
Abstract
Human papillomavirus (HPV) is the most common sexually transmitted infection in the world and the main cause of cervical cancer. Nowadays, the virus-like particles (VLPs) based on L1 proteins have been considered as the best candidate for vaccine development against HPV infections. Two commercial HPV (Gardasil and Cervarix) are available. These HPV VLP vaccines induce genotype-limited protection. The major impediments such as economic barriers especially gaps in financing obstructed the optimal delivery of vaccines in developing countries. Thus, many efforts are underway to develop the next generation of vaccines against other types of high-risk HPV. In this study, we developed DNA constructs (based on L1 and L2 genes) that were potentially immunogenic and highly conserved among the high-risk HPV types. The framework of analysis include (1) B-cell epitope mapping, (2) T-cell epitope mapping (i.e., CD4+ and CD8+ T cells), (3) allergenicity assessment, (4) tap transport and proteasomal cleavage, (5) population coverage, (6) global and template-based docking, and (7) data collection, analysis, and design of the L1 and L2 DNA constructs. Our data indicated the 8-epitope candidates for helper T-cell and CTL in L1 and L2 sequences. For the L1 and L2 constructs, combination of these peptides in a single universal vaccine could involve all world population by the rate of 95.55% and 96.33%, respectively. In vitro studies showed high expression rates of multiepitope L1 (~57.86%) and L2 (~68.42%) DNA constructs in HEK-293T cells. Moreover, in vivo studies indicated that the combination of L1 and L2 DNA constructs without any adjuvant or delivery system induced effective immune responses, and protected mice against C3 tumor cells (the percentage of tumor-free mice: ~66.67%). Thus, the designed L1 and L2 DNA constructs would represent promising applications for HPV vaccine development.
Similar content being viewed by others


Introduction
Human papillomavirus (HPV) is the most common sexually transmitted infection in the world and the main cause of cervical cancer. Globally, 4.5% of all cancers worldwide (60,000 cases per year in men and 570,000 cases per year in women) are attributable to HPV1. More than 150 viral types of HPV have been identified whose classification is based on their association with cervical cancer and precursor lesions. HPV types were classified as high-risk (16, 18, 31, 33, 35, 39, 45, 51, 52, 56, 58, 59, and 68) and low-risk (6, 11, 40, 42, 43, 44, and 54) types consistent with the generation of squamous cell carcinomas in the uterine cervix2. The papillomavirus double-stranded circular DNA genome encodes roughly eight open-reading frames (ORFs), which is responsible for viral replication, maintenance, and cell transformation. These gene products have been classified into six nonstructural regulatory proteins (E1-E6) and two structural capsid proteins (L1 and L2)3. Nowadays, the virus-like particles (VLPs) based on L1 proteins have been considered as the best candidate for vaccine development against HPV infections. Two commercial HPV vaccines are both prophylactic. Gardasil which protects against HPV genotypes 6, 11, 16 and 18, and Cervarix which protects against HPV genotypes 16 and 18; Both of them prevent cervical cancer with almost 100% efficacy4. These HPV VLP vaccines confer protection against limited genotypes5. Although a variable level of cross-protection has also been observed against phylogenetically related HPV genotypes6, major impediments such as economic barriers especially gaps in financing obstructed the optimal delivery of vaccines in developing countries. This might be approached via locally produced generic vaccines. Storage and transportation, the so-called cold chain, is another hindrance, which should be mitigated through lyophilization or protectants, and also it should be noted that many recombinant vaccines rely on multiple immunizations; however, promising results could be obtained with a single dose and certainly 2-dose regimens7.
Recently, a possible approach to broader cross-type protective immunity at a lower cost is to consider L2-based vaccination compared to L1 VLP immunization8. Indeed, the current HPV L2 vaccines improved a type-specific protection. Recent studies showed that some regions in the N-terminus of L2 can neutralize antibodies generated during various types of HPV infections9. To overcome the intrinsically low immunogenicity of the recombinant L2 protein, its potency could be increased by various formulations such as the multivalent L2 epitopes (peptide vaccine)10,11,12, fusion with L1 and other immunogenic proteins13,14,15 and multiepitope DNA-based vaccines16,17.
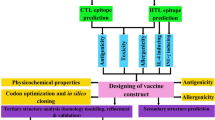
As a major field of science, bioinformatics has brought together the concepts of in silico analyses of biological queries, mathematics and statistics18. Immunoinformatics tools could help researchers to screen multiple HPV genome and predict high immunogenic epitopes, which provide a T or B cell response against HPV infection19,20,21. In this study, the combination of in silico/in vivo approaches was used to evaluate L1 and L2 proteins of high-risk HPV types (16, 18, 31, 33, 35, 39, 45, 51, 52, 56, 58, 59, and 68), and to design a pan genotype L1 and L2 constructs for development of DNA-based vaccines.
Results
Protein conservancy analysis
To select conserved epitopes between HPV subtypes, L1 and L2 protein sequences were aligned using muscle algorithms. Based on the degree of the conservancy, five regions of L1 proteins (8–22, 95–132, 307–342, 398–425 and 449–473) and four regions of L2 proteins (11–40, 54–76, 96–120, 278–305) were selected for further immune-bioinformatics analysis such as B- and T-cell epitope prediction. Among them, region 449–473 of HPV-16 L1 protein and region 54–76 of HPV-16 L2 protein had the highest score of conservancy between all high-risk HPV types. In addition, based on sequence variability of conserved regions, the L1 and L2 proteins from two main types of HPV (16 and 18) were selected as a reference for calculation of conservancy by IEDB epitope conservancy analysis tool (Tables 1 and 2).
Prediction of linear B-cell epitopes
B-cell epitopes are recognized by B-cell receptors or antibodies in their native structure. Continuous B-cell epitope prediction is very similar to T-cell epitope prediction, which has mainly been based on the amino acid properties such as hydrophobicity, exposed surface area, charge and secondary structure. At first step, the conserved region sequences were analyzed by BepiPred-2 server to predict potential B-cell epitopes (Table 3). In L1 protein, L18–22 (EATVYLPPVPVSKVV-type16), L1408–421 (PPPGGTLEDTYRFV-type16) and L1404–417 (NFGVPPPPTTSLVD-type 18) epitopes had the best B cell epitope identification scores. For L2 protein, L222–35 (KQSGTCPPDVVPKV-type18), L2100–113 (PSDPSIVSLVEETS-type16), L294–107 (EPVGPTDPSIVTLI-type18) and L257–70 (GLGIGTGSGTGGRT-type16) epitopes showed the highest epitope identification score between their own protein sequences.
Prediction of T-cell epitopes
Since a linear form of T-cell epitopes are bound to MHCs, the interface between T-cells and ligands can be accurately modeled. In this study, we used three different algorithms (published motifs, ANN and quantitative matrix) for MHC-I and two algorithms for MHC-II (ANN and quantitative matrix).
Prediction of MHC-I
At first step, the L1 and L2 conserved regions were analyzed to find the most immunodominant peptides using NetMHCpan 4.0, syfpeithi and ProPred I. In each protein, peptides with the highest binding affinity scores were determined as high-potential CTL epitope candidates (Tables 4 and 5). The analysis showed that L112–21 (YLPPVPVSKV-type 16 and YLPPPSVARV-type 18), L1460–470 (DQFPLGRKFLL-type 16), L1461–471(DQYPLGRKFLV-type 18), L211–20 (KRASATQLYK-type 16 and KRASVTDLYK-type 18), L2280–291 (DPDFLDIVALHR-type 16) and L2273–284 (DSDFMDIIRLHR-type 18) epitopes had the highest binding affinity among their own protein sequences. In general, the results of three different algorithms confirmed each other. Conservancy and allergenicity analyses were done on the selected epitopes. The sequence of all the epitopes were well conserved among high-risk HPV types and none of them were allergens (Tables 4 and 5). In addition, there was no cross-reactivity between peptide and human proteome.
Prediction of MHC-II
In this study, we used NetMHCIIpan and Propred servers for MHC-II epitope identification analysis (Table 6). Since a suitable T-cell epitope should be predicted to bind to different HLA alleles, epitopes with the maximum number of binding HLA-DR alleles were selected as high-potential helper T-cell epitope candidates. Among predicted epitopes, L18–22 (EATVYLPPVPVSKVV-type 16), L195–111 (TQRLVWACVGVEVGRGQ-type 16 and TQRLVWACAGVEIGRGQ-type 18), L1416–430 (DTYRFVTSQAIACQK-type 16), L1417–431 (DTYRFVQSVAITCQK-type 18), L2100–118 (DPSIVTLIEDSSVVTSGAP-type 16), L2281–297 (PDFLDIVALHRPALTSR-type 16) and L2274–290 (SDFMDIIRLHRPALTSR-type 18) had the highest scores of binding affinity. Also, the sequence of all the epitopes were well conserved among high-risk HPV types and none of them were allergen (Tables 6 and 7). Also, there was no cross-reactivity between peptide and human proteome.
Tap transport/proteasomal cleavage
The generation of antigenic peptides and their transport across the membrane of the endoplasmic reticulum for assembly with MHC class I molecules are essential steps in antigen presentation to cytotoxic T lymphocytes. Thus, investigating the proteasomal cleavage, Tap transport and affinity prediction of binding is essential in MHC-1 presentation pathway. The NetCTL2.1 server was used to predict TAP transport efficiency and proteasomal cleavage scores (Table 8). Between all epitopes, L112–21 (YLPPVPVSKV-type 16 and YLPPPSVARV-type 18), L1460–470 (DQFPLGRKFLL-type 16), L1461–471 (DQYPLGRKFLV-type 18), L211–20 (KRASATQLYK-type 16 and KRASVTDLYK-type 18), L2293–303 (DPDFLDIVALHR-type 16) and L2273–284 (DSDFMDIIRLHR-type 18) epitopes had the highest epitope identification scores.
Population coverage analysis
HLA distribution varies among the diverse geographic regions around the world. Thus, while designing an effective vaccine, population coverage must be taken into consideration to cover the maximum possible populations. In this study, population coverage was estimated separately for each putative epitope in 16 specified geographic regions of the world (Tables 9 and 10). For CTL epitopes, the highest population coverage of world’s population was calculated for L112–21 (84.71%), L1411–421 (90.87%), L211–20 (73.89%) and L2280–291 (67.72%). For helper T-cell epitopes, the highest population coverage was calculated 86.18% for L18–22, 91.18% for L1327–342, 98.90% for L1416–430, 83.47% for L2100–118 and 97.68% for L2281–297. Overall, these results indicated that high-potential helper T-cell epitopes and CTL epitopes can specifically bind to the prevalent HLA molecules in the target populations where the vaccine will be employed.
Peptide-protein flexible Docking
Peptides are promising candidates for different types of biological applications such as vaccine design. In recent years, a variety of approaches have been revealed for ‘protein-peptide docking, which is, predicting the structure of the protein-peptide complex, starting from the protein structure and the peptide sequence, including variable degrees of information about the peptide binding site and/or conformation. In this study, two different algorithms (Template-based and global docking) were used to calculate docking scores between MHC allele and peptides. At first, structure data of MHC-I and MHC-II were downloaded from RCSB PDB server (https://www.rcsb.org/). Then, all potential epitopes and MHC PDB files were submitted to the server separately. Top model with the highest cluster density (number of elements divided by average cluster RMSD, obtained from CABS-dock server) and interaction similarity score (similarity of the amino acids of the target complex aligned to the contacting residues in the template structure to the template amino acids, obtained from GalexyPepDock server) were selected for each peptide and its MHC (Tables 11 and 12). The results in each MHC allele might vary but in average scores, similarity score and cluster density confirmed each other. For CTL epitope, L112–21, L1104–115, L1460–470, L211–20, L2280–291, L2273–284 had the highest average docking scores on both servers. For helper T-cell epitope, L18–22, L195–111, L1417–431, L2100–118, L259–49 and L2274–290 had the highest average docking scores on both servers. Figure 1 represents a sample of successful docking model between peptide and MHC protein (successful docking means epitope binding to an MHC molecule through interaction between their R group of side chains and pockets located on the floor of the MHC molecule). Moreover, Table 13 shows MHC allele used for peptide-protein docking.

(A1) Successful peptide-protein Docking between L112–21 (YLPPVPVSKV) and HLA 0301 with cluster density scores of 214.7; (A2) Successful peptide-protein Docking between L112–21 (YLPPVPVSKV) and HLA 0301 with interaction similarity scores of 215; (B1) Successful peptide-protein Docking between L2100–118 and HLA DRB1–0101 with cluster density of 121.06; (B2) Successful peptide-protein Docking between L2100–118 and HLA DRB1–0101 with interaction similarity scores of 146.0.
Construct design
After performing the analysis, top-ranked epitopes were selected according to these parameters: (1) binding affinity between peptide and MHC (for both MHC-I and II alleles), (2) epitope identification scores for T- and B-cells, (3) proteasomal cleavage and tap transport scores, (4) conservancy degree between HPV subtypes, (5) population coverage, and (6) scores of peptide-protein docking. Based on L1 and L2 top-ranked epitopes, two different constructs were designed (Fig. 2). For L1 structure, L112–21 (type 16 & 18), L1460–470 (type 16), L1461–471 (type 18), L18–22 (type 16 & 18), L1416–430 (type 16) and L1417–431 (type 18) epitopes were selected. For L2 structure, L211–20 (type 16 & 18), L2280–291 (type 16), L2273–284 (type 18), L2281–297 (type 16), L2274–290 (type 18) and L254–69 (type 16) epitopes were presented (Table 14). For both structures, two repeats of each epitope were placed together with AAY proteolytic linker (alanine, alanine, and tyrosine). Physicochemical properties of L1 and L2 constructs (molecular weight, instability index, antigenicity, solubility and estimated half time) were summarized in Table 15.

(A) L1 construct, (B) L2 construct, (C) Percentage of population coverage in the combination of peptide candidates in one single universal construct.
Validation of the L1 and L2 DNA constructs
The designed HPV L1 and L2 genes were correctly cloned in pcDNA3.1 and pEGFP-N1 eukaryotic vectors. The presence of L1 and L2 genes were confirmed by digestion as a clear band of ~765 bp and ~700 bp on agarose gel for L1 and L2, respectively (data not shown). The recombinant endotoxin-free plasmids (i.e., pcDNA-L1 and pcDNA-L2) had a concentration range between 1.5 and 3.5 mg/mL.
Evaluation of L1 and L2 DNA expression in HEK-293T cells
In vitro DNA delivery of L1 and L2 into the eukaryotic cell line (HEK-293T) was performed by TurboFect as a transfection reagent. The levels of DNA expression were evaluated using fluorescence microscopy and flow cytometry at 48 h post-transfection. The data indicated that pEGFP-L1 and pEGFP-L2 can effectively penetrate into HEK-293T cells in vitro. The cellular uptake of the L1 and L2 genes into the HEK-293T cells was ~57.86% and ~68.42%, respectively. The delivery of pEGFP-N1 as a positive control was detected in approximately ~92.10% of HEK-293T cells (Fig. 3). Moreover, the spreading green regions were observed for L1 and L2 DNA delivery using TurboFect carrier by fluorescent microscopy in HEK-293T cells. On the other hand, western blot analysis indicated the successful expression of L1 and L2 proteins fused to GFP (i.e., L1-GFP and L2-GFP) using anti-GFP antibody. The data indicated the clear bands of ~52, ~50 and ~27 kDa for L1-GFP, L2-GFP and GFP, respectively using DAB substrate (Fig. 4).

Evaluation of GFP (B), L2-GFP (C) and L1-GFP (D) DNA delivery into HEK-293T non-cancerous cells using TurboFect. Transfection efficiency was monitored by fluorescent microscopy (above) and flow cytometry (bottom) at 48 h post-transfection as compared to the negative control (A).

Identification of protein expression in HEK-293T cells using western blot analysis. The clear bands were observed for L1-GFP (lane 1, ∼52 kDa), L2-GFP (lane 2, ~50 kDa) and GFP (lane 4, ~27 kDa) proteins, respectively. Any clear band was not detected in un-transfected cells as a negative control (lane 3). MW is molecular weight marker (prestained protein ladder, 10–170 kDa, Fermentas).
Measurement of tumor growth
To evaluate the prophylactic effects of the designed L1 and L2 DNA constructs, tumor growth and survival percentage were assessed in all groups for 60 days after challenging with C3 tumor cells. As shown in Fig. 5A, all test groups immunized with DNA constructs (G1, G2 & G3) demonstrated significantly lower tumor growth than that in control groups (PBS and empty vector, G4 & G5, p < 0.05). Our data showed progressive tumor growth in control groups on approximately 7–21 days (survival rate or tumor-free mice percentage: 0%). It was interesting that groups vaccinated with L1 DNA, L2 DNA and L1 + L2 DNA constructs similarly reduced the tumor growth (p > 0.05). As shown in Fig. 5B, group vaccinated with the mixture of L1 + L2 DNA constructs showed a higher survival rate (G3, ~66.67%) than L1 and L2 DNA constructs, alone (G1 & G2, ~33.33%).

Tumor growth curve and survival percentage in different groups: The mice were challenged with 1 × 105 C3 tumor cells two weeks after the last immunization: Tumor volumes were measured twice a week (A), the percentage of tumor-free mice (or survival rate) was evaluated in different groups (B)
Antibody assay
The levels of total immunoglobulin G (IgG), IgG2a and IgG2b in mice immunized with the mixture of L1 + L2 DNA constructs (G3) were significantly higher than other groups (p < 0.05, Fig. 6A,C,D). Moreover, our data showed that the levels of IgG1 were similar in all groups vaccinated with DNA constructs (G1, G2 & G3, p > 0.05, Fig. 6B). There are no significant differences in the secretion of IgG2a and IgG2b isotypes between groups receiving the L1 and L2 DNA constructs, alone (G1 & G2, p > 0.05, Fig. 6C,D). No significant anti-(L1 + L2) antibody responses could be detected in the sera of control groups, thus, the seroreactivities were completely L1 + L2 antigen-specific responses in mice.

Antibody responses against the mixture of L1 + L2 peptides as an antigen in different groups: (A) total IgG, (B) IgG1, (C) IgG2a and (D) IgG2b; Mice sera were prepared from the whole blood samples of each group (n = 6) two weeks after the last immunization. All analyses were performed in duplicate for each sample shown as mean absorbance at 450 nm ± SD.
Cytokine assay
The results of cytokine assay in each group showed that the levels of (L1 + L2)-specific IFN-γ, IL-10 and IL-5 secretions in groups immunized with L1 (G1), L2 (G2) and L1 + L2 (G3) DNA constructs were significantly higher than control groups (p < 0.05, Fig. 7). In contrast, there was no significant difference between mice vaccinated with L1 and L2 DNA constructs, alone (G1 & G2) for secretion of IFN-γ, IL-5 and IL-10 cytokines (p > 0.05). Among all the test groups, the group immunized with the L1 + L2 DNA construct (G3) showed the significant IFN-gamma, IL-5 and IL-10 responses compared to other groups (G1 & G2, p < 0.05, Fig. 7). Furthermore, our data indicated that the ratios of IFN-γ/IL-10 and IFN-γ/IL-5 were higher in all test groups as compared to control groups; therefore, they could trigger Th1 immune response.

The levels of IFN-γ (A), IL-10 (B) and IL-5 (C) in vaccinated groups with different formulations: The pooled splenocytes were prepared from three mice in each group (n = 3 per group) and re-stimulated with the mixture of L1 + L2 peptides in vitro. The levels of cytokines were measured in the supernatant with ELISA as mean absorbance at 450 nm ± SD for each sample. All analyses were performed in duplicate for each sample.
Granzyme B secretion
The secretion of Granzyme B in all test groups was significantly higher than the control groups (p < 0.05, Fig. 8). The group immunized with the L1 + L2 DNA construct (G3) produced significantly higher concentrations of Granzyme B than other groups (G1 & G2, p < 0.001). The level of Granzyme B in group receiving L1 DNA construct was similar to that in group receiving L2 DNA construct (p > 0.05).

Granzyme B concentration measured by ELISA using the pooled splenocytes from three mice in each group (n = 3 per group). All analyses were performed in triplicate for each sample.
Discussion
In recent years, development of bioinformatics tools applied in vaccine researches could potentially save time and resources. Indeed, the immunoinformatics tools help to identify antigenic domains for designing a multi-epitope vaccine. With sequence-based technology advancement, now we have enough information about the genomics and proteomics of different viruses22. Thus, using various bioinformatics tools, we can design peptide vaccines based on a neutralizing epitope. For example, in silico design of an epitope-based vaccine against human immunodeficiency virus23,24, coronavirus25, dengue virus26, and Saint Louis encephalitis virus27 has already been reported.
While around 13 high-risk HPVs were recognized, current vaccines just protect humans from few types. An important limitation of the current vaccines is their narrow coverage. The accessibility of fully sequenced proteome from high-risk HPV strains provides a prospect for in silico screening of reliable peptide-based therapeutic vaccine candidates among billions of possible immunogenic peptides. In silico approaches are intended to reflect the possibilities for overcoming the above-mentioned difficulties in HPV multi-type vaccine. Gupta and coworkers designed prophylactic multiepitopic DNA vaccine using all the consensus epitopic sequences of HPVs L2 capsid protein. They also evaluated how engineering CpG motifs by bioinformatics tools could increase immunogenicity of DNA vaccines28. Hosseini et al. applied in silico analysis of L1 and L2 protein of HPV 11,16,18,31 and 45 types to identify universal peptide vaccine in order to protect against mentioned types29. In 2016, Singh et al. analyzed E1, E2, E6 and E7 proteins of high-risk HPV types to identify CD8+ T-cell epitopes. They suggested a pool of 14 peptides (9 to 43 amino acids) to provide the protection against high-risk HPV types30. Panahi and colleagues used a two-step method (consist of molecular docking and sequence-based approach) to determine immunogenic epitopes for induction of immune system against the oncoproteins of HPV 16, 18, 31 and 45 types31. In 2016, Wang and coworkers suggested the regions 51–58, 87–97, 214–220, 290–296, 335–341, 351–366, 408–418, 430–442 and 475–496 as putative B-cell epitopes for HPV16 L1 protein32. Sabah et al. used in silico immunoinformatics tools and reported a conserved 9 mer epitope (ESTVHEIEL) among all HPV58 types33. Bristo et al. designed MHC-I/MHC-II hybrid ras oncopeptide that could elicit T-cell reponse in an animal model34.
In this research, we designed a framework for the comprehensive analysis of L1 and L2 conserved regions of high-risk HPV types containing both MHC-I and MHC-II epitopes. The framework begins with conservancy analysis of all 13 high-risk HPV strains following with (1) B-cell epitope mapping, (2) T-cell epitope mapping (CD4+ and CD8+), (3) allergenicity assessment, (4) tap transport and proteasomal cleavage, (5) population coverage, (6) global and template-based docking and (7) data collection, analysis, and design of the L1 and L2 DNA constructs. For experimental analysis, the final L1 or L2 DNA constructs were cloned into mammalian expression vector with green fluorescent tag (pEGFP vector) and their expression was evaluated in the eukaryotic cells using flow cytometry, fluorescent microscopy and western blotting. Moreover, the L1/L2-specific antibody and T-cell immune responses induced by L1 and L2 DNA constructs were assessed in mouse tumor model.
At first, L1 and L2 sequences obtained from high-risk HPV types were aligned using MUSCLE algorithms. Conservancy analysis showed that five regions of HPV16,18 L1 protein (8–22, 95–132, 307–342, 398–425 and 449–473) and four regions of HPV16,18 L2 protein (11–40, 54–76, 96–120 and 278–305) were more conserved among other subtypes and could be analyzed as an immunoinformatics input. In B-cell epitope prediction, L18–22, L1408–421, L1404–417, L222–35, L2100–113, L294–107 and L257–70 had the highest epitope prediction scores. Unfortunately, a reliable method for prediction of B-cell epitope has not been revealed up to now and the sensitivity and specificity of existing methods were very low (the specificity and sensitivity of this method were 0.57 and 0.58, respectively). In the case of T-cell epitope prediction, in silico analysis has been significantly improved, thus, the results are more reliable. In this study, for MHC-I epitopes, L112–21 (YLPPVPVSKV-type16 and YLPPPSVARV-type18), L1460–470 (DQFPLGRKFLL-type16), L1461–471 (DQYPLGRKFLV-type18), L211–20 (KRASATQLYK-type16 and KRASVTDLYK-type18), L2280–291 (DPDFLDIVALHR-type16) and L2273–284 (DSDFMDIIRLHR-type18) epitopes had the highest binding affinity scores. In addition, above-mentioned epitopes had the highest T-cell epitope prediction scores which were obtained from proteasomal cleavage and tap transport analysis. High degree of conservancy was observed between subtypes for these epitopes (Table 4) especially in L1460–470 (DQFPLGRKFLL-type16) and L2280–291 (DPDFLDIVALHR-type18).
L1460–470 sequences were identical with HPV 16, 31, 33, 35, 39, 52, 58, 59, 68 types and had high similarity rate with HPV 51, 56, 18 and 45. In addition, L2280–291 sequences had higher degree of conservancy with HPV 16, 31, 33, 35, 51, 52, 58, and 56. For MHC-II prediction, L18–22 (EATVYLPPVPVSKVV-type16), L1416–430 (DTYRFVTSQAIACQK-type16), L1417–431 (DTYRFVQSVAITCQK-type18), L2281–297 (PDFLDIVALHRPALTSR-type16), L2274–290 (SDFMDIIRLHRPALTSR-type18) and L254–69 (FFGGLGIGTGSGTGGR-type16) epitopes had the highest binding affinity scores. Among them, L254–69 had the greatest degree of conservancy (high similarity with all of the high-risk HPV types). One of the remarkable points is that L18–22 and L257–70 epitopes are the same (or overlapping with little difference (among B-cell and MHC-II selected epitopes. Due to a limitation of MHC-peptide binding prediction such as the gap between the peptides that are predicted to bind to MHC and those that experimentally bind35, flexible molecular docking has been employed to address this problem and raise the accuracy of MHC-peptide prediction. In the current study, template-based docking and also global docking were performed on the selected peptides to determine which peptide would get into the groove of MHC with the highest modeling scores. For MHC-I epitope, L112–21, L1460–470 and L2280–291 sequences had the highest interaction similarity and cluster density scores. For MHC-II epitopes, L195–111, L1417–431, L2100–118 and L2281–297 sequences had the highest docking scores. In this study, MHC-I-peptide docking scores confirmed MHC-I-peptide binding affinity scores because the same epitopes had the highest scores in both methods but in MHC-II molecular docking, the results were slightly different. One of the reasons is the significant conformational changes during the process due to the longer epitope length. As a general rule: the longer the length of the query peptide, the more torsions and conformational flexibilities36. Herein, due to longer peptide sequences, docking results in MHC-II were less accurate than MHC-I. For example, average similarity score in MHC-I was variable (171.8–259.7), but in MHC-II was 115.4–136. After the completion of the analysis and according to all of the above-mentioned parameters, two separate constructs were designed. In addition, accumulative population coverage of helper T-cell and CTL epitopes for the designed constructs were estimated. For the L1 and L2 constructs, the combination of 8 epitope candidates for helper T-cell and CTL in a single universal vaccine could involve all world population by the rate of 95.55% and 96.33%, respectively (Fig. 3). In previous studies, YLPPVPVSKV (HPV16 L1)37 and KRASVTDLYK (HPV18 L2)21 have been reported as potentially immunogenic epitopes. The ability of in vitro expression of the designed L1 and L2 DNA constructs was determined in HEK-293T cells using flow cytometry and western blot analysis. The transfection efficiency of the L1 and L2 DNA constructs was ~57.86% and ~68.42%, respectively indicating their high potency for delivery into the eukaryotic cells. As known, the use of a polytope DNA vaccine containing multiple T-cell and B-cell epitopes is an attractive strategy for developing a therapeutic and prophylactic vaccine against HPV infections. After in vitro assay, immunological experiments were performed in mice to determine the efficiency of the designed L1 and L2 DNA constructs without the use of adjuvant or delivery system for vaccine development. Similarly, some studies used the pcDNA vector harboring the gene of interest for immunization without any adjuvant38,39. Our data indicated that the groups immunized with L1, L2 and L1 + L2 DNA constructs increased antibody and T-cell responses as compared to control groups. Furthermore, the (L1 + L2)-specific immunity in mice receiving the mixture of L1 + L2 DNA constructs (G3) resulted in higher secretion of total IgG, IgG2a, IgG2b, IFN-γ, IL-5 and IL-10 cytokines as well as Granzyme B than other groups. The higher levels of IgG2a and IgG2b as well as IFN-gamma (as a Th1 cytokine) in this group drive T-cell responses toward Th1-type immunity. The studies showed that immunoglobulin G1 (IgG1) is related to a Th2-type response, while a Th1 response is associated with the induction of IgG2a and IgG2b in mice40. Regarding to our observations in protective studies, this regimen (L1 + L2 DNA construct: G3) could confer further protection against C3 tumor-challenged mice (survival rate: ~66.67%) depending on stimulation of CD4+ T cell-dominated Th1 responses as well as Granzyme B secretion (indicating CTL activity) as compared to the L1 or L2 DNA constructs, alone (survival rate: ~33.33%). These data showed high potency of the combined L1 + L2 DNA constructs versus each DNA construct alone as a prophylactic HPV vaccine. Taken together, immunoinformatics approaches have been emerged as a critical field for accelerating immunological researches. Yet, the immunoinformatics techniques applied to T-cells have more advancement than those dealing with B-cells30. Moreover, recently, due to the limited options for choosing an adjuvant in clinical trials, bioinformatics analyses have been developed to predict the best adjuvant. In this way, in silico studies help researchers saving time and resources, and also can guide the experimental work with higher probabilities of finding the desired solutions and with fewer trial and error repeats of assays. The accessibility of HPV genomic sequences and functional characterization of the genes involved in the virulence has significantly improved our understanding of the molecular foundation for the pathogenesis of HPV and offered a wealth of data that can be used to design new plans for vaccine design. Nowadays, powerful immune system simulators have been developed using bioinformatics tools which predict artificial immunity provided by the vaccine. These approaches could predict the best adjuvant for using in human vaccine studies. There is a multi-scale computational infrastructure approach which can stimulate the dynamics of the immune response induced by several vaccination formulations and predict optimal combination in terms of adjuvant type, dosage and timing. NetLogo is an agent-based modeling of the immune system running different simulations with different parameter settings. It also can interact with different modeling strategies including the investigation of pathogen growth, life cycle modeling environment for simulation complex phenomena41,42,43. Therefore, using these methods can increase efficiency and reduce costs in vaccine studies. In this study, for the first time, comprehensively integrated methods (using sequence-based tools in combination with flexible peptide-protein docking) were used to design highly immunogenic and protective vaccine candidates which were able to boost both humoral and cellular immune responses against all high-risk HPV types. In addition, in vivo analysis demonstrated high potency of the designed L1 and L2 constructs as combined in DNA-based vaccines without the use of adjuvant or delivery system. However, we will improve the efficiency of these DNA-based vaccines using a delivery system and also will compare their efficacy with the designed peptide-based vaccines along with adjuvants in near Future.
Methods
In silico analysis
Protein sequences retrieval
The reference L1 and L2 protein sequences of 13 high-risk HPV strains [16 (GI: 333031.L1 and GI: 333031.L2), 18 (GI: 60975.L1 and GI: 60975.L2), 31 (GI: 333048.L1 and GI: 333048.L2), 33 (GI: 333049.L1 and GI: 333049.L2), 35 (GI: 396997.L1 and GI: 396997.L2), 39 (GI: 333245.L1 and GI: 333245.L2), 45 (GI: 397022.L1 and GI: 397022.L2), 51(GI: 333087.L1 and GI: 333087.L2), 52(GI: 397038.L1 and GI: 397038.L2), 56 (GI: 397053.L1 and GI: 397053.L2), 58 (GI: 222386.L1 and GI: 222386.L2), 59 (GI: 557236.L1 and GI: 557236.L2, and 68(GI: 71726685.L1 and GI: 71726685.L2)] were extracted from PaVE database (https://pave.niaid.nih.gov/) and used as input for future bioinformatics analysis.
Protein alignments and conservancy analysis
To determine conserved epitopes between different subtypes, L1 and L2 sequence datasets were first aligned using SnapGene software 4.2.2 (From GSL Biotech; available at snapgene.com). After protein alignments analysis using muscle algorithms, the conserved epitopes of each protein were selected for immune-bioinformatics analysis such as B- and T-cell epitope prediction. Also, to calculate the degree of variability and conservancy of each epitope, IEDB epitope conservancy tools (http://tools.immuneepitope.org/tools/conservancy/) were used.
Linear B-cell epitope prediction
A successful vaccine must elicit a strong T-cell and B-cell immune response, but above all, provide protection against the disease being targeted. Therefore, it is essential to show that constructed immunogens are able to induce protective cellular and humoral immunity. Since the antibodies are induced against linear B-cell epitopes, it would be very difficult to synthesize long peptides with the native protein conformation resembling for the induction of protective antibodies. However, optimal peptide-based vaccines should be presented in a desired secondary structure of peptides in order to induce a specific humoral response41,42. For the B-cell epitope prediction of conserved regions in L1 and L2 proteins, BepiPred-2.0 server (http://www.cbs.dtu.dk/services/BepiPred-2.0/) was employed. In this study, epitope threshold value was set as 0.5 (the specificity and sensitivity of this method are 0.57 and 0.58, respectively)41.
T-cell epitope prediction
MHC-I epitope prediction: The initial step on applying bioinformatics to vaccine researches is to assess potentially immunoprotective epitopes. T-cell epitopes presented by MHC molecules are typically in a linear form containing 12 to 20 amino acids. This fact facilitates accurate modeling for the interaction of ligands and T-cells44. Thus, the most selective step in the presentation of antigenic peptide to T-cell receptor (TCR) is the binding of the MHC molecule45. In this study, we tried to use three different algorithms including Artificial Neural Networks (NetMHCpan 4.0 server43 (http://www.cbs.dtu.dk/services/NetMHCpan/), Quantitative matrix (Propred I43 (http://crdd.osdd.net/raghava/propred1/) and Published motifs (syfpeithi server46 (http://www.syfpeithi.de) to predict high-potential T-cell epitopes. For NetMHCpan, percentile rank was set at 0.5% for strong binders and 2% for weak binders and for Propred I threshold was set at 4%.
MHC-II epitope prediction: For MHC class II, NetMHCIIpan 3.2 server47 (http://www.cbs.dtu.dk/services/NetMHCIIpan/) and ProPred48 (http://crdd.osdd.net/raghava/propred/) were employed to predict potential interaction of helper T-cell epitope peptides and MHC-II. In this case, the threshold for strong and weak binders was set at 2% and 10%, respectively.
Prediction of MHC-I peptide presentation pathway
Investigating the Tap transport and proteasomal cleavage as well as affinity prediction of binding is essential in MHC-I presentation pathway. In this study, we used NetCTL 1.2 server combined with Tap transport/proteasomal cleavage tools (http://www.cbs.dtu.dk/services/NetCTL/) to access the prediction of antigen processing through the MHC class I antigen presentation pathway. In this method, parameters of weight on the C-terminal cleavage, Tap transport efficiency, and epitope identification were set to default (0.15, 0.05 and 0.75, respectively)49.
Population coverage
Since the response to T-cell epitopes is restricted by MHCs, the selection of epitopes with multiple HLA-binding increases population coverage in defined geographical regions where the peptide-based vaccine might be employed. The coverage rate of population for each epitope was computationally validated using the IEDB population coverage tool50 (/population/iedb_input). In this study, individual epitope and its binding to HLA alleles were analyzed, and different geographic areas were also selected.
Allergenicity and cross-reactivity assessment
Since proteins are very important in inducing allergenic reactions, the prediction of potential allergenicity is an important item in the safety assessment especially in the field of genetically modified foods, therapeutics, bio-pharmaceuticals etc.51. The food and agriculture organization (FAO) and world health organization (WHO) protocol includes three terms to evaluate the allergenicity of proteins which are defined as following: the term sensitivity refers to correctly predicted allergens (%), whereas specificity refers to correctly predicted non-allergens (%), and also accuracy refers to the proportion of correctly predicted proteins19. The allergenicity of the epitopes was analyzed by the PA3P (http://lpa.saogabriel.unipampa.edu.br:8080/pa3p/pa3p/pa3p.jsp) using Allergen online (8aa and 80 wordmatch) and AFDS-motif algorithms based on amino acid composition. The specificity of these methods is 95.43% (8aa), 92.88% (80aa) and 88.1% (ADFS)52. To assess cross-reactivity between peptide and human proteome, top-ranked epitope were analyzed by peptide matching program (https://research.bioinformatics.udel.edu/peptidematch/index.jsp)53.
Peptide-protein flexible Docking
Computational docking methods have been known as an important tool for drug design54. With the rapid development of peptide therapeutics in rational drug design, the use of new techniques such as protein-peptide docking is inevitable. In this study, two different algorithms (template-based docking and global docking) were performed by GalexyPepDock server55 (http://galaxy.seoklab.org/cgi-bin/submit.cgi?type=PEPDOCK) and CABS Dock server56 (http://biocomp.chem.uw.edu.pl/CABSdock). To estimate the formation of MHC-peptide complex, the GalaxyPepDock server effectively models the structural 3D peptide-protein complexes from input peptide and protein sequences using the structure database and energy-based optimization (Template-based Docking). CABS-Dock server performs Global docking procedure which at first explicit fully flexible docking simulation and then clustering-based scoring. Receptor flexibility was limited by default to small backbone fluctuation but could be increased to include selected receptor fragments56,57. This study presented an example of MHC-peptide docking performed by each individual epitope and available PDB file (Table 13) of HLA alleles, separately.
Physicochemical properties of the designed L1 and L2 constructs
Based on L1 and L2 top-ranked epitopes, two different constructs were designed. The physicochemical properties of top-ranked epitopes such as solubility, molecular weight, estimated half-time, instability index and antigenicity were determined by ProtParam (https://web.expasy.org/protparam/) tools58, VaxiJen59 (http://www.ddg-pharmfac.net/vaxijen/VaxiJen/VaxiJen.html) and Protein-Sol (https://protein-sol.manchester.ac.uk/) server60.
Experimental studies
Construction of the recombinant plasmids
After bioinformatics analysis, the selected peptides were assembled in two separated constructs (Fig. 2). The pUC57-L1 and pUC57-L2 constructs were synthesized by Biomatik Company. For in vitro experiments, the pUC57-L1 and pUC57-L2 vectors were digested by XhoI/HindIII, and the L1 and L2 genes were subcloned into XhoI/HindIII sites of pEGFP-N1 vector, individually (i.e., pEGFP-L1 and pEGFP-L2). All the recombinant vectors were transformed into Escherichia coli (E. coli) DH5α strain. After extraction of plasmids from single colonies using Mini-Kit (Qiagen), the presence of inserted L1 and L2 fragments was confirmed by digestion with restriction enzymes and sequencing. For in vivo immunological assessment, the pUC57-L1 and pUC57-L2 vectors were digested by BamHI/HindIII and the L1 and L2 genes were subcloned into BamHI/HindIII sites of pcDNA3.1 (-) vector containing cytomegalovirus early promoter and enhancer sequence, individually (i.e., pcDNA-L1 and pcDNA-L2). Indeed, we used the pcDNA vector harboring CpG motif for in vivo studies. As a final point, the recombinant DNA vectors harboring L1 and L2 genes were purified by an endotoxin-free plasmid Extra EF kit (Macherey Nagel, Germany). The concentration and purity of the recombinant L1 and L2 DNA constructs were determined by NanoDrop spectrophotometry61.
In vitro expression of L1 and L2 DNA constructs in HEK-293T cells
Human embryonic kidney cells (HEK-293T) were cultured in RPMI supplemented with 10% fetal bovine serum (FBS) at 37 °C and 5% CO2 atmosphere. After some passages, the cells were seeded in a 12-well plate. The optimal cell confluency for effective transfection was considered 70–80%. For the generation of TurboFect-plasmid DNA complex, 10 μl of TurboFect (Thermo Scientific) and 2 μg of each plasmid (pEGFP-L1, pEGFP-L2 and pEGFP-N1 as a positive control) were mixed and incubated for 15 min at room temperature. Then, the complex was added to each well in serum-free media. In addition, the non-transfected HEK-293T cells were used as negative control. After six hours, the media was replaced with the completed RPMI medium. Finally, the cells were harvested, washed and resuspended in PBS buffer, to analyze the expression of L1 and L2 DNA constructs using flow cytometry, fluorescent microscopy and western blotting at 48 hr after transfection61.
Western blot analysis
HEK-293T cells were scraped from their plates and washed with PBS1X. After washing steps, the cells were lysed in whole-cell lysis buffer (10% glycerol, 1 nM DTT, 2 mM natrium fluoride, 0.2% Triton X-100, 0.5 EDTA in PBS pH = 7.4). The extracted protein samples (L1-GFP, L2-GFP and GFP) were separated by SDS-PAGE in 12.5% (w/v) polyacrylamide gel and transferred to nitrocellulose membrane (Millipore). The membrane was equilibrated with TBST (Tris-buffered saline Tween-20) solution containing 2.5% BSA (Bovine albumin serum) overnight. The anti-GFP polyclonal antibody (1:5000 v/v; Acris antibodies GmbH) was used to recognize the expressed proteins under standard procedures. The immunoreactive protein bands were visualized by detection of peroxidase activity using a substrate named as 3, 3′-diaminobenzidine (DAB, Sigma)61.
Peptide constructs synthesis
For immunological assay (i.e., secretion of antibody, cytokine and Granzyme B), two peptide constructs (L1 and L2 peptides, Fig. 2) were synthesized by BioMatik Co. with more than 85% purity.
Mice immunization
Five groups of six female C57BL/6 mice (obtained from the breeding stocks maintained at Pasteur Institute of Iran; MHC haplotype B/H-2Kb/H-2Db) were immunized on days 0, 14, and 28 (i.e., three times with a 2-week interval) with 50 µg of each plasmid DNA (pcDNA-L1 or pcDNA-L2: G1 or G2) or their combination (pcDNA-L1+ pcDNA-L2: G3) at the right footpad as shown in Table 16. The control groups (G4 and G5) received pcDNA3.1 and PBS, respectively. All mice were maintained under specific pathogen-free conditions62. Moreover, all of the animal experimental procedures were approved by Animal Care and Use Committee of Pasteur Institute of Iran and carried out according to the Animal Experimentation Regulations of Pasteur Institute of Iran (national guideline) for scientific purposes (code: 976).
Monitoring tumor growth
For in vivo protection assay, vaccinated mice were subcutaneously challenged in the right flank with C3 tumor cells (1 × 105 cells), two weeks after the last injection. The C3 tumor cells contain whole HPV16 genome, and the presence of L1 and L2 genes was confirmed in the previous studies63. Tumor growth and the percentage of tumor-free mice were monitored twice a week by palpation for 60 days post-challenge. At each time, tumor volume was calculated by this formula: V = (a2b)/2 (a = the smallest diameter and b = the biggest diameter)62.
Antibody assay secreted from B-cells
Two weeks after the last injection, serum samples were collected from each group. The levels of goat anti-mouse immunoglobulin G1 (IgG1), IgG2a, IgG2b and total IgG antibodies (diluted 1:10,000 in 1% BSA/PBS-Tween, Sigma) secreted from B-cells were measured in the pooled sera of each group by indirect ELISA. The coated antigens were the mixture of L1 and L2 synthetic peptides (5 μg/mL). Moreover, mice sera were diluted 1:100 in 1% BSA/PBS-Tween64.
Cytokine assay secreted from T-cells
Three mice from each group were sacrificed and the spleens were removed. The red blood cell-depleted pooled splenocytes (2 × 106 cells/ml) were cultured in 48-well plates for 72 h in the presence of 5 μg/mL of L1 + L2 peptides, RPMI 5% as negative control and 5 μg/mL of concanavalin A (ConA) as positive control in complete RPMI culture medium. The supernatants were harvested to assess the secretion of IFN-γ, IL-5 and IL-10 from T-cells using the sandwich-based ELISA method (R&D Systems) according to the manufacturer’s instructions. All data were represented as mean ± SD for each sample65.
Granzyme B assay (in vitro CTL activity)
To measure Granzyme B (GrB) by ELISA, the P815 target cells (T) were seeded into 96-well plates (2 × 104 cells/well) incubated with the mixture of L1 and L2 peptides (~30 μg/mL) for 24 h. Then, the prepared splenocytes (Effector cells: E, before section) were counted and added to the target cells at E: T ratio of 100: 1 in complete RPMI culture medium for 6 h incubation. Finally, the supernatants were harvested to measure the concentration of GrB by ELISA (eBioscience kit) according to the manufacturer’s instruction64.
Statistical analysis
Statistical analyses were performed by Prism 7.0 (GraphPad, San Diego, California, USA) to determine the differences between the control and test groups using one-way ANOVA and student’s t-test. Survival rate or the percentage of tumor-free mice was evaluated using the log-rank (Mantel-Cox) test. The value of p < 0.05 was considered statistically significant.
References
de Martel, C., Plummer, M., Vignat, J. & Franceschi, S. Worldwide burden of cancer attributable to HPV by site, country and HPV type. Int J Cancer 141, 664–670, https://doi.org/10.1002/ijc.30716 (2017).
Pimenta, J. M., Galindo, C., Jenkins, D. & Taylor, S. M. Estimate of the global burden of cervical adenocarcinoma and potential impact of prophylactic human papillomavirus vaccination. BMC Cancer 13, 553, https://doi.org/10.1186/1471-2407-13-553 (2013).
Sankaranarayanan, R. HPV vaccination: the promise & problems. Indian J Med Res 130, 322–326 (2009).
White, M. D. Pros, cons, and ethics of HPV vaccine in teens-Why such controversy? Transl Androl Urol 3, 429–434, https://doi.org/10.3978/j.issn.2223-4683.2014.11.02 (2014).
Wang, J. W. & Roden, R. B. Virus-like particles for the prevention of human papillomavirus-associated malignancies. Expert Rev Vaccines 12, 129–141, https://doi.org/10.1586/erv.12.151 (2013).
Hung, C. F., Ma, B., Monie, A., Tsen, S. W. & Wu, T. C. Therapeutic human papillomavirus vaccines: current clinical trials and future directions. Expert Opin Biol Ther 8, 421–439, https://doi.org/10.1517/14712598.8.4.421 (2008).
Kreimer, A. R. et al. Efficacy of fewer than three doses of an HPV-16/18 AS04-adjuvanted vaccine: combined analysis of data from the Costa Rica Vaccine and PATRICIA Trials. The Lancet. Oncology 16, 775–786, https://doi.org/10.1016/s1470-2045(15)00047-9 (2015).
Tumban, E., Peabody, J., Tyler, M., Peabody, D. S. & Chackerian, B. VLPs displaying a single L2 epitope induce broadly cross-neutralizing antibodies against human papillomavirus. PLoS One 7, e49751, https://doi.org/10.1371/journal.pone.0049751 (2012).
Buck, C. B. & Trus, B. L. The papillomavirus virion: a machine built to hide molecular Achilles’ heels. Adv Exp Med Biol 726, 403–422, https://doi.org/10.1007/978-1-4614-0980-9_18 (2012).
Jagu, S. et al. Concatenated multitype L2 fusion proteins as candidate prophylactic pan-human papillomavirus vaccines. J Natl Cancer Inst 101, 782–792, https://doi.org/10.1093/jnci/djp106 (2009).
Jagu, S. et al. Optimization of multimeric human papillomavirus L2 vaccines. PLoS One 8, e55538, https://doi.org/10.1371/journal.pone.0055538 (2013).
Rubio, I. et al. Potent anti-HPV immune responses induced by tandem repeats of the HPV16 L2 (20–38) peptide displayed on bacterial thioredoxin. Vaccine 27, 1949–1956, https://doi.org/10.1016/j.vaccine.2009.01.102 (2009).
Schellenbacher, C., Roden, R. & Kirnbauer, R. Chimeric L1-L2 virus-like particles as potential broad-spectrum human papillomavirus vaccines. Journal of virology 83, 10085–10095, https://doi.org/10.1128/jvi.01088-09 (2009).
Pineo, C. B., Hitzeroth, I. I. & Rybicki, E. P. Immunogenic assessment of plant-produced human papillomavirus type 16 L1/L2 chimaeras. Plant Biotechnol J 11, 964–975, https://doi.org/10.1111/pbi.12089 (2013).
Alphs, H. H. et al. Protection against heterologous human papillomavirus challenge by a synthetic lipopeptide vaccine containing a broadly cross-neutralizing epitope of L2. Proc Natl Acad Sci USA 105, 5850–5855, https://doi.org/10.1073/pnas.0800868105 (2008).
Kim, D. et al. Generation and characterization of a preventive and therapeutic HPV DNA vaccine. Vaccine 26, 351–360, https://doi.org/10.1016/j.vaccine.2007.11.019 (2008).
Peng, S. et al. Control of HPV-associated tumors by innovative therapeutic HPV DNA vaccine in the absence of CD4+ T cells. Cell Biosci 4, 11, https://doi.org/10.1186/2045-3701-4-11 (2014).
Lesk, A. M. (Oxford: Oxford University Press).
Kaliamurthi, S. et al. Cancer Immunoinformatics: A Promising Era in the Development of Peptide Vaccines for Human Papillomavirus-induced Cervical Cancer. Curr Pharm Des 24, 3791–3817, https://doi.org/10.2174/1381612824666181106094133 (2018).
Wei, D. Q., Selvaraj, G. & Kaushik, A. C. Computational Perspective on the Current State of the Methods and New Challenges in Cancer Drug Discovery. Curr Pharm Des 24, 3725–3726, https://doi.org/10.2174/138161282432190109105339 (2018).
Kaliamurthi, S., Selvaraj, G., Kaushik, A. C., Gu, K. R. & Wei, D. Q. Designing of CD8(+) and CD8(+)-overlapped CD4(+) epitope vaccine by targeting late and early proteins of human papillomavirus. Biologics 12, 107–125, https://doi.org/10.2147/BTT.S177901 (2018).
Soria-Guerra, R. E., Nieto-Gomez, R., Govea-Alonso, D. O. & Rosales-Mendoza, S. An overview of bioinformatics tools for epitope prediction: Implications on vaccine development. Journal of Biomedical Informatics 53, 405–414, https://doi.org/10.1016/j.jbi.2014.11.003 (2015).
Khairkhah, N., Namvar, A., Kardani, K. & Bolhassani, A. Prediction of cross-clade HIV-1 T-cell epitopes using immunoinformatics analysis. Proteins, https://doi.org/10.1002/prot.25609 (2018).
Li, Y. et al. Immune responses induced in HHD mice by multiepitope HIV vaccine based on cryptic epitope modification. Mol Biol Rep 40, 2781–2787, https://doi.org/10.1007/s11033-012-2202-y (2013).
Oany, A. R., Emran, A. A. & Jyoti, T. P. Design of an epitope-based peptide vaccine against spike protein of human coronavirus: an in silico approach. Drug Des Devel Ther 8, 1139–1149, https://doi.org/10.2147/DDDT.S67861 (2014).
Chakraborty, S. et al. A computational approach for identification of epitopes in dengue virus envelope protein: a step towards designing a universal dengue vaccine targeting endemic regions. In Silico Biol 10, 235–246, https://doi.org/10.3233/ISB-2010-0435 (2010).
Hasan, M. A., Hossain, M. & Alam, M. J. A computational assay to design an epitope-based Peptide vaccine against saint louis encephalitis virus. Bioinform Biol Insights 7, 347–355, https://doi.org/10.4137/BBI.S13402 (2013).
Gupta, S. K., Singh, A., Srivastava, M., Gupta, S. K. & Akhoon, B. A. In silico DNA vaccine designing against human papillomavirus (HPV) causing cervical cancer. Vaccine 28, 120–131, https://doi.org/10.1016/j.vaccine.2009.09.095 (2009).
Ghorban Hosseini, N. et al. In Silico Analysis of L1/L2 Sequences of Human Papillomaviruses: Implication for Universal Vaccine Design. Viral Immunol 30, 210–223, https://doi.org/10.1089/vim.2016.0142 (2017).
Singh, K. P. et al. Sequence-based approach for rapid identification of cross-clade CD8+ T-cell vaccine candidates from all high-risk HPV strains. 3 Biotech 6, 39, https://doi.org/10.1007/s13205-015-0352-z (2016).
Arshad, M., Bhatti, A. & John, P. Identification and in silico analysis of functional SNPs of human TAGAP protein: A comprehensive study. PLoS One 13, e0188143, https://doi.org/10.1371/journal.pone.0188143 (2018).
Wang, A. et al. [Prediction of B cell epitopes of human papillomavirus type 16 L1 protein]. Xi Bao Yu Fen Zi Mian Yi Xue Za Zhi 32, 442–445 (2016).
Sabah, S. N. et al. Designing of Epitope-Focused Vaccine by Targeting E6 and E7 Conserved Protein Sequences: An Immuno-Informatics Approach in Human Papillomavirus 58 Isolates. Interdiscip Sci 10, 251–260, https://doi.org/10.1007/s12539-016-0184-5 (2018).
Bristol, J. A., Orsini, C., Lindinger, P., Thalhamer, J. & Abrams, S. I. Identification of a ras oncogene peptide that contains both CD4(+) and CD8(+) T cell epitopes in a nested configuration and elicits both T cell subset responses by peptide or DNA immunization. Cellular immunology 205, 73–83, https://doi.org/10.1006/cimm.2000.1712 (2000).
Lafuente, E. M. & Reche, P. A. Prediction of MHC-peptide binding: a systematic and comprehensive overview. Curr Pharm Des 15, 3209–3220 (2009).
Blaszczyk, M. et al. Modeling of protein-peptide interactions using the CABS-dock web server for binding site search and flexible docking. Methods 93, 72–83, https://doi.org/10.1016/j.ymeth.2015.07.004 (2016).
Sauer, M. Interaction of the host immune system with tumor cells in human papillomavirus associated diseases (2016).
Kwak, K. et al. Multivalent human papillomavirus l1 DNA vaccination utilizing electroporation. PLoS One 8, e60507, https://doi.org/10.1371/journal.pone.0060507 (2013).
Yang, A. et al. Enhancing antitumor immunogenicity of HPV16-E7 DNA vaccine by fusing DNA encoding E7-antigenic peptide to DNA encoding capsid protein L1 of Bovine papillomavirus. Cell Biosci 7, 46, https://doi.org/10.1186/s13578-017-0171-5 (2017).
Lefeber, D. J. et al. Th1-directing adjuvants increase the immunogenicity of oligosaccharide-protein conjugate vaccines related to Streptococcus pneumoniae type 3. Infect Immun 71, 6915–6920, https://doi.org/10.1128/iai.71.12.6915-6920.2003 (2003).
Pennisi, M., Russo, G., Ravalli, S. & Pappalardo, F. Combining agent based-models and virtual screening techniques to predict the best citrus-derived vaccine adjuvants against human papilloma virus. BMC Bioinformatics 18, 544, https://doi.org/10.1186/s12859-017-1961-9 (2017).
Pappalardo, F. et al. A computational model to predict the immune system activation by citrus-derived vaccine adjuvants. Bioinformatics (Oxford, England) 32, 2672–2680, https://doi.org/10.1093/bioinformatics/btw293 (2016).
Escobar Ospina, M. E. & Perdomo, J. G. A growth model of human papillomavirus type 16 designed from cellular automata and agent-based models. Artif Intell Med 57, 31–47, https://doi.org/10.1016/j.artmed.2012.11.001 (2013).
Malherbe, L. T-cell epitope mapping. Annals of Allergy, Asthma &. Immunology 103, 76–79, https://doi.org/10.1016/S1081-1206(10)60147-0 (2009).
Yewdell, J. W. & Bennink, J. R. Immunodominance in major histocompatibility complex class I-restricted T lymphocyte responses. Annu Rev Immunol 17, 51–88, https://doi.org/10.1146/annurev.immunol.17.1.51 (1999).
Rammensee, H., Bachmann, J., Emmerich, N. P., Bachor, O. A. & Stevanovic, S. SYFPEITHI: database for MHC ligands and peptide motifs. Immunogenetics 50, 213–219 (1999).
Jensen, K. K. et al. Improved methods for predicting peptide binding affinity to MHC class II molecules. Immunology 154, 394–406, https://doi.org/10.1111/imm.12889 (2018).
Singh, H. & Raghava, G. P. ProPred: prediction of HLA-DR binding sites. Bioinformatics (Oxford, England) 17, 1236–1237, https://doi.org/10.1093/bioinformatics/17.12.1236 (2001).
Larsen, M. V. et al. Large-scale validation of methods for cytotoxic T-lymphocyte epitope prediction. BMC Bioinformatics 8, 424, https://doi.org/10.1186/1471-2105-8-424 (2007).
Bui, H. H. et al. Predicting population coverage of T-cell epitope-based diagnostics and vaccines. BMC Bioinformatics 7, 153, https://doi.org/10.1186/1471-2105-7-153 (2006).
Saha, S. & Raghava, G. AlgPred: prediction of allergenic proteins and mapping of IgE epitopes. Nucleic acids research 34, W202–W209 (2006).
Chrysostomou, C. & Seker, H. Prediction of protein allergenicity based on signal-processing bioinformatics approach. Conf Proc IEEE Eng Med Biol Soc 2014, 808–811, https://doi.org/10.1109/EMBC.2014.6943714 (2014).
Chen, C. et al. A fast Peptide Match service for UniProt Knowledgebase. Bioinformatics 29, 2808–2809, https://doi.org/10.1093/bioinformatics/btt484 (2013).
Diller, D. J., Swanson, J., Bayden, A. S., Jarosinski, M. & Audie, J. Rational, computer-enabled peptide drug design: principles, methods, applications and future directions. Future Med Chem 7, 2173–2193, https://doi.org/10.4155/fmc.15.142 (2015).
Lee, H., Heo, L., Lee, M. S. & Seok, C. GalaxyPepDock: a protein-peptide docking tool based on interaction similarity and energy optimization. Nucleic Acids Res 43, W431–435, https://doi.org/10.1093/nar/gkv495 (2015).
Kurcinski, M., Jamroz, M., Blaszczyk, M., Kolinski, A. & Kmiecik, S. CABS-dock web server for the flexible docking of peptides to proteins without prior knowledge of the binding site. Nucleic Acids Res 43, W419–424, https://doi.org/10.1093/nar/gkv456 (2015).
de Oliveira, L. M. F. et al. Design, Immune Responses and Anti-Tumor Potential of an HPV16 E6E7 Multi-Epitope Vaccine. PLoS ONE 10, e0138686, https://doi.org/10.1371/journal.pone.0138686 (2015).
Gasteiger, E. et al. In The Proteomics Protocols Handbook (ed John M. Walker) 571-607 (Humana Press, 2005).
Doytchinova, I. A. & Flower, D. R. VaxiJen: a server for prediction of protective antigens, tumour antigens and subunit vaccines. BMC Bioinformatics 8, 4, https://doi.org/10.1186/1471-2105-8-4 (2007).
Hebditch, M., Carballo-Amador, M. A., Charonis, S., Curtis, R. & Warwicker, J. Protein-Sol: a web tool for predicting protein solubility from sequence. Bioinformatics (Oxford, England) 33, 3098–3100, https://doi.org/10.1093/bioinformatics/btx345 (2017).
Namvar, A., Bolhassani, A. & Hashemi, M. HPV16 L2 improves HPV16 L1 gene delivery as an important approach for vaccine design against cervical cancer. Bratisl Lek Listy 117, 179–184 (2016).
Bolhassani, A. et al. Whole recombinant Pichia pastoris expressing HPV16 L1 antigen is superior in inducing protection against tumor growth as compared to killed transgenic Leishmania. Hum Vaccin Immunother 10, 3499–3508, https://doi.org/10.4161/21645515.2014.979606 (2014).
Hitzeroth, I. I. et al. Immunogenicity of an HPV-16 L2 DNA vaccine. Vaccine 27, 6432–6434, https://doi.org/10.1016/j.vaccine.2009.06.015 (2009).
Milani, A. et al. Small heat shock protein 27: An effective adjuvant for enhancement of HIV-1 Nef antigen-specific immunity. Immunol Lett 191, 16–22, https://doi.org/10.1016/j.imlet.2017.09.005 (2017).
Salehi, M. et al. Recombinant Leishmania tarentolae encoding the HPV type 16 E7 gene in tumor mice model. Immunotherapy 4, 1107–1120, https://doi.org/10.2217/imt.12.110 (2012).
Author information
Authors and Affiliations
Contributions
A.N. and A.B. conceptualized the work. A.N. performed the experiments. A.N., A.B., G.J. and Z.N. analyzed the data. A.N. wrote the manuscript. A.B. edited the manuscript. All authors approved the final version of the paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Namvar, A., Bolhassani, A., Javadi, G. et al. In silico/In vivo analysis of high-risk papillomavirus L1 and L2 conserved sequences for development of cross-subtype prophylactic vaccine. Sci Rep 9, 15225 (2019). https://doi.org/10.1038/s41598-019-51679-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-51679-8
This article is cited by
-
Phylogeny and amino acid analysis in single and mixed bovine papillomavirus infections in Southern Brazil, 2016-2020
Archives of Virology (2023)
-
Current and future direction in treatment of HPV-related cervical disease
Journal of Molecular Medicine (2022)
-
Genetic variability in minor capsid protein (L2 gene) of human papillomavirus type 16 among Indian women
Medical Microbiology and Immunology (2022)
-
A Comprehensive in Silico Analysis for Identification of Immunotherapeutic Epitopes of HPV-18
International Journal of Peptide Research and Therapeutics (2021)
-
In Silico Design and Immunological Studies of Two Novel Multiepitope DNA-Based Vaccine Candidates Against High-Risk Human Papillomaviruses
Molecular Biotechnology (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
