
Abstract
Cerebral cavernous malformation (CCM) is a vascular malformation characterized by clustered enlarged capillary-like channels in the central nervous system. The genes harboring variants in patients with CCM include CCM1/Krev interaction trapped-1, CCM2/MGC4607, and CCM3/programmed cell death protein 10. We aimed to identify pathogenic variants in an ethnic Chinese population in Taiwan. We recruited 95 patients with multiple CCMs or a single lesion with a relevant family history. Sanger sequencing was performed for 41 patients. Variants were identified using sequence alignment tools, and the clinical significance of these variants was determined using American College of Medical Genetics and Genomics standards and guidelines. Several pathogenic variants were found in six patients, including three unrelated patients and three affected members of one family. Two novel pathogenic variants leading to early truncation comprised a deletion variant in exon 18 of CCM1 (c.1846delA; p.Glu617LysfsTer44) and an insertion variant in exon 4 of CCM2 (c.401_402insGCCC; p.Ile136AlafsTer4). One novel pathogenic splice site variant was c.485 + 1G > C at the beginning of intron 8 of CCM1. In this study, we identified novel variants related to CCM in an ethnically Chinese population in Taiwan.
Similar content being viewed by others


Introduction
Cerebral cavernous malformation (CCM, OMIM 116860) is a vascular malformation characterized by clustered enlarged capillary-like channels and the absence of the intervening neural tissue. The prevalence of CCM in the general population is estimated to be 0.5%1. CCM can be sporadic or familial, and familial cases usually have multiple lesions2. The symptoms of CCM include hemorrhage-related focal neurological deficits, seizure, and headache; however, patients with CCM are usually asymptomatic1. In familial individuals, three related genes, namely CCM1 (Krev interaction trapped-1, OMIM 604214), CCM2 (MGC4607, OMIM 607929), and CCM3 (programmed cell death protein 10, OMIM 609118), have been reported3,4,5. The inheritance is autosomal dominant with incomplete clinical and neuroradiological penetrance2. Multiple pathogenic variants have been reported in these genes, and several studies have been conducted in ethnically Chinese populations6,7,8,9,10,11,12,13,14,15,16,17,18,19, with only one case reported in Taiwan20. Therefore, we retrospectively collected clinical and magnetic resonance imaging (MRI) data of patients who had received a diagnosis of CCM on the basis of brain MRI findings from 1998 to 2006 in a tertiary medical center. Thereafter, we performed DNA analysis to identify variants in patients with multiple lesions or with relevant family histories. We aimed to establish an epidemiological and clinical data bank and identify CCM pathological variants in an ethnic Chinese population in Taiwan.
Results
Basic demographic profile and clinical presentation
We recruited 95 patients with multiple CCMs or a single CCM with a relevant family history (Table 1). Of the 95 patients, 15 (15.8%) had a relevant family history and the remaining 80 (84.2%) had a sporadic onset. 92 patients (96.8%) had multiple cerebral lesions and only 3 (3.2%) had a single lesion in their brain. The most common initial presentation was focal neurological signs (54 patients, 56.8%), including weakness, numbness, diplopia, and dysarthria, followed by seizure (28 patients, 29.5%; 3.2% with concurrent focal neurological signs) and headache (21 patients, 22.1%; 13.7% with concurrent focal neurological signs or seizure); however, only eight patients (8.4%) were asymptomatic.
In addition, 46 (48.4%), 26 (27.4%) and 23 (24.2%) patients had supratentorial hemangioma, infratentorial lesions, and both supratentorial and infratentorial lesions, respectively. Only two (2.1%) patients had extracranial cavernous hemangioma at the spinal cord. Among the 95 patients, 30 (31.6%) underwent surgical lesion removal and 38 (40.0%) received radiosurgery before this study because of recurrence or critical lesion sites.
DNA analysis
Of the 95 patients, we collected blood samples from 41 (43.1%), which comprised 29 patients with unrelated sporadic cases, 9 with unrelated familial cases, and 3 who were affected members of one family. The pedigree of this family is depicted in Fig. 1. We could not obtain blood samples from the remaining 54 patients because of loss to follow-up (47) or patients’ refusal (7). Five patients and one patient had pathogenic CCM1 and CCM2 variants, respectively; additionally, one patient possessed a CCM2 variant of uncertain significance. The clinical manifestations and gene analysis results are listed in Table 2. Among the patients with variants, one (Patient 20) underwent both surgical removal and radiotherapy for hemisphere lesions and another (Patient 54) received only radiotherapy because of symptomatic CCMs. One patient (Patient 20) experienced another episode of paraplegia, and a follow-up MRI study revealed a cavernous hemangioma in the spinal cord.

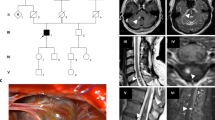
Pedigree of three affected patients with one pathogenic CCM1 variant from one family. A square represents a male patient, whereas a circle represents a female patient. A line across the symbol indicates the patient is deceased.
Radiological finding
All the patients with pathogenic CCM1 variants had multiple lesions in the bilateral hemisphere (Fig. 2A, C–F), and one patient had lesions in the spinal cord, as observed in the follow-up MRI study (Fig. 2B). Not only the patient (Patient 54) with one pathogenic CCM2 variant but also the one with one CCM2 variant of uncertain significance (Patient 69) possessed a prominent pontine lesion (Fig. 2G,H,I). Those images were reviewed by experienced radiologists based on susceptibility-weighted images21. The number of lesions among the six patients with pathogenic variants was not affected by genes (p = 0.77), sex (p = 0.827), or age (p = 0.22).

Brain MRI of patients with CCM variants. (A) Patient 20 with two CCM1 variants: multiple lesions of variable sizes in hemispheres in susceptibility weighted imaging (SWI); (B) Patient 20 with one spinal cord lesion in a T2-weight image; (C) Patient 40 with one CCM1 variant: multiple small lesions in hemispheres in SWI; (D–F) Patients 57 (D), 66 (E), and 67 (F) from one family with one CCM1 variant: multiple hemisphere lesions with variable sizes in SWI; (G,H) Patient 54 with one CCM2 variant: one prominent pontine lesion (G) and small lesions in bilateral hemispheres (H) in SWI; (I) Patient 69 with one CCM2 variant of uncertain significance: one lesion in the right pons and the other in the right basal ganglion (not shown) (arrow indicates the lesion).
Variant analysis and prediction
In the variant analysis, Patient 20 had two heterozygous CCM1 variants including one novel missense variant of uncertain significance (c.1844G > C, p.Ser615Thr) and one novel one-base pair (bp) pathogenic deletion variant (c.1846delA, p.Glu617LysfsTer44) simultaneously in the exon 18 (Fig. 3A; European Nucleotide Archive (ENA) accession number: ERZ842272). Patient 40 possessed one novel heterozygous pathogenic CCM1 splice site variant at the beginning of intron 8 (c.485 + 1G > C) (Fig. 3B; ENA accession number: ERZ842273). Patients 57, 66, and 67 had one pathogenic CCM1 3-bp deletion variant (c.1255-4_1255-2delGTA or c.1255-1_1256delGTA; previously submitted ClinVar accession number: RCV000532224.2) in intron 13, leading to a splice site variant (Fig. 3C). Patient 54 possessed one novel CCM2 pathogenic 4-bp insertion variant in exon 4 (c.401_402insGCCC, p.Ile136AlafsTer4) (Fig. 3D; ENA assessment number: ERZ842275). Finally, Patient 69 had one CCM2 missense variant of uncertain significance in exon 9 [c.970G > A, p.Glu324Lys; minor allele frequency (MAF) of A (0.000008) in Exome Aggregation Consortium (ExAC) databank] (Fig. 3E). All previously described missense variants were located in the evolutionary conservation of sequences among functional domains (Fig. 4).

Five sequencing chromatograms of patients with variants. Each part includes a chromatogram, an interpreted sequence, reference sequences (human), and an amino acid sequence (left side). The area of intron is marked within a rectangle with a dotted line, and the sites of variants are marked within the rectangle with a solid line. The underlying lines below the sequence indicate reading frames. (A) CCM1 in Patient 20: one missense (G > C) and one deletion (del-A) variant within exon 18, leading to frameshift. (B) CCM1 in Patient 40: one splice site variant (G > C) at the first base pair of intron 8. (C) CCM1 in Patients 57, 66, and 67: one 3-base pair deletion (del-GTA) variant in intron 13, alternating mRNA splicing. (D) CCM2 in Patient 54: one 4-base pair insertion variant (ins-GCCC) within exon 4, causing frameshift. (E) CCM2 in Patient 69: one missense variant (G > A) within exon 9.

Schematic representation of the gene structure and protein domains of (A) CCM1 (KRIT1) and (B) CCM2 in a Chinese population. Each part contained an upper panel and a lower panel. The upper panel contains the genetic structure and exon position and number, and the lower panel contains a corresponding protein product and its specific domain. Each variant is marked with a double-headed arrow, indicating a variant and corresponding amino acid change, respectively. All variants reported previously in the Chinese population are shown, and variants reported in these studies are marked with rectangles.
Discussion
In familial CCM, pathogenic variants in three genes have been reported3,4,5. Our results revealed five patients with CCM1 variants and two patients with CCM2 variants in 41 ethnically Chinese patients with multiple CCMs or a single CCM with a relevant family history. All the patients (100%) with variants had multiple lesions. The size and number of lesions in the brain MRI were smaller and lower, respectively, in patients with CCM2 variants than in those with CCM1 variants. This finding is compatible with those of previous studies, indicating that CCM2 variant carriers may have a milder phenotype than CCM1 variant carriers22.
In our study, for the detection rate of variants in familial CCM (7.6% [1 of 13]; three affected members of one family among 15 patients) and sporadic (3.75% [3 of 80]; 3 sporadic cases of 80 patients) was lower than that in previous studies. Studies have reported a variable detection rate of variants ranging from 16% to 60% in sporadic cases and from 70% to 90% in familial cases23,24,25. The rate may be underestimated because DNA analysis in our study could be performed only in 41 patients (43.1%). The DNA analysis technique is essential because Sanger sequencing may miss large deletions, large insertions, or duplications. Moreover, pathogenic variants of other genes causing CCM may be another factor. A case report showed that a balanced translocation between chromosome 3 and chromosome X caused decreased zona pellucida-like domain-containing protein 1 (ZPLD1) expression and led to multiple cerebral cavernous hemangiomas26. The function of ZPLD1 in pathogenesis of CCMs is unknown.
Among the patients with CCM1 variants, one sporadic case (Patient 20) had a combination of a novel missense variants (c.1844G > C, p.Ser615Thr) and a novel pathogenic deletion variant (c.1846delA, p.Glu617LysfsTer44), leading to early truncation. One SNP was present at the same location with different base pair substitutions (rs780608959: c.1844G > A, p.Ser615Asn). No information was available regarding the clinical significance of this SNP in dbSNP; therefore, this missense variant was still considered to be a variant of uncertain significance. The patients with familial CCM (Patients 57, 66, and 67) had one deletion variant (c.1255-4_1255-2delGTA), which had been reported as a pathogenic splice site variant in ClinVar. One case series revealed that six affected members of one family with the same variant presented with seizure and cutaneous vascular lesions, whereas our patients only had headache or visual disturbance. The heterogeneity of clinical symptoms in patients with the same variant may relate to underlying diseases27, immune responses28, or post-transcriptional modifications29. Another novel splice site variant (c.485 + 1G > C), which belonged to one patient (Patient 40), affected the invariant splice donor consensus sequence, leading to abnormal splicing products.
In patients with CCM2 variants, one patient with sporadic CCM (Patient 54) had a first reported insertion variant, leading to a frameshift and early truncation (c.401_402insGCCC, p.Ile136AlafsTer4). The other patient (Patient 69) had a CCM2 missense variant (c.970G > A, p.Glu324Lys) of uncertain significance, which was already recorded in the ExAC databank.
In total, 14 studies have investigated CCM1 and CCM2 variants in ethnically Chinese populations. Detailed information is listed in Table 3 6,7,8,9,10,11,12,13,14,15,16,17,18,19,20, and the locations of variants are shown in Fig. 4. Among those studies conducted in the ethnic Chinese population, most were case reports. One case series recruited five families and reported three novel variants causing early truncation15. Only two studies performed molecular screening of patients with CCMs from their brain MRI data8,9. These studies were conducted in 2004 and 2005, and one of the two reported only missense variants. In our study, we enrolled the largest number of patients from an ethnically Chinese population (41 patients) for molecular screening.
Our study has several limitations. First, because this was a retrospective study, information bias may have been present. Second, DNA analysis was performed for less than half of our recruited patients; variants may not be detected among those without DNA analysis. Third, Sanger sequencing may fail to detect large deletions, large insertions, or duplications; therefore, other detection methods, such as multiplex ligation-dependent probe amplification (MLPA), can be applied to our recruited patients to detect more variants in future studies. Finally, there were two identified variants of uncertain significance according to ACMG standards and guidelines; therefore, biological methods such as genome editing or genotyping were necessary to verify the clinical significance of those variants and to clarify the pathogenesis of familial CCMs.
In conclusion, we identified one novel pathogenic deletion and one pathogenic splice site variant in CCM1 and one novel pathogenic insertion variant in CCM2. The findings expand the knowledge related to variants present in patients with CCM, especially in the ethnic Chinese population.
Methods
Ethics approval and consent to participate
This study was approved by the Institutional Review Board of Chang Gung Memorial Hospital (CGMH) in Taiwan (IRB 96-1772B and 100-1666C). All examinations were performed after written informed consent was obtained. All samples were collected and analyzed in accordance with the relevant guidelines and regulations.
Patient recruitment
We retrospectively reviewed patients who had received a diagnosis of CCM on the basis of brain MRI findings from 1998 to 2006 at CGMH. We recruited patients who had multiple lesions or a single lesion with a relevant family history. Afterward, we collected radiological and clinical assessment data including age at onset, initial presentation, underlying diseases, family history, and surgical or radiosurgical interventions.
DNA extraction and sequencing
Venous blood samples were collected and DNA was routinely extracted using a DNA extraction kit (Stratagene La Jolla, California, United States). The extraction was monitored quantitatively and qualitatively through UV spectrophotometer absorption (ND-1000) (Nanodrop, Wilmington, United States) to verify the purity of the sample depending on the absorption at 260 nm.
Thereafter, to selectively amplify a specific DNA fragment through polymerase chain reaction (PCR), suitable primers were designed according to Primer Express version 2.0 (Applied Biosystems, United States) and online Primer3 (http://fokker.wi.mit.edu/cgi-bin/primer3/primer3_www.cgi). Our primers were short synthesized oligonucleotides, ranging from 18 to 25 bp. We used a pair of forward and reverse primers for each exon in the template DNA strand of those three genes. The primers and amplicon size of each exon are listed in Supplementary Table 1.
PCR was performed using the Fast-Run Taq Master kit (Pro Tech, Taipei, Taiwan). A typical 50-μL solution contained (1) 10 × Taq Master Mix, (2) 0.5 µM each of forward and reverse primers, (3) 100 ng of a genomic DNA template, (4) dimethyl sulfoxide for optimal performance, and (5) distilled water. The PCR reaction comprised (1) an initial denaturing step under 95 °C for 5 minutes, (2) 25–35 cycles of denaturation (95 °C, 30 seconds for each cycle), (3) primer annealing (variable, depending on the annealing temperature of the primers, 30 seconds), and (4) extension (72 °C, 30 seconds). A final extension lasted for 7 minutes at 72 °C. The final PCR products were refrigerated at 4 °C until samples were collected.
PCR products (20 ng) were purified using the Montage SEQ Sequencing Reaction Cleanup kit (Merck Millipore, Billerica, Massachusetts, United States). The resulting products were subjected to capillary electrophoresis on an ABI 3100 capillary sequencer (Applied Biosystems, United States). Sequence traces (Applied Biosystems, United States) were viewed and analyzed using the Sequencer software program.
Sequence alignment and variant identification
We used online genomic reference sequences [CCM1/KRIT1 (NG_012964.1/NC_000007.14), CCM2 (NG_016295.1/NC_000007.14), and CCM3/PDCD10 (NG_008158.1/NC_000003.12)] and mRNA transcript reference sequences [CCM1/KRIT1 (NM_194456.1), CCM2 (NM_031443.3), and CCM3/PDCD10 (NM_007217.3)] for sequence alignment. Sequence alignment was performed using the online EMBOSS Water tool (http://www.ebi.ac.uk/Tools/psa/emboss_water/), and peptide analysis and translation were conducted using ExPASy (http://web.expasy.org/translate/).
The variants were considered to be novel when they were not recorded in the in PubMed, the Human Gene Mutation Database (HGMD), ExAC, gnomAD (Genome Aggregation Database), or dbSNP (https://www.ncbi.nlm.nih.gov/projects/SNP/). The clinical significance of missense or nonsense variants was predicted using SIFT (http://sift.bii.a-star.edu.sg/), PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/index.shtml), and MutationTaster (http://www.mutationtaster.org/). MutationTaster and Ensembl Variant Effect Predictor (https://asia.ensembl.org/Tools/VEP) were used to predict the clinical significance of splice site variants. The MAF of a missense variant was searched in gnomAD or ExAC databases. Variants were considered to be pathogenic according to American College of Medical Genetics and Genomics (ACMG) standards and guidelines30. Furthermore, we analyzed evolutionary conservation in MutationTaster. Variants in evolutionary conservation may be more inclined to have clinical significance.
We followed the Human Genome Variation Society’s (HGVS) recommendations to describe sequence variants, and we used the position of coding DNA (cDNA) sequences to exhibit the position of variants. For example, a cDNA sequence with a first nucleotide corresponded to A of ATG (translation initiation codon).
Statistical methods and data analysis
Because of the small sample size, we used Mann–Whitney U test to compare the two groups. Correlations between two continuous variables were analyzed using Spearman rank correlation. We performed all analyses using SPSS version 24 (IBM, Armonk, NY, USA). A p value of < 0.05 was considered significant.
References
Robinson, J. R., Awad, I. A. & Little, J. R. Natural history of the cavernous angioma. J Neurosurg 75, 709–714 (1991).
Gunel, M. et al. Genetic heterogeneity of inherited cerebral cavernous malformation. Neurosurgery 38, 1265–1271 (1996).
Notelet, L. et al. Familial cavernous malformations in a large French kindred: mapping of the gene to the CCM1 locus on chromosome 7q. J Neurol Neurosurg Psychiatry 63, 40–45 (1997).
Dupre, N. et al. Linkage to the CCM2 locus and genetic heterogeneity in familial cerebral cavernous malformation. Can J Neurol Sci 30, 122–128 (2003).
Bergametti, F. et al. Mutations within the programmed cell death 10 gene cause cerebral cavernous malformations. Am J Hum Genet 76, 42–51 (2005).
Xu, Y. L. et al. A novel Krit-1 mutation in Han family with cerebral cavernous malformation. Zhonghua Bing Li Xue Za Zhi 32, 220–225 (2003).
Mao, Y. et al. Identification of a novel inheritable CCM1 gene mutation of 671del AT in a Chinese family with cerebral cavernous malformation. Zhonghua Yi Xue Za Zhi 83, 1572–1575 (2003).
Xie, R. et al. New mutations of the 12th exon of CCM1 gene in Chinese patients with intracranial cavernous angiomas. Zhonghua Yi Xue Yi Chuan Xue Za Zhi 21, 264–266 (2004).
Xie, R. et al. Analysis of CCM1 gene mutations in Chinese patients with intracranial cavernous malformations. Zhonghua Yi Xue Za Zhi 85, 2254–2258 (2005).
Wang, X. et al. Features of a Chinese family with cerebral cavernous malformation induced by a novel CCM1 gene mutation. Chin Med J (Engl) 126, 3427–3432 (2013).
Zhu, H. et al. Familial cerebral cavernous angiomas: clinical and genetic features in a Chinese family with a frame-shift mutation in the CCM1 gene (krit1). J Mol Neurosci 54, 790–795 (2014).
Mao, C. Y. et al. Exome capture sequencing identifies a novel CCM1 mutation in a Chinese family with multiple cerebral cavernous malformations. Int J Neurosci 126, 1071–1076 (2016).
Yang, C. et al. A Novel CCM1/KRIT1 Heterozygous Nonsense Mutation (c.1864C>T) Associated with Familial Cerebral Cavernous Malformation: a Genetic Insight from an 8-Year Continuous Observational Study. J Mol Neurosci 61, 511–523 (2017).
Wang, H. et al. A Novel KRIT1/CCM1 Gene Insertion Mutation Associated with Cerebral Cavernous Malformations in a Chinese Family. J Mol Neurosci 61, 221–226 (2017).
Yang, C. et al. Identification of a Novel Deletion Mutation (c.1780delG) and a Novel Splice-Site Mutation (c.1412-1G>A) in the CCM1/KRIT1 Gene Associated with Familial Cerebral Cavernous Malformation in the Chinese Population. J Mol Neurosci 61, 8–15 (2017).
Chen, D. H., Lipe, H. P., Qin, Z. & Bird, T. D. Cerebral cavernous malformation: novel mutation in a Chinese family and evidence for heterogeneity. J Neurol Sci 196, 91–96 (2002).
Zhao, Y. et al. A novel CCM1 gene mutation causes cerebral cavernous malformation in a Chinese family. J Clin Neurosci 18, 61–65 (2011).
Ji, B. H. et al. A Novel Deletion Mutation in CCM1 Gene (krit1) is Detected in a Chinese Family with Cerebral Cavernous Malformations. Yi Chuan Xue Bao 33, 105–110 (2006).
Huang, W. Q. et al. A Novel CCM2 Gene Mutation Associated with Familial Cerebral Cavernous Malformation. Front Aging Neurosci 8, 220 (2016).
Lan, M. Y. et al. Cavernous malformations of the central nervous system combined with cutaneous vascular lesions due to KRIT1 mutation: a case report. Clin Neurol Neurosurg 112, 729–732 (2010).
de Souza, J. M. et al. Susceptibility-weighted imaging for the evaluation of patients with familial cerebral cavernous malformations: a comparison with T2-weighted fast spin-echo and gradient-echo sequences. AJNR Am J Neuroradiol 29, 154–158 (2008).
Denier, C. et al. Genotype-phenotype correlations in cerebral cavernous malformations patients. Ann Neurol 60, 550–556 (2006).
Stahl, S. et al. Novel CCM1, CCM2, and CCM3 mutations in patients with cerebral cavernous malformations: in-frame deletion in CCM2 prevents formation of a CCM1/CCM2/CCM3 protein complex. Hum Mutat 29, 709–717 (2008).
Spiegler, S. et al. High mutation detection rates in cerebral cavernous malformation upon stringent inclusion criteria: one-third of probands are minors. Mol Genet Genomic Med 2, 176–185 (2014).
Merello, E. et al. Genetic Screening of Pediatric Cavernous Malformations. J Mol Neurosci 60, 232–238 (2016).
Gianfrancesco, F. et al. ZPLD1 gene is disrupted in a patient with balanced translocation that exhibits cerebral cavernous malformations. Neuroscience 155, 345–349 (2008).
Choquet, H. et al. Association of cardiovascular risk factors with disease severity in cerebral cavernous malformation type 1 subjects with the common Hispanic mutation. Cerebrovasc Dis 37, 57–63 (2014).
Choquet, H. et al. Polymorphisms in inflammatory and immune response genes associated with cerebral cavernous malformation type 1 severity. Cerebrovasc Dis 38, 433–440 (2014).
Choquet, H. et al. Cytochrome P450 and matrix metalloproteinase genetic modifiers of disease severity in Cerebral Cavernous Malformation type 1. Free Radic Biol Med 92, 100–109 (2016).
Richards, S. et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 17, 405–424 (2015).
Acknowledgements
We are thankful to all the patients and staff at the Department of Neurology and Neurosurgery of the Chang Gung Memorial Hospital Linkou Medical Center for their valuable support for this study. We also thank Dr. Yau-Yau Wai, a radiologist, for neuroimaging techniques and interpretation. This study was sponsored by Chang Gung Memorial Hospital, Taoyuan, Taiwan (CMRPG371273). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
Y.R.W., S.T.C. and H.C.F. conceived and designed the study. H.C.F. and C.W.C. performed the experiments. J.J.W. provided advanced techniques in magnetic resonance imaging. C.W.C. and Y.R.W. analysed the data. Y.R.W., S.T.C., P.W.H., K.C.W., M.S.H., W.C.H., L.S.R. and C.N.C. contributed reagents, materials, and analysis tools. C.W.C., Y.R.W. and S.T.C. wrote the paper. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Chang, CW., Hsu, PW., Wei, KC. et al. CCM1 and CCM2 variants in patients with cerebral cavernous malformation in an ethnically Chinese population in Taiwan. Sci Rep 9, 12387 (2019). https://doi.org/10.1038/s41598-019-48448-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-48448-y
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
