
Abstract
We investigated effects of the ryanodine receptor (RyR) modulator caffeine on Na+ current (INa) activation and inactivation in intact loose-patch clamped murine skeletal muscle fibres subject to a double pulse procedure. INa activation was examined using 10-ms depolarising, V1, steps to varying voltages 0–80 mV positive to resting membrane potential. The dependence of the subsequent, INa inactivation on V1 was examined by superimposed, V2, steps to a fixed depolarising voltage. Current-voltage activation and inactivation curves indicated that adding 0.5 and 2 mM caffeine prior to establishing the patch seal respectively produced decreased (within 1 min) and increased (after ~2 min) peak INa followed by its recovery to pretreatment levels (after ~40 and ~30 min respectively). These changes accompanied negative shifts in the voltage dependence of INa inactivation (within 10 min) and subsequent superimposed positive activation and inactivation shifts, following 0.5 mM caffeine challenge. In contrast, 2 mM caffeine elicited delayed negative shifts in both activation and inactivation. These effects were abrogated if caffeine was added after establishing the patch seal or with RyR block by 10 μM dantrolene. These effects precisely paralleled previous reports of persistently (~10 min) increased cytosolic [Ca2+] with 0.5 mM, and an early peak rapidly succeeded by persistently reduced [Ca2+] likely reflecting gradual RyR inactivation with ≥1.0 mM caffeine. The latter findings suggested inhibitory effects of even resting cytosolic [Ca2+] on INa. They suggest potentially physiologically significant negative feedback regulation of RyR activity on Nav1.4 properties through increased or decreased local cytosolic [Ca2+], Ca2+-calmodulin and FKBP12.
Similar content being viewed by others


Introduction
Skeletal muscle excitation-contraction coupling involves triggering of a Nav1.4 mediated action potential. This is detected by voltage-dependent transverse tubular Cav1.1 that undergoes transitions resulting in an allosterically-activated ryanodine receptor (RyR) mediated release of sarcoplasmic reticular (SR) Ca2+ thereby increasing cytosolic free Ca2+ concentration, [Ca2+]i, Recent findings have been suggestive of feedback mechanisms through which downstream events in this feedforward sequence might conversely affect Nav1.4 function. However, many of these reports provided structural rather than functional evidence, or involved experiments in cell lines or isolated cultured cells. They thus provide necessary rather than sufficient evidence for the actual operation of Ca2+ mediated feedback mechanisms in working muscle in vivo. Few available reports examined the existence and properties of RyR-mediated feedback actions on Nav1.4 function in intact in situ skeletal myocytes particularly those directly targeting the RyR mediated Ca2+ release process.
First, Nav1.4 possesses several potential Ca2+ and calmodulin (CaM) binding or modulation sites. These include: (a) one or more Ca2+-binding C-terminal EF-like hand motifs1; (b) a C-terminal isoleucine-glutamine (IQ) domain that could bind CaM following combination of Ca2+ with CaM’s own EF-hand motifs2; (c) a III-IV loop Ca2+/CaM-binding site3; (d) sites phosphorylatable by CaM kinase II (CaMKII) themselves regulated by Ca2+/CaM4 and (e) a phosphorylation site for phosphokinase C (PKC)5.
Secondly, single-cell patch-clamp studies reported that both rapid photo-release of caged Ca2+, raising [Ca2+]i to ~2 µM, and Ca2+ overspill from neighbouring Ca2+ channels, reduced peak Na+ currents, INa, in Nav1.4-transfected HEK293 cells and skeletal muscle cell lines. These effects were abrogated by intracellular BAPTA6. Additionally, CaM expression in the transgenic cells negatively shifted the voltage-dependences of Nav1.4 inactivation. Both the reduction of peak INa, and this negative shift in voltage dependence, were abolished by mutations in either the Ca2+-binding-EF hands on CaM or the Nav1.4 C-terminal IQ domain2,6,7,8. These results implicated CaM binding to the IQ domain in Nav1.4 modulation by [Ca2+]i.
Thirdly, increases in free intracellular concentrations of FK506 binding protein, [FKBP12]i resulting from reductions in its affinity for RyR subunits associated with RyR opening9,10, positively shifted activation and inactivation voltage dependences in cardiomyocyte Nav1.511, thought closely homologous to Nav1.44. This likely involves an indirect effect; co-immunoprecipitation assays appear to exclude direct FKBP12–Nav1.5 interactions11.
However, fewer reports are available on such [Ca2+]i-dependent Nav1.4 modulation in intact in situ skeletal myocytes. Loose-patch clamp studies reported that carbonyl cyanide-3-chlorophenylhydrazone12 and 8-(4-chlorophenylthio)adenosine-3′, 5′-cyclic monophosphate challenge13 reduced INa. These effects were reversed by the Ca2+ buffer BAPTA and RyR blocker dantrolene respectively. However, these agents affected [Ca2+]i indirectly through modifying mitochondrial Ca2+ release or altering RyR activity through CAMKII-dependent pathways via Exchange protein directly activated by cAMP (Epac).
We here test the hypothesis that the direct RyR-SR Ca2+ channel modulator caffeine14 modifies INa in intact mammalian skeletal muscle fibres. We utilised experimental protocols previously shown to accomplish contrasting effects of 0.5 and >1.0 mM caffeine challenge on spectrofluometrically measured resting skeletal myocyte [Ca2+]i15,16,17. Thus, in isolated rabbit skeletal SR or murine skeletal muscle fibre preparations, caffeine (0.5 mM) caused increased [Ca2+]i (to ~300 nM) from resting levels (typically ~106 ± 2 nM in rat gastrocnemius and soleus muscle fibres18). This was persistent over the subsequent 3–10 min intervals investigated15,16. In contrast, ≥1.0 mM caffeine induced early [Ca2+]i peaks followed by reductions to persistently below-resting [Ca2+]i within 80–90 sec16,17. The latter was attributed to a slow RyR inactivation reducing channel open probabilities taking place over seconds. This property was previously demonstrated in sheep and rabbit skeletal muscle RyRs reconstituted in lipid bilayers under levels of steady [Ca2+]i that would initially produce RyR activation (~10–100 µM)19.
The present experiments correspondingly investigated effects of 0.5 mM and 2.0 mM caffeine on INa in intact murine skeletal muscle using the loose-patch clamp approach. This employed relatively low-resistance seals (<2 MΩ) with an undisrupted surface membrane thus leaving intracellular, particularly [Ca2+]i homeostasis unperturbed. In contrast, conventional tight-patch seals often include the Ca2+-sequestrating ethylene glycol-bis(βaminoethyl ether)-N, N, N’, N’-tetraacetic acid (EGTA) in intracellular pipette solutions. In addition, the loose-patch pipettes could be re-used between patches, permitting sequential and multiple recordings from the same cell before and after pharmacological challenge. This permitted standardized comparisons between successive results with the same pipette20. Test results were compared with effects of control challenges using the known specific RyR blocker dantrolene21 to identify caffeine effects specifically attributable to its direct RyR action.
Results
Currents obtained using the double pulse protocol
The experiments employed a loose-patch clamp preparation and recording layout fully described in the Materials and Methods section (Fig. 1). Voltage-dependences of Na+ current, INa, including its activation and inactivation, were examined using a double test pulse protocol (Fig. 1D). In each sweep of the full protocol, the membrane was first held at resting membrane potential (RMP) for 5 ms. A hyperpolarising pre-pulse, duration 50 ms, imposed at times 5–55 ms from the beginning of the sweep to voltage V0 = (RMP–40) mV, first removed any residual INa inactivation at the RMP. This standardized the proportion of activatable Na+ channels prior to application of the subsequent depolarising V1 pulse, itself of duration 10 ms, at time 55–65 ms.

(A) Plan view of the muscle preparation after being pinned down at both ends in the bath. The soleus muscle (in grey) is overlaid by gastrocnemius. The shaded box in the centre represents the region of gastrocnemius fibres used for patch clamping. (B) Loose patch clamp experiment: Muscle preparation pinned down in KH solution. Pipette mounted at 45° to allow 90° contact of the bent tip with the fibre surface. The pipette is connected to a suction syringe13. (C) Equivalent circuit of loose patch clamp electrode on muscle membrane. Solution in the pipette clamped at Vpip. The voltage error arising from the flow of currents through the patch resistance (Rpatch) and the cell resistance (Rcell), along with the pipette resistance (Rpip) corrected mainly using a bridge circuit in the loose patch clamp amplifier13. (D) Complete set of pulse protocols carried out on a patch being investigated. This double test pulse protocol first superimposed a −40 mV hyperpolarising pre-pulse, lasting 50 ms, on the resting potential, RMP. This was followed by a depolarising V1 pulse varying in amplitude to test voltages between RMP and (RMP + 80) mV in +5 mV increments. This was followed by a V2 pulse to a strongly depolarised potential of (RMP + 80) mV. (E) The corresponding current trace during a single sweep in the protocol under control conditions where V1 = (RMP + 35) mV, following P/4 protocol correction, giving peak currents I1 and I2.
At early intervals immediately following solution change, a simplified protocol examined INa in individual patches. This averaged results from 5 sweeps of a pulse protocol imposing a single voltage step to a single voltage V1 at (RMP + 80) mV between 55 and 80 ms. INa features at later time intervals following solution change were studied using the full procedure. This examined effects of a family of V1 amplitudes varied between sweeps in +5 mV increments through test voltages between RMP and (RMP + 80) mV. This elicited currents reflecting the voltage dependence of INa activation. A further V2 pulse was then made to a constant, depolarised voltage of (RMP + 80) mV. This would activate all the Na+ channels within the patch not inactivated by the preceding V1 pulse. This made it possible to explore the voltage dependence of Na+ current inactivation. For each value of V1, currents from 10 sweeps were averaged before examining the next value of V1. Each complete protocol applied to a single patch therefore involved collection of currents from 170 sweeps.
Figure 1E exemplifies currents obtained by a single sweep at a single voltage, V1 = (RMP + 35) mV following P/4 correction under control conditions. Downward, negative deflections denote inward while upward, positive deflections represent outward membrane currents. The V0 pulse typically yielded no currents. The V1 and associated V2 steps each elicited transient inward currents representing the opening and inactivation of Na+ channels. The magnitudes of their peaks, i1 and i2, were measured relative to the currents measured just before the respective V1 and V2 steps.
Each patch was only subjected to a single application of the complete set of pulse protocols. This made differences between results arising from prolonged changes in the patch such as membrane blebbing unlikely22. Furthermore, following every set of sweeps a further bracketing protocol was then imposed using a single V1 step to RMP. The resulting average i2 value from the succeeding V2 step was then compared with the i2 value obtained by the first 10 sweeps of the preceding test protocol, for which V1 was also RMP. Patch stability was confirmed, permitting further analysis, if the bracketing i2 value was within 10% of the test i2 value. Thus, only patches with a stable seal, INa generation, and activation and inactivation characteristics were selected for further analysis.
Early effects of caffeine on Na+ currents
The initial experiments explored for alterations in INa at the earliest times (1–4 min) following introduction of caffeine. The simplified protocol employing a single V1 test voltage of (RMP + 80) mV permitted prompt assessments of INa at times immediately following solution change. This made it additionally possible to explore the effects of using differing patch-clamping protocols either excluding or permitting caffeine access to membrane beneath the patch seal. All these manoeuvres first obtained control currents in the absence of caffeine (Fig. 2(A–D)(i)). The INa records showed increasing followed by decaying phases, conforming to previously established expectations of a voltage-dependent activation followed by more gradual inactivation of INa. In a first procedure, the patch pipette was then withdrawn, 0.5 mM caffeine was introduced, and the patch seal re-established 30 sec later. This procedure permitted caffeine access to membrane both beyond and within the patch seal, prior to further applications of the pulse procedures (Fig. 2A(i)). The challenge with 0.5 caffeine then produced reductions in INa from the first minute of exposure (Fig. 2A(ii)).

Results of simple pulse protocols to examine INa at early times following caffeine challenge using two experimental procedures. Following obtaining control records prior to caffeine challenge (A–D) panel (i), in (A), the patch electrode was withdrawn prior to introduction of 0.5 mM caffeine, and then restored prior to imposition of test steps (ii) in the following 1–3 min. In (B) the patch seal was maintained before, during and following introduction of 0.5 mM caffeine through imposition of the test steps (ii). The latter procedure was repeated following pipette withdrawal and restoration of the patch seal (iii). (C,D) adopted identical respective procedures as (A,B) using a 2 mM caffeine concentration.
A second procedure on a different patch employing a differing procedure yielded contrasting results. Following acquisition of control records (Fig. 2B(i)), 0.5 mM caffeine-containing extracellular solution was introduced whilst the pipette seal was maintained intact. Currents were similarly obtained at timed subsequent intervals. Here, the patch seal was accordingly continually maintained before, during and following introduction of caffeine-containing extracellular solution. This gave caffeine immediate access to membrane beyond but not within the patch seal. Challenge by 0.5 mM caffeine now produced little subsequent, early change in peak currents even with repeated INa determinations made at 1 min intervals between 1 to 4 minutes (Fig. 2B(ii)). In contrast, the effect of 0.5 mM caffeine in reducing INa could be finally restored in subsequent recordings made after lifting the patch pipette and restoring the patch seal 30 s later (Fig. 2B(iii)).
Separate experiments using 2.0 mM caffeine challenge gave concordant, complementary, results. Relative to the initial recordings in the absence of caffeine (Fig. 2C(i)), 2.0 mM caffeine initially produced a reduced INa, but this was rapidly succeeded by increased INa if the latter test recordings were made after lifting then restoring the patch seal (Fig. 2C(ii)). These effects were attenuated when the 2.0 mM caffeine was added then test recordings made whilst contrastingly maintaining an intact patch seal (Fig. 2D(i,ii)).
These findings suggested that both 0.5 and 2 mM caffeine reduced INa within the first minute of exposure. This reduction persisted over 1, 2 and 3 min with 0.5 mM caffeine, but was succeeded by a marked increase in INa with 2 mM caffeine. However, such effects required a direct access of the caffeine to the area of membrane beneath the patch pipette in which INa was recorded. The subsequent explorations of caffeine effects on INa using the full pulse procedure at the later times accordingly challenged with caffeine prior to formation of the patch seal.
Effects on INa magnitudes of 0.5 mM caffeine challenge
The INa recordings showed patterns that paralleled the previously reported contrasting [Ca2+]i changes produced by 0.5 and 2.0 mM caffeine concentrations. Figure 3 illustrates current records from families of voltage steps at differing V1 between RMP and (RMP + 80) mV under control conditions (A) and at different times following introduction of 0.5 mM caffeine (B). Initial rates of rise of current increased non-linearly with degree of depolarization elicited by the V1 steps. So did the peak INa which then reached a maximum at V1 ≈ (RMP + 75) mV before decreasing with further depolarisation. The subsequent V2 steps yielded currents whose peak INa decreased with increasingly positive V1 consistent with prior voltage-dependent channel inactivation reducing the proportion of remaining activatable Na+ channels. The pulse protocols were repeated on different patches over successive time intervals following the introduction of caffeine. Figure 3B((i–iv)) illustrates typical families of traces obtained from 8, 20, 37 and 45 min following introduction of caffeine. Comparison of the successive sets of records suggested that peak INa initially showed marked, ~80%, reductions within the 10-minute interval following challenge (Fig. 3B(i–ii)). This was followed by a recovery which then returned INa to pre-treatment levels over a >40 min time-course (Fig. 3B(iii–iv)).

Families of normalized current traces obtained from the double pulse protocol in response to caffeine challenge at a concentration of 0.5 mM in the absence (A,B) or presence (C,D) of 10 μM dantrolene. (A,C) Control traces obtained in KH solution, prior to caffeine challenge in the absence (A) or presence of dantrolene (C). (B,D) Families of records obtained at successive (i) 7–8 min, (ii) 20–21 min, (iii) 35–37 min and (iv) 45–46 min intervals after addition of 0.5 mM caffeine-containing KH solution into the bath in the absence (B) or presence of dantrolene (D).
Changes in the voltage dependence of INa resulting from 0.5 mM caffeine challenge
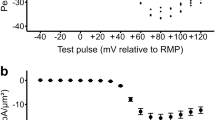
Findings of this kind yielded INa current-voltage activation (I-V) and inactivation curves plotting dependences of peak INa on V1 in response to the respective V1 and V2 voltage steps. These are plotted for families of records obtained at different intervals following onset of caffeine challenge (Fig. 4). These curves demonstrated that compared to pre-treatment controls (Fig. 4A(i)), INa was reduced at 1–10, 11–25 and 26–40 min (Fig. 4A(ii–iv) respectively). However INa had recovered by the final (41–55 min) time interval (Fig. 4A(iv)). The maximum values of peak INa could be determined within the voltage range examined in the I-V curves obtained in the controls at 1–10 and 41–55, but not 11–25 and 26–40 min. Accordingly, the same determinations were made under control conditions and at 11–25, and 26–40 minutes with a modified pulse protocol (Fig. 5) that explored an increased V1 range extending to (RMP + 120) mV and using a V2 step to (RMP + 100) mV. This encompassed the V1 at which maximum peak INa was observed, both under the control conditions (Fig. 5(i)) and at the selected times following addition of caffeine (Fig. 5(ii,iii)). Together the I-V plots suggested that the greatest peak INa occurred with depolarisations to (RMP + 105) mV at 11–25 min and at (RMP + 95) mV at 26–40 min respectively.

Activation (left panels) and inactivation I-V curves (right panels of each pair) (mean of I1 or I2 ± standard error of mean (SEM) against each V1 excursion) in response to caffeine challenge at a concentration of 0.5 mM in the absence (A) or the presence (B) of 10 μM dantrolene. Experiments first obtained families of INa records before addition of caffeine in (A(i)), (B(i)). Families of INa records then obtained in intervals (ii) 1–10 minutes, (iii) 11–25 minutes (iv) 26–40 minutes and (v) 41–55 min intervals after adding 0.5 mM caffeine.

Activation and inactivation I-V curves (mean of I1 or I2 ±standard error of mean (SEM) against each V1 excursion) for the (i) control condition prior to addition of caffeine, and (ii), 11–25 minutes and (iii) 26–40 minutes after addition of 0.5 mM caffeine using an expanded range of V1 in the absence of dantrolene.
Figure 6A plots values of these parameters in successive patches with time following application of caffeine. Tables 1A and 2A list these parameters as means ± SEM from explorations grouped in successive time intervals as adopted in Fig. 3. This made it possible to test for statistically significant differences from the control findings obtained before addition of 0.5 mM caffeine. These demonstrated that within 1–10 min after adding caffeine, there was a sharp reduction in Imax. In addition, there was a negative shift in the V* value describing INa inactivation. In contrast, V* values for INa activation and k values for both activation and inactivation then remained unchanged. There then followed marked positive shifts in the V* of both INa activation and INa inactivation. The final INa recovery to pretreatment values, was then accompanied by more gradual negative shifts in the voltage dependence of both INa activation and INa inactivation, with control Imax, and V* values regained at 26–40 and 41–55 min respectively. These changes in Imax and V* contrasted with the constant k values through these manoeuvres. Furthermore, they were abolished, as described fully below, by dantrolene-mediated RyR block.

Alterations in (i) Imax, (ii) V* and (iii) k derived from current-voltage (I-V) activation and inactivation curves followed from successive patches studied at different times before (t < 0 min) and following addition of 0.5 mM caffeine in the absence (A) and presence (B) of 10 μM dantrolene.
Changes in magnitude and voltage dependence of INa resulting from 2 mM caffeine challenge
Caffeine challenge at a 2 mM concentration gave contrasting INa changes. Following the initially decreased peak INa within 2 min described above (Fig. 2C), the pattern assumed a transient increase in INa followed by its recovery to control levels. Figure 7 shows families of INa records obtained using the full pulse protocol as in Fig. 3 at the different times subsequent to 2.0 mM caffeine challenge. The current waveforms increased to a peak INa followed by an inactivating decline (Fig. 7A). However, the families of currents demonstrated markedly greater INa at 2–15 min following exposure to 2 mM caffeine, in contrast to the reduction in INa produced by 0.5 mM caffeine. INa then progressively declined to pre-treatment levels over the succeeding time intervals (Fig. 7B(i–iii)). Figure 8A plots the resulting I-V and inactivation curves obtained at different time intervals, from which it was possible to obtain Imax, V* and k values for the I-V and inactivation curves. Figure 9 plots these values for successive patches with time following application of caffeine and Tables 3A and 4A summarise the statistical analysis of their means ± SEM from explorations in the successive time intervals, comparing these against the control pretreatment results.

Families of normalized current traces obtained from the double pulse protocol in response to caffeine challenge at a concentration of 2 mM in the absence (A,B) or the presence (C,D) of 10 μM dantrolene. (A,C) Control traces obtained in KH solution, prior to caffeine challenge. (B,D) Families of records obtained at successive (i) 3 or 5 min, (ii) 19 or 21 min and (iii) 29 or 31 min after addition of 2 mM caffeine-containing KH solution into the bath in (B) the absence or (D) presence of dantrolene.

Activation (left panels) and inactivation I-V curves (right panels) (mean of I1 or I2 ± standard error of mean (SEM)) against each V1 excursion before (i) and following (ii) introduction of 2 mM caffeine in the absence (A) or the presence (B) of 10 μM dantrolene. Families of INa records were obtained 2–15 min (ii), 16–25 min (iii), and 26–40 min (iv) after adding 2 mM caffeine.

Alterations in (i) Imax, (ii) V* and (iii) k derived from current-voltage (I-V) activation (left panels) and inactivation (right panels) followed from successive patches studied at different times before (t < 0 min) and following addition of 2 mM caffeine in the absence (A) and presence (B) of 10 μM dantrolene.
The INa amplitudes, Imax, increased threefold over the 2–15 min following addition of caffeine when compared to its control values (Compare Fig. 8A(i,ii)). Imax then recovered in subsequent intervals to regain pre-treatment values at 26–40 minutes (Fig. 8A(iii,iv)). There were delayed negative shifts in the V* of both INa activation and INa inactivation at 16–25 min followed by smaller positive shifts at 26–40 min ((Fig. 9A(ii)); Tables 3A and 4A). Again, in common with findings using 0.5 mM caffeine, values of k showed relatively little change through these protocols.
Effect of dantrolene on the INa responses to 0.5 and 2.0 mM caffeine
The final sequence of experiments examined the effects of the specific RyR inhibitor dantrolene both by itself and on these divergent actions of 0.5 and 2.0 mM caffeine on INa using otherwise identical pulse protocols. Each experiment first obtained families of INa traces in the absence of all test agents in two successive patches studied within a first 10 min interval. The bath solution was then substituted for one containing 10 µM dantrolene and 2 further patches investigated within the next 10 minutes. The final INa records were then obtained after further replacing the bath solution with one containing both 0.5 or 2.0 mM caffeine and 10 µM dantrolene-containing solution. Patches were investigated using the same full pulse protocols as shown in Fig. 1.
Including dantrolene alone slightly increased peak INa (Figs. 3C and 7C). Analysis of the I-V relationships (Figs. 4B and 8B) accordingly demonstrated small but significant increases in activation and inactivation Imax compared to controls, but no effect on activation and inactivation V* and k values (Tables 5 and 6).
Simultaneous addition of dantrolene and 0.5 mM (Fig. 3C,D) or 2.0 mM caffeine (Fig. 7C,D) (Tables 1–4) resulted in currents whose magnitudes were similar to those obtained in the presence of dantrolene alone, at all the time intervals following the addition of caffeine. Dantrolene thus abrogated both the effects of 0.5 mM caffeine in producing transient decreases (Fig. 3D(i–iv)), and the effects of 2 mM caffeine in producing transient increases in INa (Fig. 7D(i–iii)). In parallel with the previous test experiments, I-V and inactivation relationships were plotted for fibres studied in the presence of dantrolene and either 0.5 mM (Figs. 4B) or 2.0 mM caffeine (Fig. 8B) through the same time intervals as those used after challenge with 0.5 mM or 2.0 mM caffeine alone, respectively.
Figures 6B and 9B plot the resulting Imax, V* and k values for the I-V and inactivation curves for successive patches with time before, and following addition of either 0.5 (Figs. 6B) or 2 mM caffeine (Fig. 9B) in successive individual patches exposed to dantrolene. None of these parameters showed the contrasting pattern of time-dependent changes in activation and inactivation parameters shown with 0.5 and 2 mM caffeine challenge in the absence of dantrolene. Tables 1–4 summarise the statistical analysis of their means ± SEM from explorations in the successive time intervals comparing these (B) against the control pretreatment results (A). In both analyses the activation and inactivation Imax, V* and k values were respectively statistically indistinguishable from those obtained under dantrolene challenge alone when caffeine was absent. Again values of k remained constant through the experiments.
Neither challenge by dantrolene, nor that by caffeine, whether by itself or in combination with dantrolene, noticeably affected INa kinetics. When quantified by times to peak current at voltages yielding maximum INa in the I-V curves, control experimental groups subsequently used to test effects of dantrolene alone, or 0.5 or 2 mM caffeine in the absence of or following further dantrolene challenge, gave times of 0.49 ± 0.010, 0.48 ± 0.0093 and 0.50 ± 0.012 ms respectively. Dunnett multiple comparison tests demonstrated that none of the subsequent interventions produced significantly alterations for all the time intervals assessed in Tables 1–4 (all P ≫ 0.05).
These control experiments thus demonstrate that (a) the RyR inhibitor dantrolene abrogated the contrasting actions of either 0.5 mM or 2.0 mM caffeine in transiently decreasing or increasing INa with accompanying changes in V* to previously established altered RyR-mediated changes in [Ca2+]. (b) Their demonstration of an increased INa in the presence of dantrolene alone complements the demonstrations of increased INa produced by 2 mM caffeine in suggesting some background inhibition of INa at even background resting [Ca2+]i.
Discussion
The present experiments explored for the existence of physiologically effective feedback loops between ryanodine receptor (RyR) activation by tubular depolarisation and Nav1.4 channel function that would thereby modify surface membrane excitability in skeletal muscle in vivo. This mechanism would complement feedforward, excitation-contraction-coupling, processes by which Nav1.4-mediated surface membrane depolarisation triggers RyR-mediated release of intracellularly stored sarcoplasmic reticular (SR) Ca2+. Feedback alterations in Nav1.4 availability and/or voltage sensitivity are potentially physiologically important particularly under conditions of intense and prolonged muscle activation including tetanic stimulation and metabolic exhaustion. The latter changes could compromise SR Ca2+-ATPase activity through ATP depletion, thereby elevating [Ca2+]i. Any consequent effects of Ca2+ on Nav1.4 function would complement its other actions promoting slow RyR inactivation19 and increasing sarcolemmal Ca2+-activated large conductance K+ channel (KCa1.1) conductance in downregulating excitation-contraction coupling23. Such feedback mechanisms would also bear on clinical conditions affecting skeletal muscle excitability. K+-aggravated myotonia is associated with slowed INa kinetics and impaired INa inactivation that accompany Nav1.4 mutations in its EF-hand-like domain24. Other genetic, K+ and cold-aggravated myotonias are associated with reduced Ca2+-dependent Na+ channel inhibition similarly attributed to Nav1.4 mutations6. Finally, the weakness observed in dystrophic muscle is associated with elevated [Ca2+]i25.
The experiments here examined effects of direct pharmacological manipulation of RyR-mediated SR Ca2+ release by the RyR agonist caffeine14 on both Nav1.4 channel availability and its voltage-dependent activation and inactivation properties in intact skeletal myocytes. They utilised previously established contrasting effects of different applied caffeine concentrations on spectrofluometrically measured resting [Ca2+]i15,16. Low concentrations of caffeine (0.5 mM) then increased [Ca2+]i persistently over the subsequent 3–10 min recording intervals in isolated rabbit skeletal SR or murine skeletal muscle fibre preparations15. In contrast, ≥1.0 mM caffeine induced early [Ca2+]i peaks rapidly followed by [Ca2+]i decreases below even resting-levels within 80–90 sec16,17.
Here, Na+ currents, INa were measured in intact mammalian gastrocnemius skeletal muscle using the loose patch clamp technique. This avoided membrane disruption and perturbations of [Ca2+]i homeostasis inherent in other experimental measurement procedures involving conventional cell-attached patch clamp studies, which additionally often include the Ca2+-sequestrating EGTA in the pipette solutions. Furthermore, the latter can require study of isolated or cultured cells with enzymatically treated membranes as opposed to the intact in situ skeletal myocytes examined here. The loose-patch configuration also permitted sequential and multiple recordings with the same standardized pipette in successive patches before and after pharmacological challenge as required in the present experiments26. The features of the resulting INa concurred with those obtained on earlier occasions26 and with other voltage clamp methods applied to mammalian skeletal muscle27. The magnitude, voltage-dependence and quantified activation (current-voltage, I-V) and inactivation properties of INa were compared at different times following introducing caffeine. Further experiments blocking these effects with further applications of the specific RyR inhibitor dantrolene attributed the observed changes specifically to altered RyR-mediated SR Ca2+ release.
The observed contrasting alterations in INa closely paralleled the previously reported [Ca2+]i changes following caffeine challenge. Thus, INa was reduced early (1–2 min) following either 0.5 or 2 mM caffeine challenge. This effect was abrogated by reducing access of caffeine to tubular and therefore tubular-sarcoplasmic reticular triad regions in which RyRs are concentrated, in regions within the patch from which the recordings were obtained. The latter was accomplished by establishing patch seals before rather than after introduction of caffeine to the extracellular space. This would alter direct access of caffeine over a surface membrane area close to the 30 μm2 cross sectional area of the electrode tip in contact with the membrane within which INa was recorded.
Subsequent recordings following the 0.5 and 2 mM caffeine challenge obtained by identical pulse protocols respectively demonstrated contrasting increases and decreases in peak INa followed by their recovery to pretreatment levels. These findings corroborate and extend previous independent reports that INa was reduced by increased RyR activity following 8-CPT induced Epac pathway activation13. The INa changes were quantified by systematically exploring a consistent range of V1 test voltages thereby deriving families of INa records. These in turn yielded activation, I-V, and inactivation curves giving Imax, V* and k values. These respectively quantified total available INa and the position and steepness of their voltage dependences. Sustained reductions in Imax induced by 0.5 mM caffeine paralleled the sustained [Ca2+]i increases previously reported following caffeine mediated activation of RyR-mediated SR Ca2+ release15. The increased INa observed in the presence of 2 mM caffeine similarly paralleled reported contrastingly decreased [Ca2+]i attributed to slow RyR inactivation16,17. The latter findings further suggested significant inhibition of INa by even normal background [Ca2+]i.
These alterations in total available INa were accompanied by time-dependent changes in properties of the I-V and inactivation curves. With 0.5 mM caffeine challenge, the initial decreases in INa were accompanied by negative shifts in inactivation V* over 1–10 min. This was followed, over 11–25 min, by positive shifts in both INa activation and INa inactivation V*. The final INa recovery to pretreatment values between 26–55 min was accompanied by opposite, negative, shifts in activation and inactivation V*. With 2 mM caffeine challenge, the contrasting, declining phases of the transient INa increases accompanied delayed transient negative shifts in both activation and inactivation V*. In contrast, values of k of either activation or inactivation little changed throughout these protocols.
Finally, the effects of the specific RyR inhibitor dantrolene were investigated both by itself and in combination with identical caffeine concentrations and pulse protocols. Dantrolene specifically stabilises RyR closed states by selectively decreasing Ca2+ affinities of RyR activation sites21. Previous reports had demonstrated that it inhibited caffeine-induced increments in mammalian SR Ca2+ permeability28 and caffeine-induced contractures in murine diaphragm muscle29.
In the present experiments dantrolene abrogated all the effects of either 0.5 or 2 mM caffeine, whether on Imax, or activation or inactivation INa or V*. In contrast, dantrolene by itself produced only small increases in Imax without affecting activation and inactivation, V* and k values. Together with the increased INa produced by 2 mM caffeine this latter finding suggests that [Ca2+]i exerts significantly inhibits INa even at background RyR activity. These explorations using dantrolene thus trace all the present actions of caffeine on INa specifically to its effects on RyR-mediated SR Ca2+ release.
These findings together demonstrate and characterise RyR-Nav1.4 feedback phenomena complementing their established feedforward interactions underlying excitation contraction coupling (Fig. 10A). The observed contrasting effects of caffeine challenge on Imax and V* combined with previously reported [Ca2+]i changes under similar experimental conditions15,16,17 were incompatible with straightforward combined negative shifts in Na+ channel activation and inactivation V* resulting from simple screening effects arising from an increased [Ca2+]i30,31. However, they directly correlate with previous in vitro biochemical and biophysical reports on skeletal muscle Nav1.4 and RyR. They establish for these a physiological context, possibly occurring within a common microdomain, in working muscle, as opposed to in vitro systems for the first time.

Correlation between observed ryanodine receptor (RyR) agonist and antagonist actions and (A) Feedforward Nav1.5 mediated depolarisation triggering DHPR-RyR coupling causing SR Ca2+ release that is (B) inhibited by RyR-antagonist dantrolene, and previous studies reporting: (C,D) Challenge by (C) 0.5 mM caffeine increasing RyR-mediated Ca2+ release and FKBP12 dissociation reducing Nav1.4 availability thereby modifying Nav1.4 properties and (D) 2 mM caffeine, where Ca2+ release causes sustained RyR inactivation with net SR Ca2+ uptake, reduced FKBP12 dissociation, and contrasting Nav1.4 effects. Blue arrows: fluxes; green and red dotted arrows: excitatory and inhibitory actions.
First, the early INa reductions with 0.5 and 2 mM caffeine, persistent with 0.5 mM caffeine, were consistent with the reported effects of either direct Ca2+-binding to a Nav1.4 EF hand site or indirect Ca2+ binding to Nav1.4 via its binding to CaM in reducing INa (Fig. 10C). Secondly, the accompanying early negative V* shifts 1–10 min following 0.5 mM caffeine challenge paralleled reported negative shifts in voltage-dependent inactivation produced by the indirect Ca2+ binding process2,6,7,8. This was selective for Nav1.4 as opposed to Nav1.5, reflecting Nav1.4 lacking the CaM N-lobe binding site present in Nav1.51. Thirdly, the subsequent (11–25 min) positive shifts in INa activation and inactivation V* directly paralleled reported reductions in FKPB12 binding affinity for RyR following RyR activation. An increased free [FKBP12]i would then accompany the resulting SR Ca2+ release elevating [Ca2+]i. in intact fibres9,10; FKBP12 is known to positively shift V* activation and inactivation in Nav1.511, thought closely homologous to Nav1.44. This action is probably indirect and delayed: co-immunoprecipitation assays appeared to exclude direct FKBP12–Nav1.5 interactions11. Fourthly, negative shifts in both INa activation and inactivation V* values eventually accompanied the reductions in INa and recoveries from INa elevation, 26–55 or 16–40 min following 0.5 and 2.0 mM caffeine challenge respectively. These were compatible with a subsequent, previously reported, slow RyR inactivation19. This would reduce RyR open channel probabilities and Ca2+ release whilst permitting continued SR Ca2+ re-uptake and possible surface membrane Ca2+ expulsion by Na+/Ca2+ exchange32,33. These would reverse the increases in [Ca2+]i and [FKBP12]i19 (Fig. 10D). Finally, the effects of the RyR inhibitor dantrolene (Fig. 10B) abrogating all these contrasting reductions and increases in INa associated with different caffeine concentrations through the identical pulse protocols associated all these findings with feedback effects arising specifically from the RyR.
Materials and Methods
Experiments were performed under the Animals (Scientific Procedures) Act 1986 Amendment Regulations 2012 following ethical review by the University of Cambridge Animal Welfare and Ethical Review Body (AWERB). C57BL6 WT mice aged 3–6 months were used in all experiments. Mice were housed at a stable temperature of 21 °C in a licensed facility, fed sterile rodent chow (RM3 Maintenance Diet; SDS, Witham, United Kingdom) and subjected to 12-hour light/dark cycles with free access to water, bedding and environmental stimuli. Krebs–Henseleit (KH) solution (mmol/L: NaCl, 130; KCl, 4.0; HEPES, 1.2; MgCl2, 1.0; CaCl2, 1.8; glucose, 10; and Na-pyruvate, 2.0; pH adjusted to 7.4) was used in all experiments. Chemical agents were purchased from Sigma-Aldrich (Poole, UK) unless otherwise stated. The following variants of the basic KH solution containing caffeine and/or dantrolene including 0.1% dimethyl sulfoxide vehicle were prepared and used in experiments: (1) KH + 0.5 mM caffeine, (2) KH + 2.0 mM caffeine, (3) KH + 10 µM dantrolene, (4) KH + 10 µM dantrolene + 0.5 mM caffeine, and (5) KH + 10 µM dantrolene + 2.0 mM caffeine. Solutions were filtered to remove particles greater than 10 μm in diameter with standard filtration paper (Millipore, Bedford, MA, USA).
Mice were killed by cervical dislocation by Home Office-licensed personnel in accordance with Schedule 1 of The Animals (Scientific Procedures) Act 1986. The gastrocnemius and soleus muscles were then immediately isolated, with the distal (Achilles) tendon secured by a knot to a ligature and the proximal tendon still attached to its bony origin. Overlying connective tissue was excised to allow access to the myocyte surface. The isolated muscles were then immediately placed in a Sylgard-bottomed experimental bath containing KH solution with the soleus muscle facing down maximizing electrode access to gastrocnemius fibres. Experiments were carried out at stable temperatures around 20 °C. The muscle was secured in a taut and fixed position by pinning down the bony origin and the ligature with A1 insect pins (Fig. 1A).
Loose patch clamp pipettes were pulled from borosilicate glass capillaries (GC150‐10; Harvard Apparatus, Cambridge, UK) using a Brown–Flaming microelectrode puller (Model P‐97; Sutter Instrument Co. Novato, CA). They were mounted under a microscope with ×250 magnification with a calibrated eyepiece graticule, and a small indentation made on one side of the pipette using a diamond knife. A transverse force was then applied distal to the indentation, giving a fracture perpendicular to the long axis of the pipette. Squarely-broken tips were then fire‐polished under visual guidance at ×400 magnification using an electrically-heated nichrome filament to smooth the edges of the tip. The internal diameters of the pipettes were measured under ×1000 magnification. Pipettes of internal tip diameters between 28–38 µm were used in the experiments. Such loose patch recordings thus provide a complete representation of the properties of both surface and tubular membrane directly beneath these seals. Finally, the pipettes were bent at about 1 mm from the tip to make a 45° angle with the pipette’s long axis. This allowed the tips to contact the muscle surface at 90° when mounted at an angle of 45° below the horizontal on the recording amplifier head stage (Fig. 1B). The pipettes were mounted onto a pipette holder with an Ag/AgCl electrode held within the inside of the pipette. The distal half of the pipette was filled with solution from the bath, using suction provided by a syringe via an air-filled connection to the pipette holder. The bath was actively grounded at reference potential, using an Ag/AgCl electrode. These Ag/AgCl electrodes maintained electrical connection between the organ bath and pipette with the electronic circuit.
The pipette was lowered onto the gastrocnemius muscle membrane surface and gentle suction applied to allow seal formation around the resulting membrane patch. The central regions of the muscle (crosshatched region in scheme shown in Fig. 1A) were targeted for each experiment as this region was the most horizontal, permitting optimal (~90°) contact between pipette tip and muscle. The loose patch clamp configuration clamps the voltage on the extracellular side of the membrane within the pipette seal. Therefore, positive and negative potentials applied within the pipette respectively hyperpolarise and depolarise the membrane potential of the patch relative to the cell resting membrane potential (RMP). In this report, the signs of the voltage steps are inverted and membrane potentials expressed relative to RMP, a convention adopted by previous papers using this technique12,13,20,26. The Ag/AgCl electrode within the pipette both measured the intra-pipette potential relative to the ground reference potential of the bath and passed the clamp current.
Figure 1C shows the equivalent circuit of a typical patch. The loose patch technique involves a relatively low seal resistance (typically < 2 MΩ). This ohmic leakage and series resistance error [(seal resistance + patch resistance) and pipette resistance], along with additional pipette capacitance currents were largely corrected by a bridge circuit within the loose patch clamp amplifier26. The remaining linear leakage and capacitive currents were corrected using a P/4 protocol in which the test steps were followed by four control voltage steps a quarter the amplitude and of opposite signs to the test step. These were of a sign and/or amplitude that did not activate the voltage conductances, hence only eliciting leak currents. These responses were measured, summed and subtracted from the current values obtained from the original test step within the loose patch clamp26.
An IBM-compatible computer connected to the loose patch clamp amplifier, which was connected to the headstage that held the recording pipette was used to deliver voltage clamp steps relative to the RMP. Study of each patch commenced with strongly depolarising +80 mV, 15 ms pulses in order to confirm the presence of ion channels. Only patches showing clear, resolvable inward currents with kinetics characteristic of voltage-gated Na+ channels were investigated with the full set of pulse protocols. The patch clamp currents were sampled by the computer at a 50 kHz digital sampling rate and filtered with a DC-10 kHz bandwidth, using a 10 kHz Bessel low pass filter.
Experiments were initially performed with the muscle preparation immersed in control KH solution. Around 2–3 different patches were investigated using the complete set of pulses within 20 minutes after transferring the muscle preparation into the bath. Only muscles that gave stable control results were further studied in the presence of pharmacological challenge. For each pharmacological condition, data from patches collected from at least 3 muscle preparations were used. To achieve conditions comparable to previous studies on [Ca2+]i, the bathing solution was then rapidly replaced with KH solution containing either 0.5 mM and 2.0 mM caffeine. The resulting effects on Na+ current amplitude, and activation and inactivation characteristics were then examined at increasing times from a sequence of different patches following onset of caffeine challenge. Averaged currents from the 10 sweeps obtained for each pulse voltage within the experimental protocol were then normalised to the pipette tip cross-sectional area to give current densities, using the formula relating current density (in pA/µm2), measured current (in nA) and pipette diameter (in µm):
The resulting peak I1 and I2 current densities obtained from the V1 and V2 steps in the double pulse protocols generated activation and inactivation current-voltage (I-V) curves, respectively, for each patch. The rising phase of the activation I-V curve for each patch was fitted to a two-state Boltzmann function relating the peak current density for any V1 excursion (I1) to its maximum value (Imax), the voltage at half maximal peak current (V*), the excursion corresponding to the V1 step (V) and the steepness factor (k): I1 = Imax/[1 + exp{(V* − V)/k}]). The inactivation I-V curves were fitted to a similar Boltzmann function relating the peak current density I2 for the V2 step following any given V1 excursion to its maximum value (Imax), the voltage at half maximal current (V*), the excursion of the V1 step (V) and a steepness factor (k): I2 = Imax(1 − 1/[1 + exp{(V* − V)/k}]).
Successive families of ionic currents revealed alterations in current characteristics with time following caffeine challenge. These are represented by displaying currents, current-voltage curves and computed values of Imax, V* and k as obtained from each patch averaged to give means ± SEM for currents obtained within successive time intervals following introduction of pharmacological agents. Curve fitting procedures of the I-V curves used fitting algorithms in the open source programme SciDAVis (Version 1.25-mac). Derived parameters obtained under different conditions were tested for significant differences to a P < 0.05 significance level by ANOVA with a post-hoc Dunnett’s test.
Ethical approval
This research has been regulated under the Animals (Scientific Procedures) Act 1986 Amendment Regulations 2012 following ethical review and approval by the University of Cambridge Animal Welfare and Ethical Review Body (AWERB). All procedures were completed by Home Office-licensed personnel and fell within the scope of Schedule 1 of the UK Animals (Scientific Procedures) Act (1986).
Data availability
The datasets generated during and/or analysed during the current study are available from the corresponding author on reasonable request.
References
Yoder, J. B. et al. Ca 2+ -dependent regulation of sodium channels Na V 1.4 and Na V 1.5 is controlled by the post-IQ motif. Nat. Commun. 10, 1514, https://doi.org/10.1038/s41467-019-09570-7 (2019).
Young, K. A. & Caldwell, J. H. Modulation of skeletal and cardiac voltage-gated sodium channels by calmodulin. J. Physiol. 565, 349–70 (2005).
Potet, F. et al. Functional Interactions between Distinct Sodium Channel Cytoplasmic Domains through the Action of Calmodulin. J. Biol. Chem. 284, 8846–54 (2009).
Loussouarn, G. et al. Physiological and pathophysiological insights of Nav1.4 and Nav1.5 comparison. Front. Pharmacol. 6, 314, https://doi.org/10.3389/fphar.2015.00314 (2016).
Bendahhou, S., Cummins, T. R., Potts, J. F., Tong, J. & Agnew, W. S. Serine-1321-independent regulation of the mu 1 adult skeletal muscle Na+ channel by protein kinase C. Proc. Natl. Acad. Sci. USA 92, 12003–12007 (1995).
Ben-Johny, M. et al. Conservation of Ca2+/calmodulin regulation across Na and Ca 2+ channels. Cell 157, 1657–1670 (2014).
Biswas, S. et al. Calmodulin regulation of Nav1.4 current: role of binding to the carboxyl terminus. J. Gen. Physiol. 131, 197–209 (2008).
Deschênes, I. et al. Isoform-Specific Modulation of Voltage-Gated Na + Channels by Calmodulin. Circ. Res. 90, E49–E57 (2002).
Jayaraman, T. et al. FK506 binding protein associated with the calcium release channel (ryanodine receptor). J. Biol. Chem. 267, 9474–9477 (1992).
Jones, J., Reynolds, D., Lai, F. & Blayney, L. Ryanodine receptor binding to FKBP12 is modulated by channel activation state. J. Cell Sci. 118, 4613–4619 (2005).
Maruyama, M. et al. FKBP12 is a critical regulator of the heart rhythm and the cardiac voltage-gated sodium current in mice. Circ. Res. 108, 1042–52 (2011).
Filatov, G. N., Pinter, M. J. & Rich, M. M. Role of Ca(2+) in injury-induced changes in sodium current in rat skeletal muscle. Am. J. Physiol. Cell Physiol. 297, C352–C359 (2009).
Matthews, H. R. et al. Sodium current inhibition following stimulation of exchange protein directly activated by cyclic-3′, 5′-adenosine monophosphate (Epac) in murine skeletal muscle. Sci. Rep. 9, 1927, https://doi.org/10.1038/s41598-018-36386-0. (2019).
Herrmann-Frank, A., Lüttgau, H. C. & Stephenson, D. G. Caffeine and excitation-contraction coupling in skeletal muscle: A stimulating story. Journal of Muscle Research and Cell Motility 20, 223–237 (1999).
Fryer, M. W. & Neering, I. R. Actions of caffeine on fast- and slow- twitch muscles of the rat. J. Physiol. 416, 435–454 (1989).
Dettbarn, C., Gyorke, S. & Palade, P. Many agonists induce ‘quantal’ Ca2+ release or adaptive behavior in muscle ryanodine receptors. Mol Pharmacol 46, 502–7 (1994).
Pagala, M. K. & Taylor, S. R. Imaging caffeine-induced Ca2+ transients in individual fast-twitch and slow-twitch rat skeletal muscle fibers. Am. J. Physiol. 274, C623–32 (1998).
Head, S. I. Membrane potential, resting calcium and calcium transients in isolated muscle fibres from normal and dystrophic mice. J. Physiol. 469, 11–19 (1993).
Laver, D. R. & Curtis, B. A. Response of ryanodine receptor channels to Ca 2+ steps produced by rapid solution exchange. Biophys. J. 71, 732–741 (1996).
Almers, W., Roberts, W. M. & Ruff, R. L. Voltage clamp of rat and human skeletal muscle: measurements with an improved loose- patch technique. J. Physiol. 347, 751–768 (1984).
Zhao, F., Li, P., Chen, S., Louis, C. & Fruen, B. Dantrolene inhibition of ryanodine receptor Ca2+ release channels. Molecular mechanism and isoform selectivity. J. Biol. Chem. 276, 13810–13816 (2001).
Milton, R. L. & Caldwell, J. H. Na current in membrane blebs: implications for channel mobility and patch clamp recording. J. Neurosci. 10, 885–893 (1990).
Fitts, R. H. Cellular mechanisms of muscle fatigue. Physiol. Rev. 74, 49–94 (1994).
Kubota, T. et al. New mutation of the Na channel in the severe form of potassium- aggravated myotonia. Muscle Nerve 39, 666–673 (2009).
Lamb, G. D., Junankar, P. R. & Stephenson, D. G. Raised intracellular [Ca2+] abolishes excitation-contraction coupling in skeletal muscle fibres of rat and toad. J. Physiol. 489, 349–362 (1995).
Almers, W., Stanfield, P. R. & Stühmer, W. Lateral distribution of sodium and potassium channels in frog skeletal muscle: measurements with a patch-clamp technique. J. Physiol. 336, 261–84 (1983).
Adrian, R. H. & Marshall, M. W. Sodium currents in mammalian muscle. J. Physiol. 268, 223–250 (1977).
Ohnishi, S. T. Effects of halothane, caffeine, dantrolene and tetracaine on the calcium permeability of skeletal sarcoplasmic reticulum of malignant hyperthermic pigs. BBA - Biomembr. 897, 261–268 (1987).
Okwuasaba, F., Ejike, C. & Parry, O. Comparison of the skeletal muscle relaxant properties of Portulaca oleracea extracts with dantrolene sodium and methoxyverapamil. J. Ethnopharmacol. 20, 85–106 (1987).
Hille, B. Charges and potentials at the nerve surface. Divalent ions and pH. J. Gen. Physiol. 51, 221–236 (1968).
Hille, B., Woodhull, A. M. & Shapiro, B. I. Negative surface charge near sodium channels of nerve: divalent ions, monovalent ions, and pH. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 270, 301–318 (1975).
Balnave, C. D. & Allen, D. G. Evidence for Na+/Ca2+ exchange in intact single skeletal muscle fibers from the mouse. Am. J. Physiol. Cell Physiol. 274, C940–C946 (1998).
Hoya, A. & Venosa, R. A. Characteristics of Na(+)- Ca2+ exchange in frog skeletal muscle. J. Physiol. 486, 615–627 (1995).
Acknowledgements
We thank the Medical Research Council (MR/M001288/1), Wellcome Trust (105727/Z/14/Z), and British Heart Foundation (PG/14/79/31102 and PG/15/12/31280) for their generous support. We also thank Mr Chris Cardinal, Ms. Julie Gautrey and Ms. Vicky Johnson for technical assistance.
Author information
Authors and Affiliations
Contributions
H.R.M. designed the techniques and conceived and built the apparatus, S.S.S.-S., C.L.H.-H. and H.R.M. performed the experiments. C.L.-H.H. and H.R.M. supervised experimental execution. C.L.-H.H. conceived and supervised the project and wrote the paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Sarbjit-Singh, S.S., Matthews, H.R. & Huang, C.LH. Ryanodine receptor modulation by caffeine challenge modifies Na+ current properties in intact murine skeletal muscle fibres. Sci Rep 10, 2199 (2020). https://doi.org/10.1038/s41598-020-59196-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-59196-9
This article is cited by
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
