
Abstract
Leaf pathogens are limiting factors in banana (Musa spp.) production, with Pseudocercospora spp. responsible for the important Sigatoka disease complex. In order to investigate cellular processes and genes involved in host defence responses, quantitative real-time PCR (RT-qPCR) is an analytical technique for gene expression quantification. Reliable RT-qPCR data, however, requires that reference genes for normalization of mRNA levels in samples are validated under the conditions employed for expression analysis of target genes. We evaluated the stability of potential reference genes ACT1, α-TUB, UBQ1, UBQ2, GAPDH, EF1α, APT and RAN. Total RNA was extracted from leaf tissues of Musa acuminata genotypes Calcutta 4 (resistant) and Cavendish Grande Naine (susceptible), both subjected to P. musae infection. Expression stability was determined with NormFinder, BestKeeper, geNorm and RefFinder algorithms. UBQ2 and RAN were the most stable across all M. acuminata samples, whereas when considering inoculated and non-inoculated leaf samples, APT and UBQ2 were appropriate for normalization in Calcutta 4, with RAN and α-TUB most stable in Cavendish Grande Naine. This first study of reference genes for relative quantification of target gene expression in the M. acuminata-P. musae interaction will enable reliable analysis of gene expression in this pathosystem, benefiting elucidation of disease resistance mechanisms.
Similar content being viewed by others

Introduction
Banana (Musa spp.) is an important monocotyledonous fruit crop in over 100 tropical and sub-tropical countries, contributing significantly as the world’s most consumed fruit towards global food security and poverty alleviation.
Leaf pathogens are important limiting factors in global banana production, with reduced photosynthetic capacity causing negative impact on crop yield, especially in tropical growing regions1. Numerous leaf diseases are caused by fungal pathogens that include Cladosporium musae, Cloridium musae, Cordana musae, Curvularia sp., Drechslera gigantean, Hendersonia toruloides, Helminthosporium sp., Phyllacora musicola, Phyllosticta spp, and the Dothideomycete Pseudocercospora sp. responsible for the important Sigatoka disease complex in Musa. Pseudocercospora fijiensis, Pseudocercospora musae and Pseudocercospora eumusae cause necrotic leaf lesions that can reduce functional leaf area to such an extent that crop yields can be reduced by 20–50% in affected areas2,3.
The majority of commercial banana cultivars lack genetic resistance to pathogens and, in situations where disease pressure is high, are dependent upon integrated disease management strategies that employ programmed applications of systemic or protective fungicides. Such dependence on agrochemical disease control, however, can increase production costs, with a negative impact on the environment, and increased selective pressure for emergence of resistant strains in pathogen populations4,5.
In order to advance the genetic improvement and the development of resistant Musa cultivars, elucidation of the cellular mechanisms and identification of genes involved in resistance responses to foliar pathogens is of fundamental importance. For this, high-throughput next-generation sequencing approaches (NGS) are applicable for RNA sequencing (RNAseq) and transcriptome analysis. To date, a number of large scale studies have been conducted in Musa to further understanding of defense responses and genes involved in response to different phytopathogens and pests, such as Fusarium oxysporum f. sp. cubense tropical race 46,7,8 and the root-knot nematode Meloidogyne incognita9. In relation to foliar pathogens, to date, Illumina-based RNAseq transcriptome profiling of the host-pathogen interaction has been conducted with the Sigatoka leaf spot pathogen P. fijiensis10, with a characterization of unigenes expressed during interaction with P. musae determined through 454 sequencing of cDNA11.
In order to accurately quantify and validate gene expression derived from in silico NGS data, the reverse transcription quantitative real-time polymerase chain reaction (RT-qPCR) is today recognized as the benchmark approach for such work, given its’ specificity, sensitivity and rapidity12,13. For this, however, RNA sample quality, cDNA preparation and dilution, and PCR sample preparation and reaction execution must be standardized. In accordance with the Minimum Information for Publication of Real-Time Quantitative PCR Experiments (MIQE) guidelines14, normalization of expression of each target gene to that of a reference gene is also required to correct data for variations in samples that occur during cDNA preparation, with reference genes also subject to the same effect of cDNA quality15,16. For such normalization, the employment of appropriate reference genes for each organism, tissue type and time point is therefore essential. The ideal characteristic of a reference gene is a stable expression for a tissue type, regardless of developmental stage or experimental treatment conditions15,17. Genes that are involved in basal cell activities in plants have been assumed to offer constitutive expression across tissues and independent of experimental conditions. Commonly employed genes have included actin and tubulin, given their role in the cell cytoskeleton, genes involved in protein synthesis (elongation factor) and protein degradation (ubiquitin), and genes involved in glucose metabolism (glyceraldeide-3-phosphate dehydrogenase). With such housekeeping genes, however, a stable transcript response is not necessarily guaranteed across different experimental conditions, as observed in numerous plant species18,19,20 and can lead to inaccuracies in results, masking true interpretation of gene expression16,21.
In order to facilitate the identification of stable reference genes, a number of mathematical algorithms have been developed, including the programs geNorm, NormFinder and BestKeeper22,23,24. These have been widely employed in reference gene development across monocotyledonous plant species, including, for example, rice25, maize26, sugarcane27 and Setaria28.
In the case of Musa, Chen and colleagues29 conducted a study towards validation of reference genes for use in Cavendish bananas, with focus on fruit and peel at different developmental stages, after hormone treatment, and following fruit exposure to abiotic and biotic stress29. Genes were also validated using pooled plant tissues comprising root, leaf, flower, peel and pulp. Similarly, Podevin et al.30 investigated the stability of reference genes for analysis of gene expression in leaf and meristem tissues across varieties grown in vitro, under greenhouse conditions and in leaf disc material30. Zhang et al.31 also identified stable reference genes for root tissues in cultivars during interaction with Fusarium oxysporum f. sp. cubense race 1 and race 4 strains31. These data all highlight the importance of selection of multiple reference genes for normalization in accord with plant genotype, tissues, development stage and specific experimental conditions.
In this study, the stability of eight candidate reference genes for transcript normalization in whole plant leaf tissues was analyzed in two M. acuminata genotypes, contrasting in resistance to the foliar pathogen P. musae. The reliability of the most and least stable genes was determined through normalization of the relative expression of the target gene encoding RuBisCO activase (RCA). The selected reference genes will enable accurate gene expression analysis in this important pathosystem.
Results
Analysis of primer specificity and efficiency
A total of eight candidate genes were selected as potential reference genes in this study. Information on the candidate genes, in relation to accession number, primer sequences and amplification are summarized in Table 1. Specificity of each of the primers to a single gene locus was confirmed by dissociation curve analysis, with a single peak observed for each primer pair at the expected primer annealing temperature (Fig. 1), together with an absence of any signal in negative controls lacking template cDNA. PCR amplification efficiency values ranged from 90.55 to 98.21% across the candidate genes, as calculated by LinRegPCR software.

Dissociation curves of the candidate reference genes. Primers for genes (ACT) actin 1; (APT) adenine phosphoribosyltransferase; (EF1α) elongation factor 1 alpha; (GAPDH) glyceraldehyde-3-phosphate dehydrogenase; (α-TUB) alpha-tubulin; (RAN) GTP-binding nuclear protein; (UBQ1) ubiquitin 1 and (UBQ2) ubiquitin 2, revealing single peaks, each obtained from three technical replicates of different cDNA samples. No amplification was observed in negative controls lacking template cDNA.
Analysis of expression levels of candidate reference genes
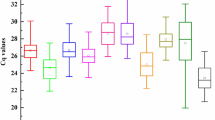
For each candidate reference gene, quantification cycle (Cq) values were presented for the expression data from each sample. Cq values ranged from 19.6 to 29.6, with broad differences observed between the candidate reference genes, highlighting the importance of statistical approaches for stability ranking in identification of accurate reference genes. The ACT1 gene (amplified with primer pair 2 [pp2]) displayed the lowest Cq value, indicating that it was more expressed than the other candidate genes. Conversely, the UBQ1 gene displayed the highest Cq values, indicating lowest gene expression. The Cq value ranges for the reference genes evaluated in the different Musa samples are displayed in Fig. 2, with a summary of all Cq values obtained for each treatment for the potential reference genes provided in Supplementary Table S1.

RT-qPCR quantification cycle (Cq) values for tested reference genes. Expression data is displayed as Cq values for each reference gene in leaf material from Musa acuminata genotypes Calcutta 4 and Cavendish Grande Naine, non-inoculated or inoculated with Pseudocercospora musae.
Expression stability
The performance of the candidate reference genes was assessed in the four biological samples, divided into three experimental sets; global pooled M. acuminata Calcutta 4 and Cavendish Grande Naine leaf material (from non-inoculated and inoculated plants); pooled M. acuminata Calcutta 4 leaf material (from non-inoculated and inoculated plants); and pooled M. acuminata Cavendish Grande Naine leaf material (from non-inoculated and inoculated plants). Stability in expression and ranking of the eight candidate reference genes, amplified with nine primer pairs, was determined using mathematical algorithms geNorm, NormFinder and BestKeeper. Re-ranking of genes was conducted using RefFinder. Summarized results from all analyses are presented in Table 2.
On the basis of geNorm analysis, many candidate reference genes showed stability values below the default limit of 0.5 when employed on data for different M. acuminata samples (Table 2, Fig. 3). UBQ2, RAN, UBQ1 and EF1α genes displayed stability values below 0.5 when employed on data for pooled M. acuminata Calcutta 4 and Cavendish Grande Naine leaf samples (from both non-inoculated and P. musae-inoculated, sample name ‘Global’), indicating greatest stability. In the case of samples from M. acuminata Calcutta 4 leaf material, either from non-inoculated or P. musae-inoculated plants (sample ‘Global C4’), all genes displayed stability values below the accepted threshold. APT and UBQ2 were highlighted as the most stably expressed genes, followed by UBQ1 and ACT1pp2. For samples from M. acuminata Cavendish Grande Naine leaf material (non-inoculated or P. musae-inoculated) (sample ‘Global CAV’), all genes once again displayed M-values below the accepted threshold, with RAN and GAPDH genes most stable, followed by α-TUB and EF1α.

GeNormPlus-derived expression stability for each candidate reference gene for Musa acuminata leaf tissues. Values of M below a 0.5 cutoff indicate high stability rate.
Calculation of pairwise variation was employed to determine the minimum number of reference genes required for stable normalization of gene expression data. According to Vandesompele and coworkers22, a conventional normalization of RT-qPCR based on a single reference gene can potentially lead to a more than 6-fold normalization error. Given this, geNorm automatically takes into account that at least two reference genes are employed for normalization. For all M. acuminata sample types (Global, pooled non-inoculated or P. musae-inoculated M. acuminata Calcutta 4 and Cavendish Grande Naine leaf samples; Global C4, pooled non-inoculated or P. musae-inoculated M. acuminata Calcutta 4 leaf samples; Global CAV, pooled non-inoculated or P. musae-inoculated M. acuminata Cavendish Grande Naine leaf samples; C4 mock, pooled non-inoculated M. acuminata Calcutta 4 leaf samples; C4 infected, P. musae-inoculated M. acuminata Calcutta 4 leaf samples; CAV mock, pooled non-inoculated M. acuminata Cavendish Grande Naine leaf samples; and CAV infected, P. musae-inoculated M. acuminata Cavendish Grande Naine leaf samples), the pairwise variation value of V2/3 was below the default cut-off value of 0.15, indicating that two reference genes are sufficient for accurate normalization of the gene expression data (Fig. 4). Values of V above the default limit, indicate that one or several extra reference genes would be required to improve RT-qPCR analysis22. Here, data revealed that the inclusion of further reference genes would not have a significant effect on accurate normalization of target gene expression.

GeNormV-derived determination of the optimal number of reference genes for accurate RT-qPCR normalization of target gene expression. For data below a cutoff value of 0.15, the inclusion of additional reference genes will not contribute significantly to the normalization of gene expression data.
NormFinder-based analysis provided gene stability ranking data with a certain overlap to that obtained with geNorm (Table 2). When considering the combined dataset ‘Global’, based on pooled M. acuminata Calcutta 4 and Cavendish Grande Naine leaf samples (non-inoculated or P. musae-inoculated), the four most stably expressed genes were in agreement, with UBQ1, UBQ2, EF1α and RAN genes ranked in order of greatest stability. In the case of data samples from M. acuminata Calcutta 4 leaf material, either from non-inoculated or P. musae-inoculated plants (sample ‘Global C4’), by contrast, only the UBQ2 gene was highly ranked with both algorithms. In the case of analysis with the algorithm NormFinder, this gene ranked as the most stably expressed, followed by α-TUB, EF1α and RAN. When data was analyzed according to samples from M. acuminata Cavendish Grande Naine leaf material (non-inoculated or P. musae-inoculated) (sample ‘Global CAV’), three of the four most highly ranked stable genes were again in agreement with geNorm data, with RAN ranked as most stable, followed by GAPDH, α-TUB and UBQ2.
On the basis of results obtained following analysis with the program BestKeeper, all candidate genes were calculated to have a standard deviation value lower than 1 (Table 2). With this algorithm, a certain overlap in most stably expressed candidate reference genes was also observed when compared to ranking positions based on geNorm and NormFinder data. In the case of data considering the ‘Global’ dataset, three of the four most highly ranked stable genes were in agreement, with the EF1α gene ranked most stable, followed by ACT1pp2, RAN and UBQ2. When considering the data samples ‘Global C4’, three of the most highly ranked stable genes were in agreement with results obtained using NormFinder and two in agreement with geNorm-derived ranking. Here, the RAN gene ranked as most stable, followed by APT, UBQ2 and EF1α. In the case of data derived from the samples ‘Global CAV’, two genes most highly ranked in terms of expression stability were in agreement with ranking data obtained using NormFinder and another two in agreement with ranking data obtained using geNorm. The four most highly ranked genes, in order of expression stability, were α-TUB, ACT1pp2, APT and GAPDH.
RefFinder was employed to enable a comprehensive re-ranking of expression stability in the candidate reference genes based on integration of the data from the previous three algorithms. When taking into account the complete ‘Global’ dataset, for both M. acuminata Calcutta 4 and M. acuminata Cavendish Grande Naine leaf material (non-inoculated and P. musae-inoculated), the four most highly ranked candidate reference genes, in order of stability, were: UBQ2, EF1α, RAN and GAPDH. In the case of the ‘Global C4’ dataset, the most stable candidate reference gene was APT, followed by UBQ2, RAN and EF1α. For the ‘Global CAV’ dataset, the most stable candidate reference gene was α-TUB, followed by RAN, UBQ2 and EF1α (Table 2).
A summary of all candidate reference gene rankings with the mathematical algorithms is provided in Supplementary Table S2.
Testing selected reference genes for normalization of M. acuminata genes expressed in leaves
For validation of reference gene ranking for leaf tissues, the relative expression of RuBisCO activase (RCA) was normalized against the reference genes. This catalytic chaperone is involved in modulating RuBisCO, a key enzyme in the plant photosynthetic pathway. Although three RCA genes were identified in the M. acuminata DH Pahang reference genome, only RCA3 could be amplified by conventional PCR in both M. acuminata Calcutta 4 and M. acuminata Cavendish Grande Naine. When considering the complete ‘global’ samples, the mathematical programs identified UBQ2 and RAN together as the most stable genes, with α-TUB and ACT1pp1 as the least stable reference genes. The former pair were evaluated for normalization of RT-qPCR expression profiles in M. acuminata Calcutta 4 and M. acuminata Cavendish Grande Naine leaf samples subjected to light and dark treatments. In both M. acuminata genotypes, RCA3 expression was down-regulated in samples from leaves covered with aluminium foil. Transcript levels of RCA3 in the three biological replicates showed that when employing the two most stable reference genes, variation between samples was reduced, whereas the same analysis using the two least stable genes introduced considerable variation in fold change in relative expression in samples across the biological replicates (Fig. 5).

Relative expression levels of the RuBisCO activase gene RCA3 in two M. acuminata genotypes using the most (UBQ2 and RAN) and least (α-TUB and ACT1pp1) stable pair of reference genes. RT-qPCR was performed in three biological replicates of light and dark treated leaf samples. Calcutta 4 (C4), Cavendish Grande Naine (CV), Light (LT) and Dark (DK). Bars represent the standard error (±SE) calculated for three technical replicates.
Discussion
In this study, genes encoding proteins involved in basal cell activities in plants were evaluated as potential reference genes for normalization of gene expression data in M. acuminata leaf material through a multi-algorithm based analysis of expression stability.
Recent investigations of gene expression in Musa have employed numerous reference genes for normalization of RT-qPCR expression data, with focus on genotypes, tissues, development stages and response to different abiotic and biotic stresses. Reference genes employed have included Actin 1 (ACT-1), clathrin adaptor complex (CAC), Elongation factor 1α (EF1α), GAPDH, pectate lyase (MWPL1), ribosomal protein 2 (RPS2), ribosomal protein 4 (RPS4), ribosomal protein L2 (L2), small nuclear RNAs (U6), ubiquitin 2 (UBQ2), ubiquitin 4 (UBQ4) and 25 S ribosomal RNA8,9,32,33,34,35,36,37,38,39,40,41,42,43,44. Such genes, however, have often been selected based on their roles in basal cell activities, without investigation of expression stability across particular experimental conditions.
The stability of reference genes in Musa has so far been evaluated across a limited number of specific experimental conditions. Chen and coworkers29 tested candidate reference genes in Cavendish subgroup material, with stability determined across plant tissues, developmental stages, and following hormone treatment, abiotic stress and biotic stress after fruit inoculation with Colletotrichum musae29. Podevin and colleagues (2012) also evaluated expression stability of reference genes in genotypes subjected to different physiological growth conditions, osmotic stress, treatment with acetone and following biotic stress with the leaf pathogen Pseudocercospora fijiensis in cultivar M. acuminata Tuu Gia30. Zhang and colleagues31 also evaluated the stability of reference genes in Cavendish Grande Naine root tissues infected with Fusarium oxysporum f. sp. cubense31. The present work is the first describing the stability of reference genes for leaf materials during the interaction with P. musae, within the context of accurate analysis of gene expression in genotypes contrasting in resistance to the pathogen.
Multi-algorithm analysis of candidate reference gene expression stability revealed differences in the top-ranked genes observed between the results generated with each algorithm, although there was greater consistency in identification, and therefore exclusion, of the least stable candidate genes. Differences in gene stability ranking between algorithms is common45,46,47,48, reflecting data analysis methods for each algorithm.
In our study, combinations of stable reference genes were identified for each condition employed. When considering all samples (Global set), analyses with all the mathematical algorithms consistently revealed UBQ2 and RAN ranked amongst the most stable genes, with α-TUB and ACT1pp1 ranked as the least stable. Previously, Chen and co-workers29 also recommended UBQ2 and RAN as the most stable general reference genes for Cavendish bananas. Although UBQ genes have been widely employed in RT-qPCR normalization across different plant species49, including monocot species, variation in their stability is apparent according to physiological conditions employed. In Oryza sativa, for example, UBQ5 was among the most stable reference gene for plants exposed to hormone treatment, saline and drought stresses, whereas UBQ10 was among the least stable genes observed50. UBQ1 was also identified as a stable reference gene for rice plants subjected to temperature, saline and hormone treatment, fungal infection and defense signaling compounds51,52. In Saccharum sp., UBQ1 also presented stable expression in leaf tissues27. RAN is a ubiquitously expressed gene that encodes a small soluble GTP-binding protein which plays roles in vegetative growth and stress tolerance in Arabidopsis53. Such high ranking of RAN as a reference gene has also been described in Cucumis melo54 and Corchorus capsularis55. According to Podevin and colleagues (2012), by contrast, ACT1, EF1α and β-TUB were identified as the most stable reference genes for leaf samples of a Cavendish genotype, with L2 and 25 S the least stable genes. In the same work ACT1 and β-TUB were identified as the most stable genes for leaf samples inoculated with P. fijiensis30. If we consider our sample groups, where leaf material was inoculated with P. musae, APT and GAPDH were always among the most stable genes for Calcutta 4 samples, while GAPDH and EF1α were the most stable genes for Cavendish Grande Naine. Such results highlight how expression stability of reference genes varies according to genotype, tissue or pathogen employed in biotic stress investigation.
Primer design must also be considered in order to avoid unspecific amplification and formation of secondary structures that may hamper the polymerase chain reaction. Since actin is commonly used as a reference gene for RT-qPCR, we designed two primer pairs for ACT1, each annealing at different positions within the target gene sequence, in order to test whether they would present different stability patterns. Both primer pairs resulted in similar size amplicons, presented similar Tms (°C), and showed similar amplification efficiencies (%) (Table 1). Results revealed, however, that gene expression following amplification with primer pair ACT1pp2 was more stable than that observed using primer pair ACT1pp1. Such results highlight that in addition to gene selection as reference for RT-qPCR analysis, target gene region for amplification, primer annealing temperature, and amplicon size can all influence the efficiency of an experiment56. In our study, we restricted amplicon size difference to a maximum of 66 bp, with all selected genes transcribed by RNA polymerase II, the same enzyme involved in gene expression of most of the target genes studied by our group. Such standardization of annealing temperature, amplicon size and consideration of RNA polymerase enzyme for reference and target gene transcription is the first for Musa reference genes for RT-qPCR.
RuBisCO is a key enzyme of the photosynthetic CO2 assimilation and photorespiratory pathways57. This enzyme comprises a large subunit, encoded by a plastidial gene, and smaller subunits, encoded by nuclear genes58,59. Activity can be regulated by RuBisCO activase (RCA), a chloroplast protein encoded by the nuclear genome. This enzyme facilitates dissociation of the sugar phosphates from RuBisCO, decreasing CO2 concentration for catalyzing the carbamylation of its catalytic lysine60,61. Previous studies have shown that RCA participates in the developmental stages of the leaves, increasing activity in mature tissues62. Silencing of an RCA isoform has also been shown to result in a reduction in photosynthesis63,64. Jiang and coworkers65 identified two RCA genes in sweet potato, where mRNA levels varied according to the intensity of light or heat65. Here, we identified three RCA genes in the M. acuminata reference (DH Pahang) genome, with RCA3 amplifiable by conventional PCR using genomic DNA and cDNA from M. acuminata Calcutta 4 and Cavendish Grande Naine genotypes. In order to validate the stability of the reference genes, we examined the relative expression of RCA3 in both genotypes subjected to light/dark treatments. When normalization of RT-qPCR was performed using the most stable reference genes (UBQ2 and RAN), a reduced relative expression of RCA3 was observed for dark treated leaf samples, with little variation between cDNA samples for biological replicates. By contrast, when normalization was conducted with the least stable reference genes (α-TUB and ACT1pp1), an over-estimation of transcript levels was observed in certain samples. These results clearly indicated that different reference genes influenced the relative expression levels of target genes under the specific conditions employed in this study. Similar such inaccurate estimation of relative expression of target genes during validation of potential reference genes has been shown across different plant taxa, for example the grass species Urochloa brizantha, and Coffea spp66,67.
In summary, this is the first report detailing validation of reference genes in M. acuminata genotypes contrasting in resistance to P. musae. As determined using the program geNorm, of the eight candidate reference genes tested, a minimum number of two genes were always sufficient to provide the necessary accuracy for RT-qPCR analysis under the experimental conditions employed. Considering the frequency of the top ranked stably expressed reference genes, UBQ2 and RAN are appropriate for normalization in studies that consider P. musae-inoculated or non-inoculated leaf samples for both M. acuminata genotypes Calcutta 4 and Cavendish Grande Naine. The various reference gene sets developed here for the genotypes contrasting in resistance to the Sigatoka leaf spot pathogen P. musae will enable accurate analysis of gene expression and pathways activated in this pathosystem, advancing understanding of host responses to this important foliar pathogen.
Methods
Plant material
M. acuminata plantlets of susceptible commercial Cavendish subgroup cultivar ‘Grande Naine’ (CAV) (AAA) (Musa International Transit Centre accession ITC0654) and the resistant wild fertile diploid Calcutta 4 (C4) (AA) (accession ITC0249), widely employed as a breeding genitor genotype, were provided by Embrapa Cassava and Tropical Fruits. In vitro-derived plantlets were grown in a mix of sterilized substrate of soil and sand (1:1), fertilizer and lime under glasshouse conditions with a 12-h light/12-h dark photoperiod, 85% relative humidity and an average temperature of 25 °C.
Leaf inoculation with P. musae
The youngest emerged leaf from 3 month old plants was artificially inoculated with a suspension comprising 2 × 104 conidiospores ml−1 of P. musae and the surfactant Tween 20 at 0.05%. Non-inoculated control plants were inoculated with only the water-surfactant mixture. A total of three independent replicates were collected for each sample.
RNA extraction and cDNA synthesis
Leaf material (2.5 cm2 area) from pathogen-inoculated and non-inoculated controls was collected at 3 and 12 days, flash frozen in liquid nitrogen and stored at −80° C. Total RNA was extracted from leaf tissues using the Concert® Plant RNA Reagent (Invitrogen, Carlsbad, CA, USA), with purification using the INVISORB Spin Plant RNA Mini Kit (Invitek, Hayward, CA, USA), according to manufacturer’s instructions. A total of 1 μg of each total RNA was treated with 2 U of DNase I (New England Biolabs, Ipswich, MA., USA) for elimination of residual DNA. Analysis of total RNA concentration and integrity was conducted following 1% agarose gel electrophoresis and Nanodrop ND-1000 spectrophotometry (Thermo Fischer Scientific, Waltham, MA, USA).
For cDNA synthesis, pools containing equimolar amounts of total RNA from three biological replicates were prepared. In total, eight pools were prepared, one for each experimental condition (Table 1). A total of 1 μg of total RNA was reverse transcribed to cDNA using Super Script II RT and Oligo(dT)16–18 primers (Invitrogen, Carlsbad, CA, USA).
Primer design
For the design of potential reference genes with stable expression in M. acuminata leaf tissues, candidate genes were selected that encode proteins involved in basal cell activites in diverse plant species, including Musa (Table 1). OligoPerfectTM Designer (Thermo Fischer Scientific, Waltham, MA, USA) was employed for design of specific primers for each candidate reference gene. Expected target amplicons varied from 80 to 150 bp, with all primers designed to specifically amplify predicted exon-exon junctions, guaranteeing amplification from cDNA rather than potential contaminant genomic DNA. Primer specificity was initially tested by electronic PCR against a local database of RNAseq data for Musa and against the reference whole genome sequence for M. acuminata, genotype DH Pahang at Phytozome (phytozome.jgi.doe.gov)10. Specificity and efficiency was subsequently tested against cDNA originating from leaf tissues of M. acuminata CAV and C4 (data not shown). Primer sequences that resulted in specific amplification are listed in Table 1.
Real-time quantitative PCR
RT-qPCR analysis of expression of candidate reference genes was conducted using a Platinum SYBR Green qPCR Super Mix-UDG w/ROX kit (Invitrogen, Carlsbad, CA, USA). PCR amplifications were performed on an ABI StepOne® Real-Time PCR thermocycler (Applied Biosystems, Foster City, USA), using three independent experimental replicates and three technical replicates per amplification. For PCR reactions, mixtures contained 2 μL of a 1:20 dilution of template cDNA, 0.2 μM of each primer (Table 1) and 5 μL Platinum® SYBR® Green qPCR Super Mix-UDG w/ROX kit (Invitrogen, Carlsbad, CA, USA), to a final volume of 10 μL. PCR amplification was conducted using an initial step of 52 °C for 2 min, 95 °C for 10 min, followed by 40 cycles of denaturation at 95 °C for 15 s, primer annealing and extension at 60 °C for 60 s. Primer specificity was verified by analyzing the Tm (dissociation) of amplified products using SDS 2.2.2 software (Applied Biosystems, Foster City, USA).
Baseline correction and efficiency determination
Raw ΔRn data was analysed using the LinRegPCR program, version 2017.1, with all calculations performed according to the manufacturer’s instructions. Baseline fluorescence was corrected by reconstruction of the log-linear phase. Data was then grouped and applied to determine RT-qPCR mean efficiency for each gene. The same program was subsequently used to calculate the average quantification cycles (Cqs) per sample68.
Expression stability and relative expression analysis
Expression data for each candidate reference gene was visualized as a quantification cycle (Cq) value from each RNA sample, as obtained from LinRegPCR analysis. This value represents the number of amplification cycles required to reach a default threshold value for detection during the exponential amplification phase of the PCR reaction.
The expression stability of each candidate reference gene was determined using the algorithms geNorm22, NormFinder23 and BestKeeper24, according to the default software parameters. GeNorm and NormFinder use the Cq data to calculate Relative Quantities (RQ) by application of the formula RQ = 2(ddCq). GeNorm evaluates expression stability by calculating an average M-value, which is defined as the pairwise variation of one gene against all other genes. Those genes presenting the lowest M-values below a threshold of 0.5 have the most stable expression. The program also calculates a V-value, a pairwise variation (Vn/n + 1) between each reference gene, to determine the optimal number of genes required for normalization. In the case of NormFinder, variation between groups of genes at both the inter- and intra-group level is calculated with ANOVA and used to generate a stability value (SV) for each reference gene, with the most stable genes presenting the lowest SV values below the default limit of 0.5. As such, this method can be more appropriate for providing estimates of expression variation from subsets of different sample types23. BestKeeper is a tool that calculates the geometric mean of Cq values, with the most stable genes indicated by high correlation coefficients and low standard deviations. Raw Cq values are used to calculate the geometric mean and rank the reference genes according to the lowest stability values. A global re-ranking of the candidate genes stability was conducted using the web-based tool RefFinder (http://www.leonxie.com/referencegene.php). RefFinder allows integration of data generated with all the algorithms geNorm, NormFinder, Bestkeeper and delta Ct method, without considering RT-qPCR efficiencies. Through assigning a weight to each individual gene, a geometric mean weight of the candidate genes can be calculated, which can then be used to rank the stability of each individual reference gene.
RCA3 expression analysis
The youngest fully emerged leaf from 3 month old plants was subjected to a photoperiod of 12/12 (L/D) at 25 °C for 7 days. In a second group, leaves were covered in aluminium foil paper and maintained at 25 °C for 7 days. After this period, leaves were flash frozen in liquid nitrogen and preserved at −80 °C until use. Three independent replicates were collected for each leaf sample. Total RNA, cDNA synthesis and real time PCR were performed as described previously.
Data Availability
All data generated and analyzed during the study is included in the published article and its Supplementary Information files.
References
CTA. Diseases of Banana, Abacá, and Enset. 544pp (CABI Pub., 2000).
Stover, R. Effect of temperature on ascospore germ tube growth of Mycosphaerella musicola and Mycosphaerella fijiensis var. difformis. Fruits. 38(9): 625–628. Francia (1983).
Crous, P. W. & Mourichon, X. Mycosphaerella eumusae and its anamorph Pseudocercospora eumusae spp. nov.: causal agent of eumusae leaf spot disease of banana. Sydowia-Horn 54, 23–34 (2002).
Gomes, L. I. S., Bibiano, L. B. J., Silva, G. F. D., Hanada, R. E. & Mizubuti, E. S. G. Baseline sensitivity of Brazilian Mycosphaerella fijiensis isolates to protectant and systemic fungicides. Tropical Plant Pathology 39, 172–177 (2014).
Brito, F. S. D., Fraaije, B. & Miller, R. N. Sigatoka Disease Complex of Banana in Brazil: Management Practices and Future Directions. Outlooks on Pest Management 26, 78–81 (2015).
Wang, Z. et al. De Novo characterization of the banana root transcriptome and analysis of gene expression under Fusarium oxysporum f. sp. Cubense tropical race 4 infection. BMC Genomics 13, 650, https://doi.org/10.1186/1471-2164-13-650 (2012).
Li, C.-y et al. Transcriptome profiling of resistant and susceptible Cavendish banana roots following inoculation with Fusarium oxysporum f. sp. cubense tropical race 4. BMC Genomics 13, 374, https://doi.org/10.1186/1471-2164-13-374 (2012).
Bai, T.-T. et al. Transcriptome and Expression Profile Analysis of Highly Resistant and Susceptible Banana Roots Challenged with Fusarium oxysporum f. sp. cubense Tropical Race 4. Plos One 8, e73945, https://doi.org/10.1371/journal.pone.0073945 (2013).
Castañeda, N. E. N. et al. Gene expression analysis in Musa acuminata during compatible interactions with Meloidogyne incognita. Annals of Botany 119, 915–930, https://doi.org/10.1093/aob/mcw272 (2017).
D’Hont, A. et al. The banana (Musa acuminata) genome and the evolution of monocotyledonous plants. Nature 488, 213, https://doi.org/10.1038/nature11241 (2012).
Passos, M. A. N. et al. Analysis of the leaf transcriptome of Musa acuminata during interaction with Mycosphaerella musicola: gene assembly, annotation and marker development. BMC Genomics 14, 78–78, https://doi.org/10.1186/1471-2164-14-78 (2013).
Gachon, C., Mingam, A., Charrier, B. & Real-time, P. C. R. what relevance to plant studies? Journal of Experimental Botany 55, 1445–1454, https://doi.org/10.1093/jxb/erh181 (2004).
Pfaffl, M. W. In A-Z of quantitative PCR (ed S A Bustin) Ch. 3, 87–112 (La Jolla, CA: International University Line, 2004).
Bustin, S. A. et al. The MIQE guidelines: minimum information for publication of quantitative real-time PCR experiments. Clinical chemistry 55, 611–622 (2009).
Bustin, S. A., Benes, V., Nolan, T. & Pfaffl, M. W. Quantitative real-time RT-PCR – a perspective. Journal of Molecular Endocrinology 34, 597–601, https://doi.org/10.1677/jme.1.01755 (2005).
Laurent, G. et al. The lack of a systematic validation of reference genes: a serious pitfall undervalued in reverse transcription-polymerase chain reaction (RT-PCR) analysis in plants. Plant Biotechnology Journal 6, 609–618, https://doi.org/10.1111/j.1467-7652.2008.00346.x (2008).
Radonić, A. et al. Guideline to reference gene selection for quantitative real-time PCR. Biochemical and Biophysical Research Communications 313, 856–862, https://doi.org/10.1016/j.bbrc.2003.11.177 (2004).
Nicot, N., Hausman, J.-F., Hoffmann, L. & Evers, D. Housekeeping gene selection for real-time RT-PCR normalization in potato during biotic and abiotic stress. Journal of Experimental Botany 56, 2907–2914, https://doi.org/10.1093/jxb/eri285 (2005).
Miranda, Vd. J. et al. Validation of reference genes aiming accurate normalization of qPCR data in soybean upon nematode parasitism and insect attack. BMC Research Notes 6, 196, https://doi.org/10.1186/1756-0500-6-196 (2013).
Reddy, D. S., Bhatnagar-Mathur, P., Cindhuri, K. S. & Sharma, K. K. Evaluation and validation of reference genes for normalization of quantitative real-time PCR based gene expression studies in peanut. PloS one 8, e78555 (2013).
Bustin, S. A. & Nolan, T. Pitfalls of Quantitative Real-Time Reverse-Transcription Polymerase Chain Reaction. Journal of Biomolecular Techniques: JBT 15, 155–166 (2004).
Vandesompele, J. et al. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biology 3, research0034.0031-research0034.0011 (2002).
Andersen, C. L., Jensen, J. L. & Ørntoft, T. F. Normalization of Real-Time Quantitative Reverse Transcription-PCR Data: A Model-Based Variance Estimation Approach to Identify Genes Suited for Normalization, Applied to Bladder and Colon Cancer Data Sets. Cancer Research 64, 5245 (2004).
Pfaffl, M. W., Tichopad, A., Prgomet, C. & Neuvians, T. P. Determination of stable housekeeping genes, differentially regulated target genes and sample integrity: BestKeeper–Excel-based tool using pair-wise correlations. Biotechnology letters 26, 509–515 (2004).
Xu, H., Bao, J.-D., Dai, J.-S., Li, Y. & Zhu, Y. Genome-Wide Identification of New Reference Genes for qRT-PCR Normalization under High Temperature Stress in Rice Endosperm. Plos One 10, e0142015, https://doi.org/10.1371/journal.pone.0142015 (2015).
Lin, Y. et al. Validation of Potential Reference Genes for qPCR in Maize across Abiotic Stresses, Hormone Treatments, and Tissue Types. Plos One 9, e95445, https://doi.org/10.1371/journal.pone.0095445 (2014).
de Andrade, L. M. et al. Reference genes for normalization of qPCR assays in sugarcane plants under water deficit. Plant Methods 13, 28, https://doi.org/10.1186/s13007-017-0178-2 (2017).
Martins, P. K. et al. Selection of reliable reference genes for RT-qPCR analysis during developmental stages and abiotic stress in Setaria viridis. Scientific Reports 6, 28348, https://doi.org/10.1038/srep28348 (2016).
Chen, L. et al. Validation of reference genes for RT-qPCR studies of gene expression in banana fruit under different experimental conditions. Planta 234, 377, https://doi.org/10.1007/s00425-011-1410-3 (2011).
Podevin, N., Krauss, A., Henry, I., Swennen, R. & Remy, S. Selection and validation of reference genes for quantitative RT-PCR expression studies of the non-model crop Musa. Molecular Breeding 30, 1237–1252, https://doi.org/10.1007/s11032-012-9711-1 (2012).
Zhang, L. et al. Identification and validation of reference genes for RT-qPCR analysis in banana (Musa spp.) under Fusarium wilt resistance induction conditions. Journal of Phytopathology 165, 746–754, https://doi.org/10.1111/jph.12614 (2017).
Asif, M. H. et al. Transcriptome analysis of ripe and unripe fruit tissue of banana identifies major metabolic networks involved in fruit ripening process. BMC Plant Biology 14, 316, https://doi.org/10.1186/s12870-014-0316-1 (2014).
Ba, L.-j et al. A ripening-induced transcription factor MaBSD1 interacts with promoters of MaEXP1/2 from banana fruit. Plant Cell Reports 33, 1913–1920, https://doi.org/10.1007/s00299-014-1668-6 (2014).
Buah, S., Mlalazi, B., Khanna, H., Dale, J. L. & Mortimer, C. L. The Quest for Golden Bananas: Investigating Carotenoid Regulation in a Fe’i Group Musa Cultivar. Journal of Agricultural and Food Chemistry 64, 3176–3185, https://doi.org/10.1021/acs.jafc.5b05740 (2016).
Elitzur, T., Vrebalov, J., Giovannoni, J. J., Goldschmidt, E. E. & Friedman, H. The regulation of MADS-box gene expression during ripening of banana and their regulatory interaction with ethylene. Journal of Experimental Botany 61, 1523–1535, https://doi.org/10.1093/jxb/erq017 (2010).
Feng, X., Lai, Z., Lin, Y., Lai, G. & Lian, C. Genome-wide identification and characterization of the superoxide dismutase gene family in Musa acuminata cv. Tianbaojiao (AAA group). BMC Genomics 16, 823, https://doi.org/10.1186/s12864-015-2046-7 (2015).
Ge, Y. et al. Hydrogen sulfide alleviates postharvest ripening and senescence of banana by antagonizing the effect of ethylene. Plos One 12, e0180113, https://doi.org/10.1371/journal.pone.0180113 (2017).
Kumaravel, M. et al. Differential proteome analysis during early somatic embryogenesis in Musa spp. AAA cv. Grand Naine. Plant Cell Reports 36, 163–178, https://doi.org/10.1007/s00299-016-2067-y (2017).
Lee, W. S. et al. Transcripts and MicroRNAs Responding to Salt Stress in Musa acuminata Colla (AAA Group) cv. Berangan Roots. PLOS ONE 10, e0127526, https://doi.org/10.1371/journal.pone.0127526 (2015).
Li, C. et al. Analysis of banana transcriptome and global gene expression profiles in banana roots in response to infection by race 1 and tropical race 4 of Fusarium oxysporum f. sp. cubense. BMC Genomics 14, 851, https://doi.org/10.1186/1471-2164-14-851 (2013).
Mbéguié-A-Mbéguié, D. et al. Use of suppression subtractive hybridization approach to identify genes differentially expressed during early banana fruit development undergoing changes in ethylene responsiveness. Plant Science 172, 1025–1036, https://doi.org/10.1016/j.plantsci.2007.02.007 (2007).
Thomas-Hall, S. et al. Phylogenetic and molecular analysis of the ribulose-1,5-bisphosphate carboxylase small subunit gene family in banana. Journal of Experimental Botany 58, 2685–2697, https://doi.org/10.1093/jxb/erm129 (2007).
Noëlani, V. D. B. et al. Tolerance in banana to Fusarium wilt is associated with early up-regulation of cell wall-strengthening genes in the roots. Molecular Plant Pathology 8, 333–341, https://doi.org/10.1111/j.1364-3703.2007.00389.x (2007).
Yang, Q.-S. et al. Comparative transcriptomics analysis reveals difference of key gene expression between banana and plantain in response to cold stress. BMC Genomics 16, 446, https://doi.org/10.1186/s12864-015-1551-z (2015).
Li, J., Han, J., Hu, Y. & Yang, J. Selection of Reference Genes for Quantitative Real-Time PCR during Flower Development in Tree Peony (Paeonia suffruticosa Andr.). Frontiers in Plant Science 7, 516, https://doi.org/10.3389/fpls.2016.00516 (2016).
Kałużna, M., Kuras, A. & Puławska, J. Validation of reference genes for the normalization of the RT-qPCR gene expression of virulence genes of Erwinia amylovora in apple shoots. Scientific Reports 7, 2034, https://doi.org/10.1038/s41598-017-02078-4 (2017).
Yang, C., Pan, H., Liu, Y. & Zhou, X. Stably Expressed Housekeeping Genes across Developmental Stages in the Two-Spotted Spider Mite, Tetranychus urticae. Plos One 10, e0120833, https://doi.org/10.1371/journal.pone.0120833 (2015).
Yan, X. et al. Reference Gene Selection for Quantitative Real-Time PCR Normalization in Reaumuria soongorica. Plos One 9, e104124, https://doi.org/10.1371/journal.pone.0104124 (2014).
Joseph, J. T., Poolakkalody, N. J. & Shah, J. M. Plant reference genes for development and stress response studies. Journal of biosciences 1, 173–187 (2018).
Jain, M., Nijhawan, A., Tyagi, A. K. & Khurana, J. P. Validation of housekeeping genes as internal control for studying gene expression in rice by quantitative real-time PCR. Biochemical and Biophysical Research Communications 345, 646–651, https://doi.org/10.1016/j.bbrc.2006.04.140 (2006).
Ueno, Y. et al. Abiotic Stresses Antagonize the Rice Defence Pathway through the Tyrosine-Dephosphorylation of OsMPK6. PLOS Pathogens 11, e1005231, https://doi.org/10.1371/journal.ppat.1005231 (2015).
Chen, F., Li, Q., Sun, L. & He, Z. The Rice 14-3-3 Gene Family and its Involvement in Responses to Biotic and Abiotic Stress. DNA Research 13, 53–63, https://doi.org/10.1093/dnares/dsl001 (2006).
Xu, P., Zang, A., Chen, H. & Cai, W. The Small G Protein AtRAN1 Regulates Vegetative Growth and Stress Tolerance in Arabidopsis thaliana. Plos One 11, e0154787, https://doi.org/10.1371/journal.pone.0154787 (2016).
Kong, Q. et al. Screening Suitable Reference Genes for Normalization in Reverse Transcription Quantitative Real-Time PCR Analysis in Melon. Plos One 9, e87197, https://doi.org/10.1371/journal.pone.0087197 (2014).
Niu, X. et al. Selection of reliable reference genes for quantitative real-time PCR gene expression analysis in Jute (Corchorus capsularis) under stress treatments. Frontiers in Plant Science 6, https://doi.org/10.3389/fpls.2015.00848 (2015).
Debode, F., Marien, A., Janssen, É., Bragard, C. & Berben, G. Influence of the amplicon length on real-time PCR results. Biotechnologie. Agronomie, Société et Environnement 21, 3–11 (2017).
Tcherkez, G. G., Farquhar, G. D. & Andrews, T. J. Despite slow catalysis and confused substrate specificity, all ribulose bisphosphate carboxylases may be nearly perfectly optimized. Proceedings of the National Academy of Sciences 103, 7246–7251 (2006).
Dhingra, A., Portis, A. R. & Daniell, H. Enhanced translation of a chloroplast-expressed RbcS gene restores small subunit levels and photosynthesis in nuclear RbcS antisense plants. Proceedings of the National Academy of Sciences 101, 6315–6320 (2004).
Yoon, M., Putterill, J. J., Ross, G. S. & Laing, W. A. Determination of the Relative Expression Levels of Rubisco Small Subunit Genes in Arabidopsis by Rapid Amplification of cDNA Ends. Analytical Biochemistry 291, 237–244, https://doi.org/10.1006/abio.2001.5042 (2001).
Portis, A. R. Rubisco activase – Rubisco’s catalytic chaperone. Photosynthesis Research 75, 11–27, https://doi.org/10.1023/a:1022458108678 (2003).
Stec, B. Structural mechanism of RuBisCO activation by carbamylation of the active site lysine. Proceedings of the National Academy of Sciences, 201210754 (2012).
Kurek, I. et al. Enhanced Thermostability of Arabidopsis Rubisco Activase Improves Photosynthesis and Growth Rates under Moderate Heat Stress. The Plant Cell 19, 3230–3241, https://doi.org/10.1105/tpc.107.054171 (2007).
Sharkey, T. D., Badger, M. R., von Caemmerer, S. & Andrews, T. J. Increased heat sensitivity of photosynthesis in tobacco plants with reduced Rubisco activase. Photosynthesis Research 67, 147–156, https://doi.org/10.1023/a:1010633823747 (2001).
Cai-Zhong, J., Paul, Q. W., Rhu, A., Daniel, K. & R., R. S. Antisense RNA inhibition of Rubisco activase expression. The Plant Journal 5, 787–798, https://doi.org/10.1046/j.1365-313X.1994.5060787.x (1994).
Jiang, Y., Wang, J., Tao, X. & Zhang, Y. Characterization and expression of Rubisco activase genes in Ipomoea batatas. Molecular Biology Reports 40, 6309–6321, https://doi.org/10.1007/s11033-013-2744-7 (2013).
Takamori, L. M., Pereira, A. V. C., Maia Souza, G., Vieira, L. G. E. & Ferreira Ribas, A. Identification of Endogenous Reference Genes for RT-qPCR Expression Analysis in Urochloa brizantha Under Abiotic Stresses. Scientific Reports 7, 8502, https://doi.org/10.1038/s41598-017-09156-7 (2017).
Martins, M. Q. et al. Selection and Validation of Reference Genes for Accurate RT-qPCR Data Normalization in Coffea spp. under a Climate Changes Context of Interacting Elevated [CO(2)] and Temperature. Frontiers in Plant Science 8, 307, https://doi.org/10.3389/fpls.2017.00307 (2017).
Ruijter, J. M. et al. Amplification efficiency: linking baseline and bias in the analysis of quantitative PCR data. Nucleic Acids Research 37, e45–e45, https://doi.org/10.1093/nar/gkp045 (2009).
Acknowledgements
This work was partially funded by Brazilian funding bodies FAPDF (Grant 0193.001725/2017), Instituto Nacional de Ciência e Tecnologia PlantStress (Grant 465480/2014-4), and CAPES (Finance Code 001). RNGM was supported by a fellowship from CNPq (Grant 305418/2017-3).
Author information
Authors and Affiliations
Contributions
R.N.G.M., F.C.A.F., G.S.C.A., M.G.C. and J.D.A. conceived and designed the study. E.C.S.R. and T.D.M.P. conducted the biotic assays and RT-qPCR analysis, contributing equally to the study. F.C.A.F., J.D.A., G.S.C.A., M.G.C. and R.N.G.M. analyzed the data, provided intellectual input, wrote and revised the manuscript. All authors have read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Rego, E.C.S., Pinheiro, T.D.M., Antonino, J.D. et al. Stable reference genes for RT-qPCR analysis of gene expression in the Musa acuminata-Pseudocercospora musae interaction. Sci Rep 9, 14592 (2019). https://doi.org/10.1038/s41598-019-51040-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-51040-z
This article is cited by
-
Evaluating the stability of host-reference gene expression and simultaneously quantifying parasite burden and host immune responses in murine malaria
Scientific Reports (2023)
-
Development of reference genes for RT-qPCR analysis of gene expression in Pleurotus pulmonarius for biotechnological applications
Scientific Reports (2023)
-
Validation of reference genes as an internal control for studying Avena sativa–Puccinia coronata interaction by RT-qPCR
Scientific Reports (2022)
-
Identification of RT-qPCR reference genes suitable for gene function studies in the pitaya canker disease pathogen Neoscytalidium dimidiatum
Scientific Reports (2022)
-
Evaluation of reference genes for transcript normalization in Fragaria chiloensis fruit and vegetative tissues
Physiology and Molecular Biology of Plants (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
