
Abstract
Equine piroplasmosis (EP) is a severe disease of horses caused by the tick-borne protozoa Theileria equi (T. equi) and Babesia caballi (B. caballi). Infectious carriers are not always symptomatic, meaning there is a risk to non-enzootic areas. Regulatory tests for EP include sero-epidemiological methods for equine babesiosis, but these lack specificity due to cross-reactivity with other Babesia species. In this study, we present a real-time quantitative recombinase polymerase amplification (qRPA) method for fast simultaneous detection of both T. equi and B. caballi. In this method, primers and probes targeting the 18S rRNA gene of both T. equi and B. caballi, the ema-1 gene of T. equi and the bc48 gene of B. caballi were designed and evaluated. The sensitivity of qRPA was evaluated using the pUC57 plasmid DNA containing the target gene. For the pUC57-bc48 gene DNA, the R2 value was 0.983 for the concentration range 0.2 ng (4.1 × 107 DNA copies) to 2.0 fg (4.1 × 101 DNA copies). For the pUC57-ema gene DNA, the R2 value was 0.993 for the concentration range 0.2 ng (5.26 × 107 DNA copies) to 2.0 fg (5.26 × 102 DNA copies). For the pUC57-Bc18S gene DNA the R2 value was 0.976 for the concentration range 2.0 ng (4.21 × 108 DNA copies) to 2.0 fg (4.21 × 102 DNA copies). For the pUC57-Te18S gene DNA, the R2 value was 0.952 (Fig. S3b) for the concentration range 2.0 ng (4.16 × 108 DNA copies) to 2.0 fg (4.16 × 102 DNA copies). Furthermore, a duplex qRPA analysis was developed and optimized and the results showed that primers and probes targeting for the bc48 gene of B. caballi and the 18S rRNA gene of T. equi is the best combination for a duplex qRPA analysis in one reaction. The developed duplex qRPA assay has good specificity, and had negative amplification for several similar parasite. For DNA extracted from real horse blood specimens, this qRPA method has comparable sensitivity to traditional qPCR, but a simpler and more rapid operating process to obtain positive amplification. The qRPA, including the duplex strategy described here, could allow fast identification of the EP-causing T. equi and B. caballi, showing great potential for on-site EP screening of horses.
Similar content being viewed by others

Introduction
Equine piroplasmosis (EP) is a haemoprotozoan infection of horses and other members of the Equidae family. It is caused via tick transmission of two intra-erythrocyte protozoa, Theileria equi (also called Babesia equi) and Babesia caballi (B. caballi)1,2,3. There are differences between B. caballi and T. equi in both the tick vector and the horse host, which is reflected in disease severity and drug susceptibility (Fig. 1a). However, making a pathogenic diagnosis is still difficult as the parasites have the same vector and a similar clinical presentation, and in addition cross-infection is possible.

Schematic description of a duplex real-time RPA strategy for the diagnosis of B. caballi and T. equi in horse blood specimens. (a) Life cycle of B. caballi and T. equi within the tick vector and within the horse host. PBMCs: peripheral blood mononuclear cells; RBCs: red blood cells. (b) Method of rapid genomic DNA identification and RPA reaction.
Infected animals present with severe acute disease characterized by high fever, lethargy, anorexia, peripheral oedema, splenomegaly, haemolysis, tachycardia, pigmenturia and occasionally death1. Animals who recover from primary infection remain recessive carriers with fluctuating levels of parasitaemia4,5. This disease is widely distributed in Asia, Europe, Africa and South America, causing economic loss and impacting the international movement of horses6,7. Importation of carrier animals with no obvious signs of disease is a major risk factor for the introduction of EP into non-enzootic areas1. Therefore, developing sensitive and specific diagnostic methods is essential for identifying asymptomatic equines carrying these parasites.
Current diagnostic methods include aetiological diagnostics8,9, immunological diagnostics10,11,12,13,14 and molecular diagnostics9,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33. Of these, molecular diagnostics is widely recognized due to its accuracy and sensitivity9,34. Cortes et al. developed a multinested PCR assay for simultaneous detection of the equine piroplasmids T. equi and B. caballi by amplification of five genetic markers (18S rRNA, β-tubulin, cytB, ema-1 and rap-1)23. To identify the species of piroplasmid in an infected horse, nested PCR assay is commonly used to amplify long fragments of 18S rRNA, followed by sequence analysis22. However, PCR-based diagnosis requires a thermocycler, skilled personnel and a long detection time, which made it inappropriate for field diagnostic applications. As an alternative, recombinase polymerase amplification (RPA) has emerged as a novel isothermal technique for molecular diagnosis of various infectious diseases35, including the protozoan parasites36 Theileria annulata37 and Babesia gibsoni38. Compared to PCR-based assay and loop-mediated isothermal amplification (LAMP), RPA is more rapid (<20 min), simpler to perform as it requires a lower temperature (37–42 °C) and has an acceptable sensitivity39,40. RPA-lateral flow assay has been used for rapid detection of Trichinella41, Perkinsus beihaiensis42, Plasmodium knowlesi43, Babesia gibsoni38, Protozoan parasites36, Theileria annulata37, Fasciola hepatica44, Schistosoma japonicum45, Schistosoma haematobium46, Leishmania donovani47, Intestinal Protozoa48, Giardia49, Plasmodium falciparum50. To avoid the “ghost band” and the false-positive results, the primers, probes and the detection procedure have to be carefully designed39.
To speed up the clearance of an imported horse at a port, or detect a piroplasmosis infection in the field or in low-resource underserved rural communities, we also adapted RPA to develop a duplex detection of both T. equi and B. caballi as an alternative to duplex qPCR26,30. Various genomic sites have been used in species identification, phylogenetic and genotype studies of both T. equi and B. caballi with PCR-based molecular techniques, including the small subunit ribosomal RNA gene (18S rRNA)11,22,51,52,53,54; and genomic sites targeted by qPCR assay have included the ema-1 gene of T. equi and the 48 kDa merozoite rhoptry protein (bc48) gene of B. caballi30. These sites have been used to create a duplex real-time PCR for simultaneous detection of both parasites using the ema-1 gene of T. equi and the bc48 gene of B. caballi30; or the 18S rRNA gene of B. caballi and the ema-1 gene of T. equi26. In this study, we designed primers and fluorescent probes to target the 18S rRNA gene of both T. equi and B. caballi, the ema-1 gene of T. equi and the bc48 gene of B. caballi. A secondary aim was to investigate the combination of target genes to construct the duplex assay.
Results and Discussion
Duplex real-time RPA strategy for the diagnosis of equine piroplasmosis
A molecular diagnostic method based on duplex real-time RPA for the diagnosis of both B. caballi and T. equi from infected horse blood was developed (Fig. 1b). To achieve rapid diagnosis, specimen DNA extraction and separation were performed using magnetic beads, as an alternative to traditional high-speed centrifugation. To simplify the steps, all reagents (including extracted DNA, primers, probes, freeze-dried enzymes and rehydration buffer) were one-time mixed and the amplification reaction was initiated by MgAc2 reagent at 37–42 °C. During RPA amplification, the fluorescent signal was created using an oligonucleotide probe flanked by a dT-fluorophore and a corresponding dT-quencher group, and was observed with a portable fluorescence detection device. Therefore, this strategy allows rapid diagnosis of piroplasmosis on-site.
Primer design
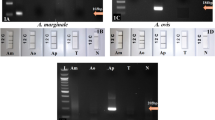
Four primers targeting the bc48 gene of B. caballi and the ema-1 gene of T. equi were tested for sensitivity and specificity. The B. caballi-bc48-F1/R2 primer could amplify pUC57-bc48 (Fig. 2a, line 1), but not T. equi genomic DNA (Fig. 2a, line 1-Te). The B. caballi-bc48-F2/R1 primer was able to amplify pUC57-bc48 (Fig. a, line 2) and T. equi genomic DNA to some extent (Fig. 2a, line 2-Te). The B. caballi-bc48-F1/R1 primer produced an amplification curve with only water as the template (Fig. 2a, line 3). The primer set B. caballi-bc48-F2/R2 has a low amplification efficiency for T. equi genomic DNA (Fig. 2a, line 4). Therefore, B. caballi-bc48-F1/R2 was chosen to test the sensitivity for pUC57-bc48 and B. caballi genomic DNA.

Real-time fluorescence curves of RPA assay using different primers and probes. (a) B. caballi-bc48-F1/R2 (line 1) and Primer B. caballi-bc48-F2/R1 (line 2), targeting the B. caballi bc48 gene for pUC57-bc48 plasmid DNA; (b) Primer T. equi-ema-F1/R1 (line 1) and T. equi-ema-F2/R2 (line 2), targeting T. equi ema-1 gene for pUC57-ema plasmid DNA. (c) Primer B. caballi-Bc18S-F1/R1 assay for pUC57-Bc18S plasmid DNA (line a), pUC57-Te18S plasmid DNA (line b) and T. equi-Bc18S-F1/R1 assay for pUC57-Bc18S plasmid DNA (line c); (d) Primer B. caballi-Bc18S-F2/R2 for pUC57-Bc18S plasmid DNA (line 1), pUC57-Te18S plasmid DNA (line 1-Te), B. caballi-Bc18S-F2/R3 (line 2-Te), B. caballi-Bc18S-F3/R2 (line 3-Te) and B. caballi-Bc18S-F3/R3 (line 4-Te) for pUC57-Te18S plasmid DNA; (e) Primer T. equi-18S-F2/R2 for pUC57-Te18S plasmid DNA (line 1-Te), pUC57-Bc18S plasmid DNA (line 1-Bc), T. equi-18S-F3/R2 for pUC57-Te18S plasmid DNA (line 2-Te), and pUC57-Bc18S plasmid DNA (line 2-Bc).
Additionally, two primer sets (T. equi-ema-F1/R1 and T. equi-ema-F2/R2) could amplify pUC57-ema DNA (Fig. 2b, lines 1&2), but not B. caballi genomic DNA (Fig. 2b, lines 1-b and 2-b). Therefore, both primers could be used for pUC57-ema DNA and T. equi genomic DNA.
In this study, we also designed the primers and probes for the 18S rRNA gene of B. caballi and T. equi. Because of the high similarity of this gene between the two vectors, neither B. caballi-18S-F1/R1 nor T. equi-18S-F1/R1 were able to specifically amplify the plasmid DNA (Fig. 2c). We found that neither B. caballi-18S-F2/R2 (Fig. 2d, line 1-Te), nor B. caballi-18S-F3/F2 (Fig. 2d, line 3-Te) nor B. caballi-18S-F3/R3 (Fig. 2d, line 4-Te) were able to amplify the T. equi 18S rRNA gene; although B. caballi-18S-F2/R3 could amplify this gene to some extent (Fig. 2d, line 2-Te). Neither T. equi-18S-F2/R2 nor T. equi-18S-F3/R2 were able to amplify the B. caballi 18S rRNA gene, although T. equi-18S-F2/R2 showed good amplification for pUC57-T.equi-18S plasmid DNA (Fig. 2e). Therefore, B. caballi-18S-F2/R2, B. caballi-18S-F3/F2, B. caballi-18S-F3/R3, T. equi-18S-F2/R2 and T. equi-18S-F3/R2 primers could be used for a pUC57-Bc18S or pUC57-Te18S plasmid DNA assay.
Duplex RPA assay
To develop the duplex RPA assay for B. caballi and T. equi, we first investigated the primers and probes targeting the bc48 gene of B. caballi and the ema-1 gene of T. equi using pUC57-bc48 and pUC57-ema plasmid DNA. A carboxy fluorescein (FAM)-signal was only detected from pUC57-bc48 plasmid DNA (Fig. 3a, lines b& d); and a ROX-signal was only detected from pUC57-ema plasmid DNA (Fig. 3a, lines a&c). This indicated good specificity of these primers and probes. Compared with the primers and probes targeting the ema-1 gene, those targeting the 18S rRNA gene of T. equi had greater amplification (Fig. 3b), possibly due to ema-1 gene sequence heterogeneity in a subset of the original isolates27. Therefore, we chose T. equi-18S-F2/R2 with a T. equi-18S-probe to amplify T. equi genomic DNA.

Duplex real-time RPA assay. (a) Assay using a mixture of primer B. caballi-bc48-F1/R2 and T. equi-ema-F1/R1 for pUC57-bc48 plasmid DNA (lines b, c) and pUC57-ema plasmid DNA (lines a, d). (b) Assay for T. equi genomic DNA using primer T. equi-18S-F3/R2 (solid line) and T. equi-ema-F1/R1 (short dashed line). (c) T. equi-18S-F3/R2/B. caballi-18S-P for pUC57-Te18S plasmid DNA (line a) and pUC57-Bc18S plasmid DNA (line b); B. caballi-18S-F3/R3/T.equi-18S-P for pUC57-Bc18S plasmid DNA (line c) and pUC57-Te18S plasmid DNA (line d). (d) T. equi-18S-F2/R2/B.caballi-18S-P for pUC57-Te18S plasmid DNA (line a) and pUC57-Bc18S plasmid DNA (line b); B. caballi-18S-F2/R2/T. equi-18S-P for pUC57-Te18S plasmid DNA (line c) and pUC57-Bc18S plasmid DNA (line d). (e) Mixture of T. equi-18S-F3/R2 and B. caballi-18S-F3/R2 for pUC57-Te18S plasmid DNA (lines a, a’) or pUC57-Bc18S plasmid DNA (lines b, b’). (f) Mixture of T. equi-18S-F2/R2 and B. caballi-Bc48-F1/R2 for B. caballi genomic DNA (lines, a, a’), T. equi genomic DNA (lines, b, b’) and water (lines, c, c’).
When the primers and probes targeting the 18S rRNA genes of B. caballi and T. equi were mixed in one reaction tube, the amplification signals showed cross interference (Fig. 3c–e), which meant that developing a duplex assay based on these primers and probes was inappropriate. Therefore, primers B. caballi-bc48-F1/R2 and T. equi-18S-F2/R2 were chosen to establish a duplex RPA strategy for B. caballi and T. equi. These primers and probes were added into one reaction tube to detect water (Fig. 3f, blue lines), B. caballi genomic DNA (Fig. 3f, black lines) and T. equi genomic DNA (Fig. 3f, red lines). Results showed specific amplification of B. caballi or T. equi genomic DNA and no cross interference. Therefore, a valid duplex RPA strategy was developed by employing a B. caballi-bc48-F1/R2/B. caballi-bc48-probe and a T. equi-18S-F2/R2/T. equi-18S-probe for the diagnosis of equine piroplasmosis.
Sensitivity and specificity of real-time RPA assay
To evaluate the sensitivity of the primers and probes for the bc48 gene of B. caballi and the ema-1 gene of T. equi, we chose B. caballi-bc48-F2/R2 and T. equi-ema-F1/R1 to evaluate the sensitivity and linearity of the real-time RPA assay. The amplification curves are shown in Fig. 4a,b. For the pUC57-bc48 gene DNA, the R2 value was 0.983 (Fig. S2a) for the concentration range 0.2 ng (4.1 × 107 DNA copies) to 2.0 fg (4.1 × 101 DNA copies). For the pUC57-ema gene DNA, the R2 value was 0.993 (Fig. S2b) for the concentration range 0.2 ng (5.26 × 107 DNA copies) to 2.0 fg (5.26 × 102 DNA copies).

Sensitivity of real-time RPA assay. (a) pUC57-Bc48 plasmid DNA with primer B. caballi-bc48-F1/R2. (b) pUC57-ema plasmid DNA with primer T. equi-ema-F1/R1; lines a-h, from 0.2 ng to 0.02 fg. (c) pUC57-Bc18S plasmid DNA with primer B. caballi-Bc18S-F3/R2. (d) pUC57-Te18S plasmid DNA with primer T. equi-18S-F3/R2/; lines a-g, from 2 ng to 2 fg. Line h, water. (e) Real-time PCR assay for serially-diluted pUC57-Bc18S plasmid DNA; (f) Real-time PCR assay for serially-diluted pUC57-ema plasmid DNA; lines a–g, from 2 ng to 2 fg. Line h, water.
The sensitivity of the primers and probes for the 18S rRNA gene of B. caballi and T. equi assay were evaluated using B. caballi-18S-F3/R2 and T. equi-18S-F3/R2 respectively (Fig. 4c,d). The linearity results indicated that for the pUC57-Bc18S gene DNA the R2 value was 0.976 (Fig. S3a) for the concentration range 2.0 ng (4.21 × 108 DNA copies) to 2.0 fg (4.21 × 102 DNA copies). For the pUC57-Te18S gene DNA, the R2 value was 0.952 (Fig. S3b) for the concentration range 2.0 ng (4.16 × 108 DNA copies) to 2.0 fg (4.16 × 102 DNA copies).
Real-time PCR is the widely-recognized gold standard of genomic DNA identification and quantification due to its good specificity and high sensitivity. In this study, we employed the reported PCR primers and probes to assay pUC57-Bc18S and pUC57-ema DNA26,28 and the results showed that 2.0 fg pUC57-Bc18S DNA (Figs. 4e) and 2.0 fg of pUC57-ema DNA (Fig. 4f) could be detected. The onset of real-time PCR was linearly fitted against the logarithm of initial pUC57-Bc18S concentration from 2.0 fg (4.21 × 102 DNA copies) to 2.0 ng (4.21 × 108 DNA copies), with an R2 value of 0.998 (Fig. S4a); and for pUC57-ema concentration from 2.0 fg (5.26 × 102 DNA copies) to 2.0 ng (5.26 × 108 DNA copies), with a R2 value of 0.997 (Fig. S4b). This shows that the sensitivity of the developed real-time RPA is comparable to real-time PCR assay.
The specificity of the developed qRPA was evaluated using several other parasite (Sample No. 16–22 in Table 1), and the results (Fig. 5f) showed that none of these parasites generated FAM or ROX fluorescence signals in the duplex RPA reaction containing the primers and probes targeting for bc48 gene of B. caballi and the 18S gene of T. equi. But the B. caballi genomic DNA from sample HQ1–4 could generate remarkable fluorescence signals of FAM Dye and T. equi genomic DNA from sample DQ1 and DQ2 produced obvious fluorescence signals of ROX Dye (Fig. 5f), indicating that the developed primers and probes have good specificity.

Fluorescence curves of horse blood assay using real-time PCR and real-time RPA methods. (a) Real-time RPA assay for B. caballi genomic DNA using primer B. caballi-bc48-F1/R2/bc48-P (solid lines, three samples) and B. caballi-18S-F3/R2/bc-18S-P (short dashed lines, three samples). Line 11, a; Line 12, b; Line 13, c. (b) Real-time PCR assay for real samples containing B. caballi genomic DNA using Bc_18SF402/ c_18SR4596/Bc_18SP. Lines 1–10, horse blood samples, numbered HQ1–10. (c) Real-time PCR assay for real samples containing T. equi genomic DNA using Te_EMA1-F/ Te_EMA1-R/Te_EMA1-P. Lines 14 and 15, horse blood samples, numbered DQ1 and DQ2. (d) Real-time RPA assay for real samples containing B. caballi genomic DNA using B. caballi-bc48-F1/R2/bc48-P. Lines 1–10, HQ 1–10. (e) Real-time RPA assay for real samples containing T. equi genomic DNA using T. equi-18S-F2/R2/T. equi-18S-P. Line 14, DQ1; line 15, DQ2; lines 1–3, HQ 1–3. (f) Real-time fluorescence curves of a duplex assay for B. caballi genomic DNA (line 1, HQ1; 2, HQ2; 3, HQ-3; 4, HQ-4), T. equi genomic DNA (line 14, DQ1; 15, DQ2), trypanosoma evansi (line 16), trypanosome brucei (line 17), toxoplasma gondii (line 18), plasmodium falciparum (line 19), theileria hirci (line 20), theileria sinensis (line 21), trichinella spiralis inorganic (line 22). Solid lines indicated the FAM signal generated by the bc48 gene of B. caballi, and short dashed lines indicated the ROX signal generated by the 18S gene of T. equi.
Applicability of RPA assay for horse blood samples
To investigate the efficacy of the developed primers and method in a real sample, genomic DNA extracted from horse blood containing B. caballi or T. equi was used as a template for an RPA assay. Horse blood samples collected from the Hongshan Military Horse Farm of the Beijing Military Region during a period of high incidence of equine piroplasmosis in April, 2018 were detected. Before detection, we evaluated the amplification efficiency of primer sets (the 18S rRNA, bc48 and ema-1 genes). For B. caballi, the bc48 gene primers had better amplification efficiency than the 18S gene primers (Fig. 5a). For T. equi, the 18S gene primers had better amplification efficiency than the ema-1 gene primers (Fig. 3b). Therefore, B. caballi-bc48-F1/R2 and T. equi-18S-F2/R2 were applied to assay the real horse blood sample.
The horse blood samples containing B. caballi or T. equi were assayed using real-time RPA (Fig. 5d,e), and the results compared to real-time PCR (Fig. 5b,c). Results of all the samples were consistent according to the onset time of the two methods. Furthermore, the chosen specimens containing B. caballi were assayed using the developed duplex real-time RPA method. The results showed that only FAM signals were generated from B. caballi samples (Fig. 5f), confirming the validity of the developed method for diagnosing EP. The new method has the advantages of being more rapid, whilst maintaining high sensitivity and specificity.
Repeatability of RPA assay
During RPA amplification, fluorescence intensity can easily be affected by the concentration of the probe and DNA template, even by the pipette due to the small volume of DNA template40. The repeatability of the RPA assay was therefore evaluated by measuring 0.02 ng (4.1 × 106 DNA copies) and 20 fg (4.1 × 103 copies) of pUC57-bc48 plasmid DNA four times (Fig. 6a). Since quantification was based on the onset time of amplification, we investigated this across the four experiments. The results showed that the deviations of onset time were 8.53% for 0.02 ng and 6.03% for 20 fg DNA (Fig. 6b), an acceptable level of repeatability.

Repeatability of the RPA assay. (a) The real-time fluorescence curves of the RPA assay for 0.02 ng (solid line, four samples) and 20 fg (short dashed lines, four samples) of pUC57-bc48 plasmid DNA. (b) The onset time of real-time amplification of four assays. The colour of the columns corresponds to the colour of the lines in part (a).
Conclusion
In view of the harm caused by EP and the inadequacy of existing rapid diagnosis methods, we developed a new real-time qRPA system for the rapid diagnosis of the typical pathogens of EP: B. caballi and T. equi. The presented qRPA assay, by amplifying the bc48 or 18S rRNA gene of B. caballi and the ema-1 gene or 18S rRNA gene of T. equi, showed strict specificity and comparable sensitivities. The developed qRPA strategy has a simpler operating process with a lower constant reaction temperature (39~42 °C) and was more suitable to assay blood specimens in the field as a rapid diagnosis method compared with traditional qPCR. Furthermore, a duplex analysis strategy based on the bc48 gene of B. caballi and the 18S rRNA gene of T. equi was established, and needed only 20 min to amplify the pathogens in horse blood specimens. The qRPA, including the duplex strategy described here, shows great potential for on-site EP screening.
Materials and Methods
Materials
Qubit® dsDNA HS Assay kit and Qubit® 2.0 Fluorometer were from Life technologies (Carlsbad, CA). A TwistAmp® exo enzyme pellet including Mg(Ac)2 solution (280 mM in water) were purchased from TwistDx Co. (Cambridge, UK). Agencourt AMPure XP beads were obtained from Beckman Coulter (California, USA). All chemicals were of reagent grade. Milli-Q grade (>18 MΩ) water from a Milli-Q water purification system (Merck KGaA, Darmstadt, Germany) was autoclaved for 20 min at 120 °C was used throughout the experiment.
The horse blood samples were obtained from the Hongshan Military Horse Farm of the Beijing Military Region in the Keshiketeng Banner (World Geopark) (Chifeng City, Inner Mongolia) and Heilongjiang Bayi Agricultural University (Daqing, Heilongjiang). EP depends on the seasonal activity of ticks and the highest incidence is at the end of April. Therefore, 30 horse blood samples were taken from tick-bitten horses during this period. The other parasites, i.e. trypanosoma evansi (strain STIB 805), trypanosome brucei (brucei TREU927), toxoplasma gondii (RH), plasmodium falciparum (3D7), theileria hirci (Dschunkowsky), theileria sinensis (Blood extract), trichinella spiralis inorganic (Blood extract) were stored in Shenyang Agricultural University (Shenyang, Liaoning). The information of all these samples were listed in Table 1. Horse blood sample collection and processing were performed according to Procedures for Veterinary Clinical Techniques published by China Agriculture Press, and Veterinary Laboratory Biosafety Guidelines approved by the Ministry of Agriculture of the People’s Republic of China.
DNA extraction procedure
Total DNA was extracted from 200 μl of whole blood using the DNeasy Blood and Tissue Kit (Qiagen, Hilden, Germany) according to the manufacturer’s protocol. Briefly, 20 μl Qiagen Protease was added into the blood sample, then 200 μl Buffer AL was added, followed by a brief vortexing. This mixture was incubated at 56 °C for 10 min with four times up-and-down mixing. After a brief vortexing, 200 μl ethanol was added to the sample, and mixed thoroughly by vortexing. Then the DNA released from the sample was purified with the DNeasy mini spin column with AW1 and AW2 washes, respectively. At last, 100 μl of elution buffer was used to elute the DNA retained on the membrane of the spin column. The eluted DNA was quantified using a Qubit® 2.0 Fluorometer and a Qubit® dsDNA HS Assay kit.
Primer design
Two genes from B. caballi were chosen as the target of the RPA assay: bc48 (GenBank No. AB017700.1) and 18S rRNA (GenBank No. Z15104.1). The two genes from T. equi were ema-1 (GenBank No. KC347577.1) and 18S rRNA (GenBank No. Z15105.1). The primers and probes are listed in Tables 2 and 3. Primers and probes for bc48 and ema-1 were listed in Table 2, and for 18S rRNA in Table 3. The forward and reverse primers had 30–35 bp. The probes were labelled with a FAM/ Black Hole Quencher 1 or ROX/Black Hole Quencher 1. All primers and probes were evaluated for biophysical properties and dimer formation by Oligoanalyzer 3.1 (IDT, Leuven, Belgium) and blasted (https://www.ncbi.nlm.nih.gov/tools/primer-blast) against the NCBI nucleotide database to make sure that there was no homology with sequences from another organism.
Construction of recombinant plasmid carrying targeting genes of B. caballi or and T. equi
Sequence of the bc48 or 18S rRNA genes from B. caballi, and the ema-1 or 18S rRNA genes from T. equi, were commercially synthesized and cloned into plasmid pUC57 (Genscript, Jiangsu, China), respectively. The prepared recombinant plasmids termed pUC57-bc48, pUC57-Bc18S, pUC57-ema and pUC57-Te18S, respectively. Then, plasmids were replicated in E. coli DH5α cells, extracted and purified using an endotoxin-free Plasmid Maxiprep Kit. After that, plasmid DNA was quantified using a Qubit® 2.0 Fluorometer and a Qubit® dsDNA HS Assay kit.
Development of RPA assay
The RPA assay was performed based on the manual instructions. In brief, each assay was performed in a test tube filled with TwistAmpTM exonuclease and 50 μl reaction mixture. In process, the enzyme pellet was rehydrated by 46.5 μL of a master-mix containing rehydration buffer (29.5 μL), nuclease-free water (12.2 μL), 10 μM forward and reverse primer (each 2.1 μL) and 10 μM probe (0.6 μL). Next, 2 μL of plasmid DNA or DNA specimen extracted from parasites was added to each reaction, with a final concentration of 420 nM for each primer and 120 nM for probe. Last, 2.5 μL of Mg(Ac)2 (280 mM) was added to the lid of each tube. All reactions were simultaneously initiated by centrifuging the magnesium acetate into the reaction mixture and transferring the tubes to a 39 °C heat block for 30 min. Fluorometric data were collected every 10 s. Referring to the previous works, the onset time of amplification was used to calculate the linearity and limit of detection, which was more practical than fluorescence intensity and helpful to improve the repeatability of the real-time RPA assay55,56.
Evaluation of specificity and sensitivity
Sensitivity of the RPA assay were tested using pure recombinant plasmid genomic DNA, which allowed accurate determination of the detection limit in terms of copy numbers. A ten-fold serial dilution of recombinant plasmid DNA, ranging from 2.0 ng to 0.02 fg, was used as standard to determine sensitivity level, establish amplification efficiency and resolve the limit of detection. The DNA copy number of 2.0 ng of the pUC57-bc48 plasmid is equal to 4.1 × 108 copies; 2.0 ng of pUC57-ema equates to 5.26 × 108 copies; 2.0 ng of pUC57-Bc18S equates to 4.21 × 108 copies; and 2.0 ng of pUC57-Te18S is 4.16 × 108 copies. For specificity, the interfering DNA samples from trypanosoma evansi (strain STIB 805), trypanosome brucei (brucei TREU927), toxoplasma gondii (RH), plasmodium falciparum (3D7), theileria hirci (Dschunkowsky), theileria sinensis (Blood extract), trichinella spiralis inorganic (Blood extract), were evaluated using the developed duplex RPA assay. The onset time of amplification was plotted against the concentration of total DNA.
As a comparison, real-time PCR assay was also performed. Primers and probes reported in a previous duplex qPCR assay of EP26,28 (Table 4) were synthesized by Sangon Biotech (Shanghai, China). The PCR reactions were run on a Light Cycler 480 (Roche, USA) using a TaqMan Gene Expression Master Mix (Thermo Fisher Scientific, USA). The cycling protocol consisted of the initial activation cycle at 95 °C for 10 min, followed by 45 cycles of 30 s at 95 °C, 30 s at 57 °C and 20 s at 72 °C with fluorescence data acquisition after the elongation step. Negative controls consisted of nuclease-free water and DNA extracted from the blood of an EP-free horse.
Ethical statement
The Ethical Committee of Shenyang Agricultural University approved the laboratory animal experiments (permit no. SYXK <Liao> 2011–0001).
References
Wise, L. N., Kappmeyer, L. S., Mealey, R. H. & Knowles, D. P. Review of equine piroplasmosis. Journal of veterinary internal medicine 27, 1334–1346, https://doi.org/10.1111/jvim.12168 (2013).
Mehlhorn, H. & Schein, E. Redescription of Babesia equi Laveran, 1901 as Theileria equi Mehlhorn, Schein 1998. Parasitology research 84, 467–475, https://doi.org/10.1007/s004360050431 (1998).
de Waal, D. T. Equine piroplasmosis: a review. The British veterinary journal 148, 6–14, https://doi.org/10.1016/0007-1935(92)90061-5 (1992).
Taboada, J. & Merchant, S. R. Babesiosis of companion animals and man. The Veterinary clinics of North America. Small animal practice 21, 103–123, https://doi.org/10.1016/s0195-5616(91)50011-5 (1991).
Uilenberg, G. Babesia-a historical overview. Veterinary parasitology 138, 3–10, https://doi.org/10.1016/j.vetpar.2006.01.035 (2006).
Friedhoff, K. T. & Soule, C. An account on equine babesioses. Revue scientifique et technique (International Office of Epizootics) 15, 1191–1201, https://doi.org/10.20506/rst.15.3.972 (1996).
Jongejan, F. & Uilenberg, G. The global importance of ticks. Parasitology 129(Suppl), S3–14, https://doi.org/10.1017/s0031182004005967 (2004).
Ribeiro, A. J., Cardoso, L., Maia, J. M., Coutinho, T. & Cotovio, M. Prevalence of Theileria equi, Babesia caballi, and Anaplasma phagocytophilum in horses from the north of Portugal. Parasitology research 112, 2611–2617, https://doi.org/10.1007/s00436-013-3429-9 (2013).
Poschl, B., Waneesorn, J., Thekisoe, O., Chutipongvivate, S. & Karanis, P. Comparative diagnosis of malaria infections by microscopy, nested PCR, and LAMP in northern Thailand. The American journal of tropical medicine and hygiene 83, 56–60, https://doi.org/10.4269/ajtmh.2010.09-0630 (2010).
Grause, J. F. et al. Efficacy of imidocarb dipropionate in eliminating Theileria equi from experimentally infected horses. Veterinary journal (London, England: 1997) 196, 541–546, https://doi.org/10.1016/j.tvjl.2012.10.025 (2013).
Vieira, M. I. B. et al. Serological detection and molecular characterization of piroplasmids in equids in Brazil. Acta tropica 179, 81–87, https://doi.org/10.1016/j.actatropica.2017.12.028 (2018).
Awinda, P. O. et al. Serum antibodies from a subset of horses positive for Babesia caballi by competitive enzyme-linked immunosorbent assay demonstrate a protein recognition pattern that is not consistent with infection. Clinical and vaccine immunology: CVI 20, 1752–1757, https://doi.org/10.1128/cvi.00479-13 (2013).
Knowles, D. P. Jr., Kappmeyer, L. S., Stiller, D., Hennager, S. G. & Perryman, L. E. Antibody to a recombinant merozoite protein epitope identifies horses infected with Babesia equi. Journal of clinical microbiology 30, 3122–3126 (1992).
Wise, L. N. et al. Verification of post-chemotherapeutic clearance of Theileria equi through concordance of nested PCR and immunoblot. Ticks and tick-borne diseases 9, 135–140, https://doi.org/10.1016/j.ttbdis.2017.08.007 (2018).
Bashiruddin, J. B., Camma, C. & Rebelo, E. Molecular detection of Babesia equi and Babesia caballi in horse blood by PCR amplification of part of the 16S rRNA gene. Veterinary parasitology 84, 75–83, https://doi.org/10.1016/s0304-4017(99)00049-7 (1999).
Ruegg, S. R., Torgerson, P., Deplazes, P. & Mathis, A. Age-dependent dynamics of Theileria equi and Babesia caballi infections in southwest Mongolia based on IFAT and/or PCR prevalence data from domestic horses and ticks. Parasitology 134, 939–947, https://doi.org/10.1017/s0031182007002405 (2007).
Alhassan, A. et al. Development of a single-round and multiplex PCR method for the simultaneous detection of Babesia caballi and Babesia equi in horse blood. Veterinary parasitology 129, 43–49, https://doi.org/10.1016/j.vetpar.2004.12.018 (2005).
Nicolaiewsky, T. B. et al. Detection of Babesia equi (Laveran, 1901) by nested polymerase chain reaction. Veterinary parasitology 101, 9–21, https://doi.org/10.1016/s0304-4017(01)00471-x (2001).
Battsetseg, B. et al. Detection of natural infection of Boophilus microplus with Babesia equi and Babesia caballi in Brazilian horses using nested polymerase chain reaction. Veterinary parasitology 107, 351–357, https://doi.org/10.1016/s0304-4017(02)00131-0 (2002).
Schwint, O. N., Knowles, D. P., Ueti, M. W., Kappmeyer, L. S. & Scoles, G. A. Transmission of Babesia caballi by Dermacentor nitens (Acari: Ixodidae) is restricted to one generation in the absence of alimentary reinfection on a susceptible equine host. Journal of medical entomology 45, 1152–1155, https://doi.org/10.1603/0022-2585(2008)45[1152:tobcbd]2.0.co;2 (2008).
Baldani et al. In vitro culture, PCR, and nested PCR for the detection of Theileria equi in horses submitted to exercise. Arquivo Brasileiro de Medicina Veterinária e Zootecnia, https://doi.org/10.1590/s0102-09352008000300005 (2008).
Wang, J. et al. The first molecular detection and genetic diversity of Babesia caballi and Theileria equi in horses of Gansu province, China. Ticks and tick-borne diseases 10, 528–532, https://doi.org/10.1016/j.ttbdis.2019.01.003 (2019).
Montes Cortes, M. G., Fernandez-Garcia, J. L. & Habela Martinez-Estellez, M. A. A multinested PCR for detection of the equine piroplasmids Babesia caballi and Theileria equi. Ticks and tick-borne diseases 10, 305–313, https://doi.org/10.1016/j.ttbdis.2018.11.008 (2019).
Alanazi, A. D., Said, A. E., Morin-Adeline, V., Alyousif, M. S. & Slapeta, J. Quantitative PCR detection of Theileria equi using laboratory workflows to detect asymptomatic persistently infected horses. Veterinary parasitology 206, 138–145, https://doi.org/10.1016/j.vetpar.2014.09.019 (2014).
Kizilarslan, F., Yildirim, A., Duzlu, O., Inci, A. & Ciloglu, A. Molecular Detection and Characterization of Theileria equi and Babesia caballi in Horses (Equus ferus caballus) in Turkey. Journal of Equine Veterinary Science 35, 830–835 (2015).
Lobanov, V. A., Peckle, M., Massard, C. L., Brad Scandrett, W. & Gajadhar, A. A. Development and validation of a duplex real-time PCR assay for the diagnosis of equine piroplasmosis. Parasites & vectors 11, 125, https://doi.org/10.1186/s13071-018-2751-6 (2018).
Bhoora, R. et al. Sequence heterogeneity in the equi merozoite antigen gene (ema-1) of Theileria equi and development of an ema-1-specific TaqMan MGB assay for the detection of T. equi. Veterinary parasitology 172, 33–45, https://doi.org/10.1016/j.vetpar.2010.04.025 (2010).
Bhoora, R. et al. Development and evaluation of real-time PCR assays for the quantitative detection of Babesia caballi and Theileria equi infections in horses from South Africa. Veterinary parasitology 168, 201–211, https://doi.org/10.1016/j.vetpar.2009.11.011 (2010).
Kim, J., Lim, J. & Lee, C. Quantitative real-time PCR approaches for microbial community studies in wastewater treatment systems: Applications and considerations. Biotechnology Advances 31, 1358–1373 (2013).
Heim, A. et al. Detection and molecular characterization of Babesia caballi and Theileria equi isolates from endemic areas of Brazil. Parasitology research 102, 63–68, https://doi.org/10.1007/s00436-007-0726-1 (2007).
Xie, J., Liu, G., Tian, Z. & Luo, J. Development of loop-mediated isothermal amplification (LAMP) for detection of Theileria equi. Acta tropica 127, 245–250, https://doi.org/10.1016/j.actatropica.2013.05.007 (2013).
Bilgiç, H. B., Karagenc, T., Bakirci, S., Eren, H. & Weir, W. Loop mediated isothermal amplification (LAMP) of theileria annulata DNA. Ankara Universitesi Veteriner Fakultesi Dergisi 64, 211–221, https://doi.org/10.1501/Vetfak_0000002801 (2017).
Alhassan, A. et al. Development of loop-mediated isothermal amplification (LAMP) method for diagnosis of equine piroplasmosis. Veterinary parasitology 143, 155–160, https://doi.org/10.1016/j.vetpar.2006.08.014 (2007).
Alhassan, A. et al. Comparative evaluation of the sensitivity of LAMP, PCR and in vitro culture methods for the diagnosis of equine piroplasmosis. Parasitology research 100, 1165–1168, https://doi.org/10.1007/s00436-006-0430-6 (2007).
Daher, R. K., Stewart, G., Boissinot, M. & Bergeron, M. G. Recombinase Polymerase Amplification for Diagnostic Applications. Clinical chemistry 62, 947–958, https://doi.org/10.1373/clinchem.2015.245829 (2016).
Castellanos-Gonzalez, A., White, A. C. Jr., Melby, P. & Travi, B. Molecular diagnosis of protozoan parasites by Recombinase Polymerase Amplification. Acta tropica 182, 4–11, https://doi.org/10.1016/j.actatropica.2018.02.002 (2018).
Yin, F. et al. Rapid diagnosis of Theileria annulata by recombinase polymerase amplification combined with a lateral flow strip (LF-RPA) in epidemic regions. Veterinary parasitology 237, 125–129, https://doi.org/10.1016/j.vetpar.2017.02.019 (2017).
Cui, J. et al. Detection of Babesia gibsoni in dogs by combining recombinase polymerase amplification (RPA) with lateral flow (LF) dipstick. Parasitology research 117, 3945–3951, https://doi.org/10.1007/s00436-018-6104-3 (2018).
Li, J., Macdonald, J. & von Stetten, F. Review: a comprehensive summary of a decade development of the recombinase polymerase amplification. Analyst 144, 31–67, https://doi.org/10.1039/c8an01621f (2019).
Lei, R. et al. Rapid detection of the pathogenic fungi causing blackleg of Brassica napus using a portable real-time fluorescence detector. Food chemistry 288, 57–67, https://doi.org/10.1016/j.foodchem.2019.02.089 (2019).
Li, T. T. et al. Rapid and Visual Detection of Trichinella Spp. Using a Lateral Flow Strip-Based Recombinase Polymerase Amplification (LF-RPA) Assay. Front. Cell. Infect. Microbiol. 9, https://doi.org/10.3389/fcimb.2019.00001 (2019).
Wu, L., Ye, L. T., Wang, Z. R., Cui, Y. Y. & Wang, J. Y. Utilization of recombinase polymerase amplification combined with a lateral flow strip for detection of Perkinsus beihaiensis in the oyster Crassostrea hongkongensis. Parasites Vectors 12, https://doi.org/10.1186/s13071-019-3624-3 (2019).
Lai, M. Y., Ooi, C. H. & Lau, Y. L. Recombinase Polymerase Amplification Combined with a Lateral Flow Strip for the Detection of Plasmodium knowlesi. Am. J. Trop. Med. Hyg. 98, 700–703, https://doi.org/10.4269/ajtmh.17-0738 (2018).
Cabada, M. M. et al. Recombinase Polymerase Amplification Compared to Real-Time Polymerase Chain Reaction Test for the Detection of Fasciola hepatica in Human Stool. Am. J. Trop. Med. Hyg. 96, 341–346, https://doi.org/10.4269/ajtmh.16-0601 (2017).
Sun, K. et al. Recombinase polymerase amplification combined with a lateral flow dipstick for rapid and visual detection of Schistosoma japonicum. Parasites Vectors 9, https://doi.org/10.1186/s13071-016-1745-5 (2016).
Rosser, A., Rollinson, D., Forrest, M. & Webster, B. L. Isothermal Recombinase Polymerase amplification (RPA) of Schistosoma haematobium DNA and oligochromatographic lateral flow detection. Parasites Vectors 8, https://doi.org/10.1186/s13071-015-1055-3 (2015).
Damasceno, J. D. et al. Functional compartmentalization of Rad9 and Hus1 reveals diverse assembly of the 9-1-1 complex components during the DNA damage response in Leishmania. Mol. Microbiol. 101, 1054–1068, https://doi.org/10.1111/mmi.13441 (2016).
Crannell, Z. et al. Multiplexed Recombinase Polymerase Amplification Assay To Detect Intestinal Protozoa. Anal. Chem. 88, 1610–1616, https://doi.org/10.1021/acs.analchem.5b03267 (2016).
Crannell, Z. A. et al. Recombinase Polymerase Amplification-Based Assay to Diagnose Giardia in Stool Samples. Am. J. Trop. Med. Hyg. 92, 583–587, https://doi.org/10.4269/ajtmh.14-0593 (2015).
Kersting, S., Rausch, V., Bier, F. F. & von Nickisch-Rosenegk, M. Rapid detection of Plasmodium falciparum with isothermal recombinase polymerase amplification and lateral flow analysis. Malar. J. 13, https://doi.org/10.1186/1475-2875-13-99 (2014).
Peckle, M. et al. Molecular epidemiology of Theileria equi in horses and their association with possible tick vectors in the state of Rio de Janeiro, Brazil. Parasitology research 112, 2017–2025, https://doi.org/10.1007/s00436-013-3360-0 (2013).
Criado-Fornelio, A., Martinez-Marcos, A., Buling-Sarana, A. & Barba-Carretero, J. C. Molecular studies on Babesia, Theileria and Hepatozoon in southern Europe. Part II. Phylogenetic analysis and evolutionary history. Veterinary parasitology 114, 173–194, https://doi.org/10.1016/s0304-4017(03)00141-9 (2003).
Braga, M. et al. Genetic diversity of piroplasmids species in equids from island of Sao Luis, northeastern Brazil. Revista brasileira de parasitologia veterinaria = Brazilian journal of veterinary parasitology: Orgao Oficial do Colegio Brasileiro de Parasitologia Veterinaria 26, 331–339, https://doi.org/10.1590/s1984-29612017046 (2017).
Kim, C. M. et al. Diagnostic real-time PCR assay for the quantitative detection of Theileria equi from equine blood samples. Veterinary parasitology 151, 158–163, https://doi.org/10.1016/j.vetpar.2007.10.023 (2008).
Yang, Y. et al. Development of real-time and lateral flow strip reverse transcription recombinase polymerase Amplification assays for rapid detection of peste des petits ruminants virus. Virology journal 14, 24, https://doi.org/10.1186/s12985-017-0688-6 (2017).
Crannell, Z. A., Rohrman, B. & Richards-Kortum, R. Quantification of HIV-1 DNA using real-time recombinase polymerase amplification. Analytical chemistry 86, 5615–5619, https://doi.org/10.1021/ac5011298 (2014).
Acknowledgements
Rong Lei and Xinyi Wang contributed equally to this work. This work was supported by the National Key R&D Program of China (2017YFD0501200), CAMS Innovation Fund for Medical Sciences (CIFMS) (2019-I2M-5-042) and Department of Science & Technology of Liaoning Province (No. 2019-MS-270).
Author information
Authors and Affiliations
Contributions
The study was designed by N.J., R.L. and X.Y.W. performed the experiment. D.Z., Y.Z.L., N.J. and Q.J.C. helped to analyze the results. Figure 1 was drawn by Di Zhang. The manuscript was written by R.L., X.Y.W. and N.J.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Lei, R., Wang, X., Zhang, D. et al. Rapid isothermal duplex real-time recombinase polymerase amplification (RPA) assay for the diagnosis of equine piroplasmosis. Sci Rep 10, 4096 (2020). https://doi.org/10.1038/s41598-020-60997-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-60997-1
This article is cited by
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
