
Abstract
China has two populations of Lentinula edodes sensu lato as follows: L. edodes sensu stricto and an unexcavated morphological species respectively designated as A and B. In a previous study, we found that the nuclear types of the two populations are distinct and that both have two branches (A1, A2, B1 and B2) based on the internal transcribed spacer 2 (ITS2) sequence. In this paper, their mitogenome types were studied by resequencing 20 of the strains. The results show that the mitogenome type (mt) of ITS2-A1 was mt-A1, that of ITS2-A2 was mt-A2, and those of ITS2-B1 and ITS2-B2 were mt-B. The strains with heterozygous ITS2 types had one mitogenome type, and some strains possessed a recombinant mitogenome. This indicated that there may be frequent genetic exchanges between the two populations and both nuclear and mitochondrial markers were necessary to identify the strains of L. edodes sensu lato. In addition, by screening SNP diversity and comparing four complete mitogenomes among mt-A1, mt-A2 and mt-B, the cob, cox3, nad2, nad3, nad4, nad5, rps3 and rrnS genes could be used to identify mt-A and mt-B and that the cox1, nad1 and rrnL genes could be used to identify mt-A1, mt-A2 and mt-B.
Similar content being viewed by others


Introduction
Lentinula edodes (Berk.) Pegler belongs to Lentinula, Omphalotaceae, Agaricales, Agaricomycetes, Basidiomycota, Fungi1. Its morphological characteristics were first described by Berkeley in 1877 based on a very poor specimen purchased from a shop in Japan2 and more than 12 names have been used to represent the species3,4. In the latter half of the 20th century, the species was mainly placed into the genus Lentinus, as was proposed by Singer5. Until 1975, Pegler found that L. edodes had a monomictic hyphal system with generative hyphae, whereas Lentinus had a dimitic system with generative hyphae and skeleton-ligative hyphae. Therefore, this species was placed into the genus Lentinula. This classification is supported by many molecular phylogenetic studies6,7.
According to the morphological characteristics and geographical distribution, two species of Lentinula (L. lateritia and L. novaezelandiae) were distinct from L. edodes. However, mating compatibility studies have demonstrated that these three-morphological species are interfertile8. Many molecular phylogenetic studies confirm that these morphological species should belong to a biological species9,10,11,12,13,14,15. Some mycologists regard this biological species as Asian-Australasian populations of Lentinula according to their geographical distribution, and this species is also known as shiitake, an informal term in Japan12,16. However, according to nomenclatural priority, the biological species should be named L. edodes. To distinguish between the two L. edodes species, the biological species has been named L. edodes sensu lato17 and the morphological species has been named L. edodes sensu stricto12.
The nuclear ribosomal internal transcribed spacer (ITS) region has been used as a universal DNA barcode marker for fungi18. Hibbett et al.12,13 and Xu et al.16 used the ITS region to study the phylogenetic relationship of L. edodes sensu lato and found that there should be four distinct lineages of L. edodes sensu lato. In addition to L. edodes sensu stricto (in North-east Asia), L. lateritia (in South-east Asia and Australasia) and L. novaezelandiae (in New Zealand), there should be another unexcavated morphological species (in south-western East Asia). In addition, L. edodes sensu stricto and L. lateritia both tend to dissimilate into two subgroups. China is the first country to begin cultivating L. edodes sensu lato19 and is an important genetic diversity center of L. edodes sensu lato16; the informal Chinese term for L. edodes sensu lato is Xianggu. According to the geographical distribution of the four distinct lineages of L. edodes sensu lato, China has two populations: L. edodes sensu stricto and an unexcavated morphological species.
The genomic ITS region contains three sequences: internal transcribed spacer 1 (ITS1), 5.8 S ribosomal DNA (5.8 S) and internal transcribed spacer 2 (ITS2), and exhibits an incomplete concerted evolution20. Compared with 5.8 S sequence, ITS1 and ITS2 sequences have comparable discriminating power and barcode gaps, and could be used to instead of the whole ITS region for discriminating many sister species21,22. In addition, because the relatively short length of ITS2 (as compared to the whole ITS regions) allows Illumina and other next-generation sequencing technologies to obtain the whole ITS2 sequence in paired-end sequencing, using ITS2 sequence alone does have an advantage22. In the previous work, we found that ITS1 and ITS2 sequences were similar for discriminating two populations of L. edodes sensu lato in China, and that ITS2 could be also used for discriminating the branches of two populations23. Based on their ITS2 sequences, L. edodes sensu stricto (marked as ITS2-A) contained two branches (ITS2-A1 and ITS2-A2), and the unexcavated morphological species (marked as ITS2-B) also contained two branches (ITS2-B1 and ITS2-B2). In addition, we found that many strains were heterozygous ITS2 types, such as A1 + A2, A2 + B1, A2 + B2 and B1 + B2, which came from their heterokaryons.
The strain of L. edodes sensu lato is a filamentous fungi, and its hyphal cell harbors two nuclei that are inherited from both parents and one cytoplasm that is inherited from mother24,25. The mitochondrion is the powerhouse of the cytoplasm and contains its own genetic material, and the mitochondrial genome (mitogenome) has been widely accepted as an effective evolutionary marker in fungi26,27,28,29. Because the ITS2 sequence is located in the nuclear genome, one question arises as to whether the two populations of L. edodes sensu lato with different ITS2 types belong to identical or different mitogenome types (mt). Therefore, we selected 20 strains with different ITS2 types to answer this question. The specific research contents are as follows: 1) Sequence the complete mitogenome of L135 as the reference genome. 2) Study the mitogenome types of the two L. edodes sensu lato populations in China by resequencing the mitogenomes of 19 strains with different ITS2 types. 3) Study the SNP and indel diversity among the different mitogenome types. 4) Screen suitable markers for identifying different mitogenome types by sequencing the complete mitogenome of one strain that belonged to different mitogenome type from that of L135, and these two complete mitogenomes were aligned with the other two complete mitogenomes of L. edodes sensu lato (NC_018365.1 and KY217797.1) from the NCBI Organelle Genome Database.
Results
The reference mitogenome of L135
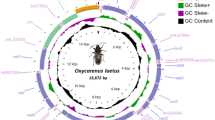
The mitogenome of L135 is a circular DNA molecule that is 119,134 bp in size with a GC content of 30.77%. It consists of 15 conserved protein-coding genes, 2 rRNA genes, 26 tRNA genes, 30 hypothetical protein-coding genes (10 in the introns of 4 conserved protein-coding genes, 4 in the introns of 2 rRNA gene and 16 in intergenic regions) and 3 miscellaneous features (Fig. 1a).

The mitogenome maps of L135 and YAASM363. (a) The mitogenome map of L135. (b) The mitogenome map of YAASM363. The protein-coding genes (wine red), rRNA genes (green), tRNA genes (bottle green), miscellaneous features (gray), introns (dotted line), hypothetical protein-coding genes (orf + number), and strand coding direction (arrow direction: clockwise-sense, anticlockwise-antisense) are shown.
The 15 conserved protein-coding genes include genes that encode NADH dehydrogenase subunits (nad1, nad2, nad3, nad4, nad4L, nad5 and nad6), apocytochrome b (cob), cytochrome c oxidase subunits (cox1, cox2 and cox3), ATP synthase A chain subunits (atp6, atp8 and atp9) and ribosomal protein S3 (rps3) (Table 1). Except for atp8, all other 14 genes were encoded on the sense strand (Fig. 1a). All 15 conserved protein-coding genes had the start codon ATG. However, 11 of the 15 genes had TAA as the stop codon, while the remaining four (cox2, cox3, nad4 and nad5) had TAG as the stop codon. In addition, 5 conserved protein-coding genes harbored 15 introns as follows: cob (3 introns), cox3 (1 intron), nad5 (1 intron), cox1 (7 introns), and nad1 (3 introns); in addition, 10 introns harbored the 10 following hypothetical protein-coding genes: 1 intron of cob, 1 intron of cox3, 7 introns of cox1 and 1 intron of nad1. The 10 intronic hypothetical protein-coding genes were encoded on the sense strand (Fig. 1a).The large-subunit rRNA (rrnL) gene had 5 introns and 2 introns contained 3 intronic hypothetical protein-coding genes. The small-subunit rRNA (rrnS) gene had 1 intron that contained 1 intronnic hypothetical protein-coding gene. The 2 rRNA genes were encoded on the sense strand. The 4 intronic hypothetical protein-coding genes of 2 rRNA were encoded on the sense strand (Fig. 1a).
The 26 tRNA genes coded for 20 common amino acids and were encoded on the sense strand (Fig. 1a). Of these 21 tRNA genes varied in size from 71 bp to 76 bp and could be folded into the typical cloverleaf structure. However, 5 other tRNA genes, including the tRNAs for the amino acids S (34080-34187), S (34964–35047), L (83281–83369), Y (107743–107826) and L (109774–109856), varied in size from 83 bp to 89 bp and had an additional variable loop (see Supplementary File 1).
The remaining regions without annotation were intergenic regions and 16 intergenic hypothetical protein-coding genes were located in these regions. Except for 6 intergenic hypothetical protein-coding genes (orf261, orf131, orf272, orf181, orf154 and orf130), the other 10 intergenic hypothetical protein-coding genes were encoded on the sense strand. In addition, the 3 miscellaneous features may encode mitochondrial plasmid DNA polymerase and RNA polymerase (Fig. 1a).
Mitogenome types of the 20 strains with different ITS2 types
According to the results of Song et al.23, the 20 strains had 38 ITS sequences (Table 2). Therefore, 38 ITS2 sequences were extracted from the 38 ITS sequences to construct their neighbor-joining (NJ) tree. The 38 ITS2 sequences were divided into two lineages (bootstrap support = 99%): ITS2-A and ITS2-B. Each lineage was subdivided into 2 branches (bootstrap support ≥95%): ITS2-A1 and ITS2-A2, and ITS2-B1 and ITS2-B2 (Fig. 2a).

Population analysis of ITS2 and the mitogenome of 20 strains. (a) A NJ tree was constructed using the ITS2 data. Different ITS2 sequences of each strain were marked as c1, c2, c3 and c4 (in order). (b) A NJ tree was constructed the using the SNP data. (b) PCA. (c) Population structure. The number of strains was the same as those listed in Table 2. Population A: purple; Population B: blue.
Based on the mitogenome of L135, the remaining 19 strains (Table 2) were resequenced, and their mitogenomes were assembled. The raw clean reads of the 19 strains varied from 6.69 million to 41.26 million with Q30 > 91%, and the average depths of the 19 strains were more than 544-fold (see Supplementary Table S1). Compared to the mitogenome coverage of L135, the mitogenome coverage of the 19 strains averaged approximately 95%. A total of 12,321 SNPs and 2,272 indels were identified (Table 3).
An NJ tree revealed that the mitogenomes of 20 strains belonged to two distinct lineages (bootstrap support = 100%): mt-A and mt-B; mt-A was subdivided into two branches (bootstrap support = 100%): mt-A1 and mt-A2 (Fig. 2b). The principal component analysis (PCA) (Fig. 2c) and population structure analyses (Fig. 2d) confirmed the results of the NJ tree. However, the 3 strains (YAASM298, YAASM301 and YAASM2323) were recombinant mitogenome types between mt-A and mt-B. Because most of their SNPs belonged to mt-B, these 3 strains were included in mt-B.
SNP and indel diversity among mt-A1, mt-A2 and mt-B
By testing the considerable differences in the SNPs and indels among mt-A1, mt-A2 and mt-B, the number of SNPs and indels in mt-B was considerably higher than that in mt-A1 and mt-A2. The number of indels in mt-A2 was considerably higher than that in mt-A1. By testing the SNP diversity in the CDS region among the mitogenomes of the 19 resequenced strains aligned with L135 (see Supplementary Table S2), the mitogenomes of 18 strains, except for EFISAAS5146, had more synonymous mutations than nonsynonymous mutations in the defined CDS regions of the 15 conserved protein-coding genes, and all 19 strains had more nonsynonymous mutations than synonymous mutations in the CDS regions of the 31 hypothetical protein-coding genes. The number of synonymous and nonsynonymous mutations in mt-B was different that those of mt-A1 and mt-A2.
To screen some markers for identifying mt-A1, mt-A2 and mt-B, the different SNPs among mt-A1, mt-A2 and mt-B were tested. There were 40 different SNPs between mt-A1 and mt-A2 (Fig. 3a) with 36 SNPs in the intergenic region, 2 SNPs in the 2 intronic hypothetical protein-coding genes of cox1 (orf202 and orf190), 1 SNP in rrnS, and 1 stoploss in the nad2 CDS. There were 304 different SNPs between mt-A and mt-B (Fig. 3b) with 137 SNPs in cob [18 SNPs in its intronic hypothetical protein-coding gene (orf240) and 119 SNPs in the intron], 99 SNPs in the intergenic region and 13 SNPs in the intergenic hypothetical protein-coding genes (6 SNPs in the orf715, 3 SNPs in the orf575, 2 SNPs in the orf138, 1 stoploss in the orf212 and 1 stoploss in the orf206), 24 SNPs in the nad1 gene [22 SNPs in the intron, 2 SNPs in its intronic hypothetical protein-coding gene (orf184)], 8 SNPs in the cox3 gene (6 SNPs in the CDS and 2 SNPs in the intron), 5 SNPs in the nad2 CDS, 4 SNPs in the rrnL gene, 4 SNPs in nad5 (2 SNPs in the CDS and 2 SNPs in the intron), 4 SNPs in the rps3 CDS, 3 SNPs in the intronic hypothetical protein-coding gene of cox1 (orf214), 3 SNPs in the rrnS gene.

SNP differences among the different mitogenome types. (a) SNP differences between mt-A1 and mt-A2. (b) SNP differences between mt-A and mt-B. Site (red): nonsynonymous SNP in the CDS; Site (green): synonymous SNP in the CDS; Site (yellow): stoploss SNP in the CDS. DNA: adenine (green), thymine (red), guanosine (purple), cytosine (blue). Oligo DNA: K (guanosine/thymine), S (guanosine/cytosine), Y (thymine/cytosine), W (adenine/thymine) and R (adenine/guanosine)./indicates intergenic regions.
Suitable markers for identifying mt-A1, mt-A2 and mt-B
To screen some markers for identifying mt-A1, mt-A2 and mt-B, the complete mitogenome of YAASM363 in mt-B was also tested. In addition, the two complete mitogenomes of L. edodes sensu lato (NC_018365.1 and KY217797.1) that were released in the NCBI database were also used in phylogenetic analysis (Fig. 4a mitogenome). The mitogenome of NC_018365.1 had a high homology with that of L135 (bootstrap support = 100%) and should belong to mt-A1. The KY217797.1 strain was collected by us in Guizhou Province and was identified as EFISAAS5052. Therefore, the mitogenome of KY217797.1 belonged to mt-A2.

NJ trees and synteny analysis of the complete mitogenomes among L135, NC_018365.1, KY217797.1 and YAASM363. (a) NJ trees of the four complete mitogenomes based on different sequences. (b) Synteny analysis of the four complete mitogenomes.
The NC_018365.1 mitogenome had a good synteny with the L135 mitogenome in gene number and arrangement (Fig. 4b and Supplementary Table S3). The total length and GC content of the NC_018365.1 mitogenome were 121,394 bp and 30.70%, respectively. Based on the mitogenome annotation of NC_018365.1 from the NCBI database, some hypothetical protein-coding genes and rrnL introns of NC_018365.1 were determined by homology comparison with the mitogenome of L135 (see Supplementary Table S3). The mitogenome of NC_018365.1 consisted of 15 conserved protein-coding genes, 2 rRNA genes, 26 tRNA genes, 30 hypothetical protein-coding genes (10 in the introns of 4 conserved protein-coding genes, 4 in the introns of 2 rRNA gene and 16 in intergenic regions) and 3 miscellaneous features (see Supplementary Table S3).
The total length and GC content of the KY217797.1 mitogenome were 116,819 bp and 30.75%, respectively. Because the mitogenome annotation of KY217797.1 given in the NCBI database was poor, we reannotated it. It consists of 15 conserved protein-coding genes, 2 rRNA genes, 26 tRNA genes, 24 hypothetical protein-coding genes (4 in the introns of 4 conserved protein-coding genes, 4 in the introns of 2 rRNA gene and 16 in intergenic regions) and 3 miscellaneous features (see Supplementary Table S3). The biggest difference between KY217797.1 and L135 mitogenomes was the cox1 gene, which missed two introns and 6 intronic hypothetical protein-coding genes in the KY217797.1 mitogenome (Fig. 4b and Supplementary Table S3).
The total length and GC content of the YAASM363 mitogenome were 115,116 bp and 31.76%, respectively (Fig. 1b). This mitogenome was found to contain 15 conserved protein-coding genes, 2 rRNA genes, 26 tRNA genes, 22 hypothetical protein-coding genes (10 in the introns of 4 conserved protein-coding genes, 3 in the introns of 1 rRNA gene and 9 in intergenic regions) and 2 miscellaneous features (Fig. 1b and Supplementary Table S3). The mitogenome of YAASM363 had many insertions, deletions and alternative splicing regions similar to those of L135 as follows: 1) cox3-exon1 of YAASM363 had an exon insertion. 2) cox1-exon2 and cox1-exon3 of L135 combined into cox1-exon2 in YAASM363, and cox1-exon5 of L135 broke up into cox1-exon4 and cox1-exon5 in YAASM363; therefore, the intronic hypothetical protein-coding gene (orf440) was lacking in YAASM363. 3) rrnS had no intron or the intronic hypothetical protein-coding gene (orf401) in YAASM363. 5) Miscellaneous features and 7 intergenic hypothetical protein-coding genes near nad4 (orf131, orf272, orf181, orf154, orf101, orf130 and orf283) were missing in YAASM363. The other insertions and deletions in the intronic and intergenic regions were not detailed in the paper.
The 15 conserved protein-coding genes and the 2 rRNA genes of these four mitogenomes were extracted for alignment (Fig. 4a). The atp6, atp8, atp9, cox2, nad4L and nad6 genes were identical in four mitogenomes. The cob, cox3, nad2, nad3, nad4, nad5, rps3 and rrnS genes, which were distinct between mt-B and two branches of mt-A and only a small degree of difference between mt-A1 and mt-A2, could be used as markers for identifying mt-A and mt-B. The cox1, nad1 and rrnL genes of four mitogenomes were all considerably different and could be used as markers for identifying mt-A1, mt-A2 and mt-B.
Discussion
There are two main strategies for acquiring a complete mitogenome. The first strategy is to isolate the mitochondrial DNA separately from the whole genomic DNA of an organism and to sequence the mitogenome directly30; the second strategy is to sequence the whole genome and to assemble the mitogenome based on a sequencing library31. In this paper, the complete mitogenomes of L135 and YAASM363 were assembled based on Illumina (450 bp) and PacBio (8–10 kb) sequencing data. Genomic resequencing is widely used to analyze the genetic diversity and structure of populations. Xiao et al.32 analyzed the genetic diversity and population structure of wild and cultivated strains of Chinese L. edodes sensu lato by resequencing their nuclear genomes based on Illumina sequencing data. The mapped rates of the nuclear genome in the whole genome varied from 85.01% to 97.63%. In this paper, we analyzed the mitogenome types of two L. edodes sensu lato populations in China by resequencing their mitogenomes with Illumina sequencing. The mapped rates of the mitogenome in the whole genome varied from 2.25% to 11.27%. The total mapped rates of the nuclear genome and mitogenome were near 100%. Therefore, the resequencing analysis in this paper was feasible.
Since the beginning of the fungal mitogenome project33, 347 fungal mitogenomes have been released in the NCBI Organelle Genome Database, 220 of which have been curated28. These fungal mitogenomes vary extensively in size and range from 12 kb to over 235 kb27; this variation is mainly caused by intronic and plasmid-derived regions34. They generally have the same set of core genes as follows: 15 conserved protein-coding genes (nad1, nad2, nad3, nad4, nad4L, nad5, nad6, cob, cox1, cox2, cox3, atp6, atp8, atp9 and rps3), 2 rRNA genes (rrnS and rrnL) and tRNA genes25,34. In the NCBI database, two complete mitogenomes of L. edodes sensu lato have been released: NC_018365.1 and KY217797.1. Because they have distinct annotations in the NCBI database, we did not select them as the reference mitogenome. In this paper, we reannotated them again by comparing them with those of L135 and YAASM363. Finally, these four mitogenomes of L. edodes sensu lato had 15 conserved protein-coding genes, 2 rRNA genes and 26 tRNA genes, and showed complete synteny with each other in the order of these genes (Fig. 4b and Supplementary Table S3).
Variations in mitochondrial genome sizes among different strains within the same species have also been reported in many fungi35. In this paper, the four complete mitogenomes of L. edodes sensu lato were very similar in size, ranging from about 115 to 121 kb. The two mt-A1 strains (L135 and NC_018365.1) had the largest mitogenome size, followed by one mt-A2 strain (KY217797.1), and then one mt-B strain (YAASM363) (Fig. 4b). Introns are the major contributors to the mitogenome size variations of these four complete mitogenomes. For example, the length of cox1 exon was identical (1602 bp) in four complete mitogenomes. But the whole cox1 genes of two mt-A1 strains (L135 and NC_018365.1) were 11088 bp in length and both had 7 introns; that of one mt-A2 strain (KY217797.1) was 8391 bp in length and missed 2 introns; that of one mt-B strain (YAASM363) was 12029 bp in length and had 7 introns with alternative splicing of 3 exons (see Supplementary Table S3).
The proportion of nonsynonymous mutations to synonymous mutations is commonly estimated in evolutionary biology for testing hypotheses related to selective pressure36. In the CDS regions of 15 conserved protein-coding genes, the proportion of nonsynonymous mutations to synonymous mutations between L135 and 18 strains, with the exception of EFISAAS5146, was less than 1. the evolution of the 15 conserved protein-coding genes between L135 and the 18 strains, except for EFISAAS5146, was due to purifying (negative) selection. The rps3 gene of EFISAAS5146 had 4 nonsynonymous mutations and 1 synonymous mutation that was different from L135 and did not fit the rule. Tracing the rps3 gene of the other 18 strains, the strains of group B had high levels of nonsynonymous mutations that were different from L135. This implied that the rps3 gene had a higher rate of evolution than the other 14 conserved protein-coding genes. In the CDS region of 30 hypothetical protein-coding genes, the proportion of nonsynonymous mutations to synonymous mutations between L135 and 19 strains was greater than 1. This suggested that the evolution of the 31 hypothetical protein-coding genes in L135 and the 19 strains was based on diversifying (positive) selection. For example, the putative protein of orf214 was similar to a homing endonuclease containing a LAGRIDADG_2 domain that promotes the mobility of the intron37.
The nuclear and mitochondrial genetic relationships of the wild L. edodes sensu lato strains are highly associated with their geographic distribution9,32. ITS2-A1 is mainly distributed in northern Asia (Russia, Korea, Japan and northeastern and northwestern China) and ITS2-A2 is mainly distributed in southern Asia (pan-mid-southern China and Thailand). ITS2-B is mainly distributed in the southwest of eastern Asia (western China, India and Nepal)12,13,16,23. Tracing the source and parent information of the 20 strains (Table 2), there should be a correspondence between ITS2 and the mitogenome types in each population. The mitogenome type (mt) of each strain is as follows: ITS2-A1 was mt-A1, ITS2-A2 was mt-A2, and ITS2-B1 and ITS2-B2 were mt-B. However, 2 strains (EFISAAS0376 and YAASM2323) with the ITS2-A2 type that belonged to mt-B did not fit the rule. The best explanation is that these 2 strains may have exchanged their mitochondria with the other mitogenome type mt-B strains. This explanation needs to be validated. In addition, during the mon-mon crossing of two compatible homokaryons mitogenome recombination is easily found at the junction of two homokaryons24,38,39. The 3 strains with recombinant mitogenomes may be the offspring of mt-A and mt-B, and their SNPs from mt-A were analyzed (see Supplementary Fig. S1). These results indicate that there are frequent genetic exchanges between the two populations of L. edodes sensu lato in China.
Many studies have shown that the classification of the same species is considerably differentiated in both the mitochondrial and nuclear genes40,41. In animal, most species have shown a greater rate of evolution in mitochondrial genes, but the opposite is true for most plants and most fungi populations35. In the paper, we also calculated the percentage of plasmorphism site (pplymorphism rate) in ITS2 and the whole mitogenome between L135 and the other 19 strains (see Supplementary Table S4): 12.04% of the 272 ITS2 sites and 1.74% of the mitogenome sites were polymorphic. In addition, the polymorphism rates of 15 conserved protein-coding genes and 2 rRNA genes were varied from 0% to 2.48% (see Supplementary Table S4). Therefore, L. edodes sensu lato showed a greater rate of evolution for nuclear genes than for mitochondrial genes. The following question is that which one between nuclear and mitochondrial genes is best for identifying strains. Zietara et al.41 suggested that both nuclear and mitochondrial genes were necessary to identify the parasite strains of Gyrodactylus salaries. In the same sense, we believe that both nuclear and mitochondrial markers are necessary to identify the strains of L. edodes sensu lato in China.
To find suitable mitochondrial genes for indentifying the strains of L. edodes sensu lato in China, we analyzed the different SNPs among mt-A1, mt-A2 and mt-B and compared four complete mitogenomes with different mitogenome types. These two studies showed some same results as follows: The cob, cox3, nad1, nad2, nad5, rps3 and rrnS genes had higher diversity between mt-A and mt-B (Fig. 3b) and could distinguish the mitogenome of YAASM363 (mt-B) from the other three mitogenomes (mt-A). In addition, the cox1 gene may be the biggest difference of 15 conserved protein-coding genes among different mitogenome types. The exon length of cox1 gene was 1602 bp, but the exon arrangement was distinct among the mitogenomes of mt-A1 (L135 and NC_018365.1), mt-A2 (KY217797.1) and mt-B (YAASM363). Except for the ITS2 sequence, the other nuclear genes should be able to be used to identify the different strains of L. edodes sensu lato in China. However, because the same gene was specific in different species, the suitable genes for identifying the two populations of L. edodes sensu lato should be determined. In a previous study, we found that the intergenic spacer 1 (IGS1) could be used as a marker for identifying internal mt-A142, and we have sequenced a nuclear genome of YAASM363 (ITS2-B). In the next work, we will determine suitable nuclear markers for strain identification by comparing the nuclear genomes of different ITS2 types.
Materials and Methods
Strains, cultivation, DNA isolation and ITS2 sequence analysis of the 20 strains
Twenty strains were assayed in this study (Table 2). These strains were provided by some professional research institutes, were identified by a fruiting test and were maintained at the Institute of Edible Fungi at Shanghai Academy of Agricultural Sciences, Shanghai, People’s Republic of China. The strains were cultured on cellophane PDA (4% potato extract, 20% dextrose and 15% agar) medium in 9 cm Petri dishes at 25 °C in the dark. When growing over the cellophane, the strains were scraped into 5 mL sterile freezing tubes and were freeze-dried by Coolsafe 55-4 (Labogene ScanLaf, Lynge, Denmark). The total DNA of each strain was isolated with the E.Z.N.A Fungal DNA Mini Kit-D3390 (Omega Bio-Tek, Inc, Norcross, United States of America) and was confirmed by 1% (w/v) agarose gel electrophoresis.
All ITS sequences of the twenty strains (Table 2) were used to extract the ITS2 sequences according to the methods described by Song et al.23. An NJ tree of all ITS2 sequences was reconstructed with MEGA version 7.0. The number of bootstrap replicates was 1000, and a Kimura 2-parameter model was used as the substitution model.
Illumina library preparation and mitogenome resequencing of the 19 strains
At least 3 μg of the total DNA from each strain was prepared for an Illumina paired-end library. The paired-end library (450 bp) was prepared following the Illumina standard genomic DNA library preparation procedure and was sequenced on an Illumina HiSeq 4000 platform (Illumina, Int, San Diego, United States of America) according to the Illumina sequencing method manual.
Raw Illumina sequencing reads of each strain were generated by CASAVA v 1.8.2 (http://support.illumina.com/sequencing/sequencing_software/casava.ilmn). After removing the adaptors, the high quality reads were obtained using Trimmomatic v 0.36 (http://www.usadellab.org/cms/?page=trimmomatic) and were aligned to the reference genome (L135) using BWA-SW (http://bio-bwa.sourceforge.net/). The BAM file was obtained using Sequence Alignment/Map (SAM) tools v 1.4 (http://samtools.sourceforge.net/). Then, Picard Tools (http://picard.sourceforge.net/) was used to remove the reads caused by PCR-amplification and to obtain the valid BAM files. The depth and coverage of the whole-genome sequencing of each of the other strains (Table 2) were generated by Custom Perl CGI Database Scripts.
SNP/indel calling, PCA and population structure analysis of the 20 strains
SNPs and indels were detected by the GATK v 3.8.0 “Unified Genotyper” function (http://www.broadinstitute.org/gatk/) based on the valid BAM files and were annotated by ANNOVAR (2017Jul16) (http://www.openbioinformatics.org/annovar/). All SNPs between the reference mitogenome (L135) and the 19 strains were utilized to compute the NJ tree and PCA. The NJ tree was constructed by EMBOSS fneighbor (http://emboss.toulouse.inra.fr/cgi-bin/emboss/fneighbor?_pref_hide_optional=1), and the PCA was performed on the R platform. Population structure analysis was performed using fast structure (https://github.com/rajanil/fastStructure) based on the needed map files, which were generated by PLINK (http://www.plink.com/).
PacBio library preparation, mitogenome assembly and annotation of L135 and YAASM36
The DNA of L135 and YAASM363 was also used for PacBio library preparation (8–10 kb). A Blue Pippin (Sage Science, Beverly, United States of America) was used for size selection, and the sequencing was performed on the Sequel Sequencer according to manufacturer’s instructions.
The mitogenomes of L135 and YAASM363 were reconstructed from the Illumina and PacBio sequencing data. First, the basic genome framework was assembled from the PacBio data using Celera Assembler v 8.0 (http://sourceforge.net/projects/wgs-assembler/files/wgs-assembler/wgs-8.1/). Second, the basic assembly was verified and the circle mitogenome was completed. Sequencing gaps were filled if there were any. Third, the assembly was corrected and aligned with the Illumina data using Burrows-Wheeler Aligner (BWA) software (http://bio-bwa.sourceforge.net/).
The two mitogenomes were annotated by homology prediction using BLASTn (https://blast.ncbi.nlm.nih.gov/Blast.cgi?PROGRAM=tblastn&PAGE_TYPE=BlastSearch&LINK_LOC=blasthome) and GeneWise (https://www.ebi.ac.uk/Tools/psa/genewise/). The transfer RNA (tRNA) and ribosomal RNA (rRNA) genes were predicted by tRNAscan-SE v 1.4 (http://lowelab.ucsc.edu/tRNAscan-SE/) and by homology prediction (identity >90% and coverage >90%). The secondary structures were predicted with tRNAscan-SE v 1.4 (http://lowelab.ucsc.edu/tRNAscan-SE/). A whole mitochondria genome BLAST search was performed against five databases, including Kyoto Encyclopedia of Genes and Genomes (KEGG), Clusters of Orthologous Groups (COG), Non-Redundant (NR) Protein Database, Swiss-Prot, and Gene Ontology (GO). The circular mitogenome map was drawn using SnapGene v 4.0 (http://www.snapgene.com).
Synteny analysis and SNP marker screening
Synteny analysis of the four mitogenomes was computed by Mauve (http://darlinglab.org/mauve/user-guide/introduction.html). The population SNP markers and the SNPs from mitochondrial group A were marked in 3 strains with recombinant mitogenomes by Adobe Photoshop CS4 v 11.0.
Phylogenetic analysis of four complete mitogenomes, conserved protein-coding and rRNA genes
The 15 conserved protein-coding genes and the 2 rRNA genes were extracted from four complete mitogenomes (L135, NC_018365.1, KY217797.1 and YAASM363) and aligned with MEGA version 7.0, respectively. The NJ trees of four complete mitogenomes, 9 conserved protein-coding and 2 rRNA genes (Fig. 4a) were constructed with MEGA version 7.0. The number of bootstrap replicates was 1000, and a Kimura 2-parameter model was used as the substitution model.
Ethical statement
This article does not contain any studies with human participants or animals performed by any of the authors.
Data Availability
The L135 strain from mitochondrial group A has been deposited in the Guangdong Microbial Culture Center (GDMCC) under the accession number 5.568, and the mitogenome sequence of L135 has been deposited in GenBank under the accession number MF774812. The YAASM363 strain from mitochondrial group B has been deposited in the GDMCC under the accession number 5.567, and the mitogenome sequence of YAASM363 has been deposited in the GenBank under the accession number MF774813. The final dataset supporting the conclusions of this study is also available, and the raw data are available upon request.
References
International Mycological Association. MycoBank Database Fungal Databases, Nomenclature and Species Banks. http://www.mycobank.org/ (2018-11-10).
Pegler, D. N. The classification of the genus Lentinus Fr. (Basidiomycota). Kavaka. 3, 11–20 (1975).
Pegler, D. N. The genus Lentinula (Tricholomataceae tribe Collybieae). Sydowia. 36, 227–239 (1983).
Hibbett, D. S. Towards a phylogenetic classification for shiitake: taxonomic history and molecular perspectives. Rept Tottori Mycol Ins. 30, 30–42 (1992).
Singer, R. Is shiitake a Cortinellus? Mycologia. 33, 449–451 (1941).
Molina, F. I., Shen, P. & Jong, S. C. Molecular evidence supports the separation of Lentinula edodes from Lentinus and related genera. Can J Bot. 70, 2446–2459 (1992).
Hibbett, D. S. & Vilgalys, R. Phylogenetic relationships of Lentinus (Basidiomycotina) inferred from molecular and morphological characters. Syst Bot. 18, 409–433 (1993).
Shimomura, N., Hasebe, K., Nakai-Fukumasa, Y. & Komatsu, K. Intercompatibility between geographically distant strains of shiitake. Rep Tottori Mycol Inst. 30, 26–29 (1992).
Fukuda, M., Nakai, Y. F., Hibbett, D. S., Matsumoto, T. & Hayashi, Y. Mitochondrial DNA restriction fragment length polymorphisms in natural populations of Lentinula edodes. Mycol Res. 98(2), 169–175 (1994).
Nicholson, M. S., Bunyard, B. A. & Royse, D. J. Phylogeny of the genus Lentinula based on ribosomal DNA restriction fragment length polymorphism analysis. Mycologia. 89(3), 400–407 (1997).
Nicholson, M. S., Bunyard, B. A. & Royse, D. J. Phylogenetic implications of restriction maps of the intergenic regions flanking the 5S ribosomal RNA gene of Lentinula species. Fungi. 2, 48–57 (2009).
Hibbett, D. S., Fukumasa-Nakai, Y., Tsuneda, A. & Donoghue, M. J. Phylogenetic diversity in shiitake inferred from nuclear ribosomal DNA sequences. Mycologia. 87(5), 618–638 (1995).
Hibbett, D. S., Hansen, K. & Donoghue, M. J. Phylogeny and biogeography of Lentinula inferred from an expanded rDNA dataset. Mycol Res. 102(9), 1041–1049 (1998).
Hibbett, D. S. Shiitake mushrooms and molecular clocks: historical biogeography of Lentinula. J Biogeogr. 28, 231–241 (2001).
Mata, J. L., Petersen, R. H. & Hughes, K. W. The genus Lentinula in the Americas. Mycologia. 93(6), 1102–1112 (2001).
Xu, X. F., Li, A., Cheng, S. M., Lin, F. X. & Lin, F. C. Reappraisal of phylogenetic status and genetic diversity analysis of Asian population of Lentinula edodes. Prog Nat Sci. 16(3), 274–280 (2006).
Hibbett, D. S. & Donoghue, M. J. Implications of phylogenetic studies for conservation of genetic diversity in Shiitake mushrooms. Conserv Biol. 10(5), 1321–1327 (1996).
Schoch, C. L. et al. Nuclear ribosomal internal transcribed spacer (ITS) region as a universal DNA barcode marker for Fungi. P Natl Acad Sci USA 109(16), 6241–6246 (2012).
Chang, S. T. & Miles, P. G. Historical record of the early cultivation of Lentinus in China. Mushroom J Trop. 7, 31–37 (1987).
Simon, U. K. & Weiβ, M. Intragenomic variation of fungal ribosomal genes is higher than previously thought. Mol Biol Evol. 25(11), 2251–2254 (2008).
Blaalid, R. et al. ITS1 versus ITS2 as DNA metabarcodes for fungi. Mol Ecol Resour. 13(2), 218–224 (2013).
Xu, J. P. Fungal DNA barcoding. Genome. 59, 913–932 (2016).
Song, X. X. et al. ITS1/5.8S/ITS2, a good marker for initial classification of Shiitake culinary-medicinal Lentinus edodes (Agaricomycetes) strains in China. Int J Med Mushrooms. 20(3), 227–242 (2018).
Fukuda, M., Harada, Y., Imahori, S., Nakai, Y. F. & Hayashi, Y. Inheritance of mitochondrial DNA in sexual crosses and protoplast cell fusions in Lentinula edodes. Curr Genet. 27, 550–554 (1995).
Xu, J. P. & Wang, P. F. Mitochondrial inheritance in basidiomycete fungi. Fungal Biol Rev. 29(3-4), 209–219 (2015).
Shen, X. Y. et al. Characterization and phylogenetic analysis of the mitochondrial genome of Shiraia bambusicola reveals special features in the order of Pleosporales. PLoS ONE. 10(3), E0116466, https://doi.org/10.1371/journal.pone.0116466 (2015).
Jelen, V., Jonge, R. D., Van, de Peer, Y., Javornik, B. & Jakše, J. Complete mitochondrial genome of the Verticillium-wilt causing plant pathogen Verticillium nonalfalfae. PLoS ONE, 11(2), e0148525; 10.137/journal.pone.0148525 (2016).
Robicheau, B. M., Young, A. P., Labutti, K., Grigoriev, I. V. & Walker, A. K. The complete mitochondrial genome of the conifer needle endophyte, Phialocephala scopiformis DAOMC 229536 confirms evolutionary division within the fungal Phialocephala fortinii s.l.-Acephala appalanata species complex. Fungal Biol-UK. 121, 212–221 (2017).
Zhang, S., Wang, X. N., Zhang, X. L., Liu, X. Z. & Zhang, Y. J. Complete mitochondrial genome of the endophytic fungus Pestalotiopsis fici: features and evolution. Appl Microbiol Biot. 101(4), 1593–1604 (2017).
Wang, Y., Zeng, F. Y., Hon, C. C., Zhang, Y. Z. & Leung, F. C. C. The mitochondrial genome of the Basidiomycete fungus Pleurotus ostreatus (oyster mushroom). FEMS Microbiol Lett. 280, 34–41 (2008).
Férandon, C., Xu, J. P. & Barroso, G. The 135 kbp mitochondrial genome of Agaricus bisporus is the largest known eukaryotic reservoir of group I introns and plasmid-related sequences. Fungal Genet Biol. 55, 85–91 (2013).
Xiao, Y. et al. Population genomic analysis uncovers environmental stress-driven selection and adaptation of Lentinula edodes population in China. Sci Rep-UK, 6, 36789, https://doi.org/10.1038/srep36789 (2016).
Paquin, B. et al. The fungal mitochondrial genome project: evolution of fungal mitochondrial genomes and their gene expression. Curr Genet. 31, 380–395 (1997).
Himmelstrand, K., Olson, Å., Durling, M. B., Karlsson, M. & Stenlid, J. Intronic and plasmid-derived regions contribute to the large mitochondrial genome sizes of Agaricomycetes. Curr Genet. 60(4), 03–313 (2014).
Sandor, S., Zhang, Y. J. & Xu, J. P. Fungal mitochondrial genomes and genetic polymorphisms. Appl Microbiol Biotechnol. 102, 9433–9448 (2018).
Arenas, M. Trends in substitution models of molecular evolution. Front Genet. 6, 319, https://doi.org/10.3389/fgene.2015.00319 (2015).
Edgell, D. R. Selfish DNA: homing endonucleases find a home. Curr Biol. 19(3), 115–117 (2009).
Baptista-Ferreira, J. L. C., Economou, A. & Casselton, L. A. Mitochondrial genetics of Coprinus: recombination of mitochondrial genomes. Curr Genet. 7, 405–407 (1983).
Matsumoto, T. & Fukumasa-Nakai, Y. Mitochondrial DNA inheritance in sexual crosses of Pleurotus ostreatus. Curr Genet. 30, 549–552 (1996).
Sorenson, M. D., Sefc, K. M. & Payne, R. B. Speciation by host switch in brood parasitic indigobirds. Nature. 424, 928–931 (2003).
Zietara, S. M., Rokicka, M., Stojanovski, S. & Lumme, J. Introgression of distant mitochondria into the genome of Gyrodactylus salaries: nuclear and mitochondrial markers are necessary to identify parasite strains. Acta Parasitol. 55(1), 20–28 (2010).
Song, X. X. et al. Intergenic spacer 1 (IGS1) polymorphism map: a marker for the initial classification of cultivated Lentinula edodes strains in China. J Integr Agr. 17(11), 2458–2466 (2018).
Acknowledgements
This paper was funded by the China Agriculture Research System (Grant No. CARS-20), the Agriculture Research System of Shanghai, China (Grant No. 201809) and SAAS Program for Excellent Research Team (Grant No. SPERT-2017-A-02).
Author information
Authors and Affiliations
Contributions
X.S., Y.Z., M.C. and Q.T. performed the plans. X.S. wrote the paper. Y.Z. and M.C. edited the paper. C.S. and D.B. provided strains. R.Y. contributed to the data analysis. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Song, X., Zhao, Y., Song, C. et al. Mitogenome types of two Lentinula edodes sensu lato populations in China. Sci Rep 9, 9421 (2019). https://doi.org/10.1038/s41598-019-45922-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-45922-5
This article is cited by
-
Multi-stage nuclear transcriptomic insights of morphogenesis and biparental role changes in Lentinula edodes
Applied Microbiology and Biotechnology (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
