did not examine intermediate phenotype cells35. We compared our data to both studies. As might be anticipated due to the low abundance and hydrophobic nature of cell surface proteins, each prior study only identified a fraction of the cell surface proteins found by selective surface biotinylation. Furthermore, for each subset of proteins previously identified as differentially expressed, there was generally poor correspondence to our data. There are a number of possible explanations for this finding, particularly including a small sample size of differentially expressed proteins that were also quantified in our study in each case. Additionally, correspondence between cell surface and whole-cell protein abundances can in any case be poor, and there may have been some confounding in one of the prior studies from isolation and measurement of protein samples in five different locations, using four different types of mass spectrometer35. Neither prior study examined intermediate-phenotype cells. We believe this shows the clear benefits of selective interrogation of the cell surface proteome, in a single multiplexed experiment that can make precise quantitative comparisons.
Although the monocyte subsets can be delineated using CD14/CD16 expression, use of further markers in parallel could aid distinction of subgroups which may appear as a near continuum if viewed in only two dimensions12. It has previously been suggested that the intermediate monocyte subset may be a transitional state between the two larger circulating monocyte populations, and the classical subset are more distinct from intermediate and non-classical cells than the latter two populations are from one another. Our data support this second observation. However, we also identified proteins that are uniquely up- or down-regulated in intermediate cells, arguing against the hypothesis that these cells may be purely transitional. Future work will be required to determine which combinations of markers can be used to definitively separate individual subsets, and will expand recent single cell transcriptomic data which has suggested that intermediate monocytes are more heterogenous than previously anticipated9,36. Additionally, further studies will also be required to examine the monocyte cell surface from individuals with diseases including asthma, rheumatoid arthritis and sarcoidosis to determine whether subpopulations exhibit phenotypic differences in these conditions. We thus provide an orthogonal analysis of monocyte subgroups to complement previous transcriptomic studies which will facilitate many future studies of monocytes. This valuable resource may also be useful to assist elucidation of the true nature of intermediate monocytes.
Materials and Methods
Monocyte isolation
For proteomic studies, leukocyte enriched blood samples were obtained from healthy UK-based blood donors via NHS blood and transplant (NHSBT, Cambridge, UK). No further information about these donors was available from NHSBT, in line with their conditions of supply. Cells were eluted and PBMC isolated by centrifugation on a Ficoll gradient (GE Healthcare) for 20 minutes at 800 g. PBMC were aspirated from the interface, and monocytes isolated using a negative selection kit (‘Pan Monocyte Enrichment’, Miltenyi Biotec, 130-096-537). Monocyte subsets were subsequently defined by flow cytometry after staining with anti-CD14, anti-CD16 and anti-CD86. For fluorescence activated cell sorting on either Influx or Aria III cell sorter (Becton Dickinson), gating strategies are shown in Fig. 1. Live cells were defined by a forward and side-scatter gate. Live CD86+ cells were sorted into three subsets defined by CD14 and CD16 expression. Samples used for flow-cytometry based phenotypic validation of cell surface marker expression (Fig. 4) were derived from healthy donors of European ancestry local to Cambridge, UK. Here, 5–10 ml of whole anticoagulated blood was collected from each donor and subjected to enrichment and staining as described above, including stains for anti-CD14, anti-CD16 and anti-CD86 in addition to individual markers of interest. Donated blood was collected with informed consent in accordance with the Declaration of Helsinki. Ethical approval was obtained from University of Cambridge Human Biology Research Ethics Committee (HBREC.2016.011).
Plasma membrane profiling
Plasma membrane profiling was performed broadly as previously described13, with the following modifications. The total number of monocytes biotinylated was dependent on the yield from enrichment procedures detailed above, typically in the range of 105–106 cells for intermediate and non-classical monocytes, and 107 for classical monocytes. Precise sorted counts were collected during each enrichment from the Influx or Aria III cell sorter. Each of the three donated samples was biotinylated and labelled separately, combining at the final stage prior to TMT analysis. Briefly, for each enriched monocyte subset sample, sialic acid residues at the cell surface were oxidized using sodium meta-periodate (Thermo) and biotinylated with aminooxy-biotin (Biotium). Following quenching, cells were incubated in 1% (v/v) Triton X-100 lysis buffer (10 mM Tris HCl, 1.6% Triton, 150 mM NaCl). Biotinylated glycoproteins were precipitated using high affinity streptavidin agarose beads (Pierce), and washed extensively. Captured protein was then reduced with dithiothreitol (DTT), alkylated with iodoacetamide (Sigma) and digested on-bead with trypsin (Promega) in 200 mM HEPES pH 8.5 for 3 hours. Trypsin cleaves C-terminal to basic residues, except when they are N-terminal to a Proline residue. Tryptic peptides were collected and the whole sample labelled using TMT reagents following dilution in 200 mM HEPES pH 8.5 adjusted to a final 30% acetonitrile concentration (v/v). To ensure analysis of each cell type in a 1:1:1 ratio, a fraction of each labelled peptide sample was combined in proportion to the number of cells collected for each subset. Labelling was as follows; classical monocytes donation 1 (126), intermediate monocytes donation 1 (127N), non-classical monocytes donation 1(127C), classical monocytes donation 2 (128N), intermediate monocytes donation 2 (128C), non-classical monocytes donation 2 (129N), classical monocytes donation 3 (129C), intermediate monocytes donation 3 (130N), non-classical monocytes donation 3 (130C). The reaction was quenched with hydroxylamine, and TMT-labelled samples combined in a 1:1:1:1:1:1:1:1:1 (all subsets) or 1:1:1 (Classical only) ratio. Labelled peptides were subjected to C18 solid-phase extraction (Sep-Pak, Waters) and vacuum-centrifuged to near-dryness.
Offline SCX fractionation
Offline fractionation was performed as previously described14,37 with the following modifications. 10 mg of PolySulfethyl A bulk material (Nest Group Inc) was loaded on to a fritted 200 μl tip in 100% Methanol using a vacuum manifold. SCX material was conditioned slowly with 1 ml SCX buffer A (7 mM KH2PO4, pH 2.65, 30% Acetonitrile), then 0.5 ml SCX buffer B (7 mM KH2PO4, pH 2.65, 350 mM KCl, 30% Acetonitrile) then 2 ml SCX buffer A. Dried peptides were resuspended in 500 μl SCX buffer A and added to the tip at a flow rate of ~150 μl/min, followed by a 150 μl wash with SCX buffer A. Fractions were eluted in 150 μl buffer at increasing K+ concentrations (10, 25, 40, 60, 90, 150 mM KCl), vacuum-centrifuged to near dryness then desalted using StageTips.
LC-MS3
LC-MS3 was performed with modifications as previously described38,39. Mass spectrometry data was acquired using an Orbitrap Lumos (Thermo Fisher Scientific, San Jose, CA). An Ultimate 3000 RSLC nano UHPLC equipped with a 300 µm ID × 5 mm Acclaim PepMap µ-Precolumn (Thermo Fisher Scientific) and a 75 µm ID × 50 cm 2.1 µm particle Acclaim PepMap RSLC analytical column was used. An unfractionated singleshot was analysed initially to ensure similar peptide loading across each TMT channel, thus avoiding the need for excessive electronic normalization. As all normalisation factors were>0.33 and <3.0, data for each singleshot experiment was analysed with data for the corresponding fractions to increase the overall number of peptides quantified. For monocyte subset analysis, 2 µl of fractionated peptide was initially analysed by mass spectrometry, followed by the remainder of the sample having observed satisfactory chromatography.
For LC/MS3, loading solvent was 0.1% trifluoroacetic acid (TFA), analytical solvent A: 0.1% FA and B: 80% acetonitrile + 0.1% FA. All separations were carried out at 55 °C. Samples were loaded at 10 µl/minute for 5 minutes in loading solvent before beginning the analytical gradient. The following gradient was used: 3–7% B over 4 minutes, 7–37% B over 58 minutes, 37–95% B over 4 minutes followed by a 2 minute wash at 95% B and equilibration at 3% B for 15 minutes. Each analysis used a MultiNotch MS3-based TMT method40,41. The following settings were used: MS1: 380–1500 Th, Quadrupole isolation, 120,000 Resolution and 2 × 105 AGC target, 50 ms maximum injection time. MS2: Quadrupole isolation at an isolation width of m/z 0.7, CID fragmentation (NCE 35) with ion trap scanning out in rapid mode from m/z 120, 1.5 × 104 AGC target, 300 ms maximum injection time in centroid mode. MS3: in Synchronous Precursor Selection mode the top 10 MS2 ions were selected for HCD fragmentation (NCE 65) and scanned in the Orbitrap at 60,000 resolution with an AGC target of 1.5 × 105 and a maximum accumulation time of 250 ms, ions were not accumulated for all parallelisable time. The entire MS/MS/MS cycle had a target time of 3 s. Dynamic exclusion was set to +/−10 ppm for 70 s. MS2 fragmentation was trigged on precursors 5 × 103 counts and above.
Data analysis
Data analysis was performed with modifications as previously described18,38. In the following description, we list the first report in the literature for each relevant algorithm. Mass spectra were processed using a Sequest-based software pipeline for quantitative proteomics, “MassPike”, through a collaborative arrangement with Professor Steve Gygi’s laboratory at Harvard Medical School. MS spectra were converted to mzXML using an extractor built upon Thermo Fisher’s RAW File Reader library (version 4.0.26). In this extractor, the standard mzXML format has been augmented with additional custom fields that are specific to ion trap and Orbitrap mass spectrometry and essential for TMT quantitation. These additional fields include ion injection times for each scan, Fourier Transform-derived baseline and noise values calculated for every Orbitrap scan, isolation widths for each scan type, scan event numbers, and elapsed scan times. This software is a component of the MassPike software platform and is licensed by Harvard Medical School.
A combined database was constructed from the human Uniprot database (26th January, 2017), and common contaminants such as porcine trypsin. The combined database was concatenated with a reverse database composed of all protein sequences in reversed order. Searches were performed using a 20 ppm precursor ion tolerance42. Fragment ion tolerance was set to 1 Da. TMT tags on lysine residues and peptide N termini (229.162932 Da) and carbamidomethylation of cysteine residues (57.02146 Da) were set as static modifications, while oxidation of methionine residues (15.99492 Da) was set as a variable modification.
To control the fraction of erroneous protein identifications, a target-decoy strategy was employed43,44. Peptide spectral matches (PSMs) were filtered to an initial peptide-level false discovery rate (FDR) of 1% with subsequent filtering to attain a final protein-level FDR of 1%45,46. PSM filtering was performed using a linear discriminant analysis, as described previously47. This distinguishes correct from incorrect peptide IDs in a manner analogous to the widely used Percolator algorithm48, though employing a distinct machine learning algorithm. The following parameters were considered: XCorr (minimum 1), ΔCn, missed cleavages, peptide length, charge state, and precursor mass accuracy. Protein assembly was guided by principles of parsimony to produce the smallest set of proteins necessary to account for all observed peptides47.
Proteins were quantified by summing TMT reporter ion counts across all matching peptide-spectral matches using”MassPike”, as described previously40,41. Briefly, a 0.003 Th window around the theoretical m/z of each reporter ion (126, 127n, 127c, 128n, 128c, 129n, 129c, 130n, 130c) was scanned for ions, and the maximum intensity nearest to the theoretical m/z was used. The primary determinant of quantitation quality is the number of TMT reporter ions detected in each MS3 spectrum, which is directly proportional to the signal-to-noise (S:N) ratio observed for each ion49. Conservatively, every individual peptide used for quantitation was required to contribute sufficient TMT reporter ions so that each on its own could be expected to provide a representative picture of relative protein abundance40. An isolation specificity filter was additionally employed to minimise peptide co-isolation50. Peptide-spectral matches with poor quality MS3 spectra (more than 9 TMT channels missing and/or a combined S:N ratio of less than 135 (9-plex, monocyte subsets) or 45 (3-plex, classical monocytes) across all TMT reporter ions) or no MS3 spectra at all were excluded from quantitation. Peptides meeting the stated criteria for reliable quantitation were then summed by parent protein, in effect weighting the contributions of individual peptides to the total protein signal based on their individual TMT reporter ion yields. Protein quantitation values were exported for further analysis in Microsoft Excel.
For protein quantitation, reverse and contaminant proteins were removed. For further analysis and display in figures, fractional TMT signals were used (i.e. reporting the fraction of maximal signal observed for each protein in each TMT channel, rather than the absolute normalized signal intensity). This effectively corrected for differences in the numbers of peptides observed per protein.
Proteins were filtered to include those most likely to be present at the cell surface with high confidence. These contained proteins with the Gene Ontology (GO) terms of ‘plasma membrane’ (PM), ‘cell surface’ (CS), ‘extracellular’ (XC) or with a short 4- or 5-part GO cellular compartment term that included ‘integral to membrane’, but with no subcellular assignment13 (ShG).
To estimate the relative abundance of each protein, a method based on iBAQ was employed. The summed MS1 maximum precursor intensity for each protein across all matching peptides was calculated. Each value was divided by the number of theoretically observable tryptic peptides 7–30 amino acids in length for the respective protein, as determined by in silico trypsin digestion of human Swissprot canonical and isoform database (2017_01_26) using the OrgMassSpecR51 package in R 3.5.152.
For Fig. 1b, the Database for Annotation, Visualisation and Integrated Discovery (DAVID) version 6.8, was used to determine pathway enrichment53. 373 proteins identified at the plasma membrane of classical monocytes were searched against a background of the whole human proteome.
Hierarchical centroid clustering based on Euclidian distance or uncentered correlation was performed using Cluster 3.054 (Stanford University) and visualised using Java Treeview55 (http://jtreeview.sourceforge.net). Principle component analysis and K-means analysis to determine the number of distinct patterns of protein expression between monocyte subsets was performed using XLSTAT v2019.1.2 (Addinsoft). Once the number of expression profiles was determined, Cluster 3.0 was used to perform K-means clustering.
Flow cytometric analysis of monocyte surface proteins
Monocytes were obtained by negative selection using a Pan-Monocyte enrichment kit as described above. Cells were incubated with Fc blocking reagent (Biolegend Human Trustain FcX) for 15 min at 4 °C. Four-colour staining was then used to validate phenotype, using anti-CD14, anti-CD16, anti-CD86 and an antibody against the marker of interest for 15 min at 4 °C. Specific antibodies used were: APC-Cy7 anti-CD14 (Biolegend, 325620), PE anti-CD14 (Biolegend, 301850), BV421 anti-CD16 (Becton Dickinson, 562874), PE-Cy7 anti-CD86 (Becton Dickinson, 561128), FITC anti-ITGA5 (Miltenyi Biotec, 130-110-592), APC anti-SIGLEC10 (Miltenyi Biotec, 130-103-731), PE anti-LTB4R (BioRad, MCA2108PET), PE anti-CD35 (Miltenyi Biotec, 130-099-913). Cells were washed in PBS/0.4% (v/v) citrate/0.5% (v/v) BSA prior to fixation and analysis on a BD LSR Fortessa (Beckton Dickinson). After gating by forward and side-scatter, ≥30,000 events were captured. Data was analysed using FlowJo V10 (FlowJo, LLC). Monocytes were defined using forward and side-scatter filters and positive CD86 expression as shown in Supplementary Fig. S1.
For Fig. 4, monocytes were gated into subsets: CD14++, CD16− (Classical), CD14++, CD16+ (Intermediate), CD14+, CD16++ (Non-classical). MFI values for each subset were normalised to a maximum of 1 prior to averaging and calculation or the standard deviation and standard error of the mean.
Experimental design
For each proteomic analysis (classical monocyte or subset analysis) three independent samples were obtained and prepared separately prior to simultaneous isobaric tag labelling and further analysis. Triplicate analysis was chosen to take advantage of the capability to study up to 10 samples simultaneously using tandem mass tags. As such, three complete sets of three subsets were chosen. Validation studies using flow cytometry were performed in biological triplicate from three independent donors as described above. Flow cytometry fluorophore staining was performed with singly stained and unstained control populations in addition to the test samples. For proteomic data, a Benjamini-Hochberg-corrected two-tailed t-test was used to estimate p-values. For flow cytometry data, a two-tailed t-test was used to estimate p-values based on an observed near-normal distribution of signal within subgroups.
Data availability
The mass spectrometry proteomics data has been deposited to the ProteomeXchange Consortium (http://www.proteomexchange.org/) via the PRIDE56 partner repository, project identifier PXD013832. Project name ‘Comprehensive cell surface proteomics defines markers of classical, intermediate and non-classical monocytes’.
References
Ziegler-Heitbrock, L. et al. Nomenclature of monocytes and dendritic cells in blood. Blood 116, e74–80 (2010).
Wong, K. L. et al. Gene expression profiling reveals the defining features of the classical, intermediate, and nonclassical human monocyte subsets. Blood 118, e16–31 (2011).
Boyette, L. B. et al. Phenotype, function, and differentiation potential of human monocyte subsets. PLoS One 12, e0176460 (2017).
Cros, J. et al. Human CD14dim monocytes patrol and sense nucleic acids and viruses via TLR7 and TLR8 receptors. Immunity 33, 375–386 (2010).
Moniuszko, M., Bodzenta-Lukaszyk, A., Kowal, K., Lenczewska, D. & Dabrowska, M. Enhanced frequencies of CD14++CD16+, but not CD14+CD16+, peripheral blood monocytes in severe asthmatic patients. Clinical immunology (Orlando, Fla.) 130, 338–346 (2009).
Rossol, M., Kraus, S., Pierer, M., Baerwald, C. & Wagner, U. The CD14(bright) CD16+ monocyte subset is expanded in rheumatoid arthritis and promotes expansion of the Th17 cell population. Arthritis and rheumatism 64, 671–677 (2012).
Wildgruber, M. et al. The “Intermediate” CD14(++)CD16(+) monocyte subset increases in severe peripheral artery disease in humans. Scientific reports 6, 39483 (2016).
Hofer, T. P. et al. slan-defined subsets of CD16-positive monocytes: impact of granulomatous inflammation and M-CSF receptor mutation. Blood 126, 2601–2610 (2015).
Ziegler-Heitbrock, L. & Hofer, T. P. Toward a refined definition of monocyte subsets. Frontiers in immunology 4, 23 (2013).
Ancuta, P. et al. Transcriptional profiling reveals developmental relationship and distinct biological functions of CD16+ and CD16− monocyte subsets. BMC genomics 10, 403 (2009).
Zawada, A. M. et al. SuperSAGE evidence for CD14++CD16+ monocytes as a third monocyte subset. Blood 118, e50–61 (2011).
Thomas, G. D. et al. Human Blood Monocyte Subsets: A New Gating Strategy Defined Using Cell Surface Markers Identified by Mass Cytometry. Arteriosclerosis, thrombosis, and vascular biology 37, 1548–1558 (2017).
Weekes, M. P. et al. Proteomic plasma membrane profiling reveals an essential role for gp96 in the cell surface expression of LDLR family members, including the LDL receptor and LRP6. J.Proteome Res. 11, 1475–1484 (2012).
Weekes, M. P. et al. Quantitative temporal viromics: an approach to investigate host-pathogen interaction. Cell 157, 1460–1472 (2014).
Dutertre, C. A. et al. Single-Cell Analysis of Human Mononuclear Phagocytes Reveals Subset-Defining Markers and Identifies Circulating Inflammatory Dendritic Cells. Immunity 51, 573–589.e578 (2019).
Naamati, A. et al. Functional proteomic atlas of HIV infection in primary human CD4+ T cells. eLife 8 (2019).
Egan, E. S. et al. Erythrocytes lacking the Langereis blood group protein ABCB6 are resistant to the malaria parasite Plasmodium falciparum. Communications biology 45, 1–9 (2018).
Ravenhill, B. J. et al. Quantitative comparative analysis of human erythrocyte surface proteins between individuals from two genetically distinct populations. Communications biology 350, 1–9 (2019).
Bausch-Fluck, D. et al. A mass spectrometric-derived cell surface protein atlas. PLoS One 10, e0121314 (2015).
Fallman, M., Andersson, R. & Andersson, T. Signaling properties of CR3 (CD11b/CD18) and CR1 (CD35) in relation to phagocytosis of complement-opsonized particles. Journal of immunology (Baltimore, Md.: 1950) 151, 330–338 (1993).
Yokomizo, T., Izumi, T., Chang, K., Takuwa, Y. & Shimizu, T. A G-protein-coupled receptor for leukotriene B4 that mediates chemotaxis. Nature 387, 620–624 (1997).
Shao, W. H., Del Prete, A., Bock, C. B. & Haribabu, B. Targeted disruption of leukotriene B4 receptors BLT1 and BLT2: a critical role for BLT1 in collagen-induced arthritis in mice. Journal of immunology (Baltimore, Md.: 1950) 176, 6254–6261 (2006).
Campbell, I.D. & Humphries, M.J. Integrin structure, activation, and interactions. Cold Spring Harbor perspectives in biology 3 (2011).
Gahmberg, C. G. et al. Regulation of integrin activity and signalling. Biochimica et biophysica acta 1790, 431–444 (2009).
Munday, J. et al. Identification, characterization and leucocyte expression of Siglec-10, a novel human sialic acid-binding receptor. The Biochemical journal 355, 489–497 (2001).
Li, N. et al. Cloning and characterization of Siglec-10, a novel sialic acid binding member of the Ig superfamily, from human dendritic cells. The Journal of biological chemistry 276, 28106–28112 (2001).
Kivi, E. et al. Human Siglec-10 can bind to vascular adhesion protein-1 and serves as its substrate. Blood 114, 5385–5392 (2009).
Yasunaga, M. & Matsumura, Y. Role of SLC6A6 in promoting the survival and multidrug resistance of colorectal cancer. Scientific reports 4, 4852 (2014).
Jhiang, S. M. et al. Cloning of the human taurine transporter and characterization of taurine uptake in thyroid cells. FEBS letters 318, 139–144 (1993).
Dornier, E. et al. TspanC8 tetraspanins regulate ADAM10/Kuzbanian trafficking and promote Notch activation in flies and mammals. The Journal of cell biology 199, 481–496 (2012).
Bradshaw, E. M. et al. CD33 Alzheimer’s disease locus: altered monocyte function and amyloid biology. Nature neuroscience 16, 848–850 (2013).
Weekes, M. P. et al. Comparative analysis of techniques to purify plasma membrane proteins. J.Biomol.Tech. 21, 108–115 (2010).
Kristiansen, M. et al. Identification of the haemoglobin scavenger receptor. Nature 409, 198–201 (2001).
Zhao, C. et al. Identification of novel functional differences in monocyte subsets using proteomic and transcriptomic methods. J Proteome Res 8, 4028–4038 (2009).
Segura, V. et al. In-Depth Proteomic Characterization of Classical and Non-Classical Monocyte Subsets. Proteomes 6 (2018).
Villani, A.C. et al. Single-cell RNA-seq reveals new types of human blood dendritic cells, monocytes, and progenitors. Science 356 (2017).
Dephoure, N. & Gygi, S. P. A solid phase extraction-based platform for rapid phosphoproteomic analysis. Methods 54, 379–386 (2011).
Soday, L. et al. Quantitative Temporal Proteomic Analysis of Vaccinia Virus Infection Reveals Regulation of Histone Deacetylases by an Interferon Antagonist. Cell Rep. 27, 1920–1933 e1927 (2019).
Caller, L.G. et al. Temporal Proteomic Analysis of BK Polyomavirus Infection Reveals Virus-Induced G2 Arrest and Highly Effective Evasion of Innate Immune Sensing. J. Virol. 93 (2019).
McAlister, G. C. et al. Increasing the multiplexing capacity of TMTs using reporter ion isotopologues with isobaric masses. Analytical Chemistry 84, 7469–7478 (2012).
McAlister, G. C. et al. MultiNotch MS3 enables accurate, sensitive, and multiplexed detection of differential expression across cancer cell line proteomes. Anal. Chem. 86, 7150–7158 (2014).
Haas, W. et al. Optimization and use of peptide mass measurement accuracy in shotgun proteomics. Mol. Cell Proteomics 5, 1326–1337 (2006).
Elias, J. E. & Gygi, S. P. Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat Methods 4, 207–214 (2007).
Elias, J. E. & Gygi, S. P. Target-decoy search strategy for mass spectrometry-based proteomics. Methods in Molecular Biology 604, 55–71 (2010).
Wu, R. et al. Correct interpretation of comprehensive phosphorylation dynamics requires normalization by protein expression changes. Mol. Cell Proteomics 10, M111 009654 (2011).
Kim, W. et al. Systematic and quantitative assessment of the ubiquitin-modified proteome. Mol Cell 44, 325–340 (2011).
Huttlin, E. L. et al. A tissue-specific atlas of mouse protein phosphorylation and expression. Cell 143, 1174–1189 (2010).
Kall, L., Canterbury, J. D., Weston, J., Noble, W. S. & MacCoss, M. J. Semi-supervised learning for peptide identification from shotgun proteomics datasets. Nat. Methods 4, 923–925 (2007).
Makarov, A. & Denisov, E. Dynamics of ions of intact proteins in the Orbitrap mass analyzer. Journal of the American Society for Mass Spectrometry 20, 1486–1495 (2009).
Ting, L., Rad, R., Gygi, S. P. & Haas, W. MS3 eliminates ratio distortion in isobaric multiplexed quantitative proteomics. Nat Methods 8, 937–940 (2011).
Dodder, N. OrgMassSpecR: Organic Mass Spectrometry. R package version 0.5-3. https://CRAN.R-project.org/package=OrgMassSpecR (2017)
Team, R.C. R: A language and environment for statistical computing. https://www.R-project.org/ (2018)
Huang da, W., Sherman, B. T. & Lempicki, R. A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nature protocols 4, 44–57 (2009).
de Hoon, M. J., Imoto, S., Nolan, J. & Miyano, S. Open source clustering software. Bioinformatics (Oxford, England) 20, 1453–1454 (2004).
Saldanha, A. J. Java Treeview–extensible visualization of microarray data. Bioinformatics (Oxford, England) 20, 3246–3248 (2004).
Vizcaino, J. A. et al. 2016 update of the PRIDE database and its related tools. Nucleic acids research 44, D447–456 (2016).
Acknowledgements
We are grateful to Prof. Steven Gygi for providing access to the “MassPike” software pipeline for quantitative proteomics. This work was supported by an Evelyn Trust research training fellowship (18/27) to BJR, a Wellcome Trust Senior Clinical Research Fellowship (108070/Z/15/Z) to MPW and a strategic award to Cambridge Institute for Medical Research from the Wellcome Trust (100140). This study was additionally supported by the Cambridge Biomedical Research Centre, UK. We would like to thank the NIHR Cambridge BRC phenotyping hub for their assistance in sorting primary monocytes into their subgroups.
Author information
Authors and Affiliations
Contributions
B.J.R. and M.P.W. designed and performed the experiments. B.J.R. and L.S. isolated monocytes from leukocyte enriched donor samples. R.A. collected proteomic data. B.J.R., J.H. and M.P.W. analysed proteomic data. B.J.R. and M.P.W. wrote the manuscript. M.P.W. supervised all research.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ravenhill, B.J., Soday, L., Houghton, J. et al. Comprehensive cell surface proteomics defines markers of classical, intermediate and non-classical monocytes. Sci Rep 10, 4560 (2020). https://doi.org/10.1038/s41598-020-61356-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-61356-w
This article is cited by
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
