
Abstract
The dysbiosis of human gut microbiota is strongly associated with the development of colorectal cancer (CRC). The dysbiotic features of the transition from advanced polyp to early-stage CRC are largely unknown. We performed a 16S rRNA gene sequencing and enterotype-based gut microbiota analysis study. In addition to Bacteroides- and Prevotella-dominated enterotypes, we identified an Escherichia-dominated enterotype. We found that the dysbiotic features of CRC were dissimilar in overall samples and especially Escherichia-dominated enterotype. Besides a higher abundance of Fusobacterium, Enterococcus, and Aeromonas in all CRC faecal microbiota, we found that the most notable characteristic of CRC faecal microbiota was a decreased abundance of potential beneficial butyrate-producing bacteria. Notably, Oscillospira was depleted in the transition from advanced adenoma to stage 0 CRC, whereas Haemophilus was depleted in the transition from stage 0 to early-stage CRC. We further identified 7 different CAGs by analysing bacterial clusters. The abundance of microbiota in cluster 3 significantly increased in the CRC group, whereas that of cluster 5 decreased. The abundance of both cluster 5 and cluster 7 decreased in the Escherichia-dominated enterotype of the CRC group. We present the first enterotype-based faecal microbiota analysis. The gut microbiota of colorectal neoplasms can be influenced by its enterotype.
Similar content being viewed by others

Introduction
A trend towards a decreased overall incidence (a decrease of 3.3% per year in men and 3.0% in women) of and mortality (a decrease of 2.5% per year in men and 3.0% in women) from colorectal cancer (CRC) was noted from 2006 to 20101 and was attributed to the use of screening tests to detect colon neoplasms at early time points and the removal of pre-malignant lesions2,3,4. Nonetheless, CRC was still the fourth most common cause of cancer-related deaths worldwide in 20125 and was the third most common cancer in the United States in 20141. Despite the availability of various methods to screen for CRC, approximately 30% of the adults in the US do not receive appropriate screenings for their age. Colonoscopy is the gold standard for the accurate diagnosis of CRC6,7. However, the invasive and unpleasant nature of colonoscopies often causes patients unwanted pain and discomfort, leading more than half to prefer non-invasive screening methods6,8. Current “non-invasive” faecal screening tests, including the faecal immunochemical (FIT) and the multi-target faecal DNA tests, have significantly improved the detection rate of CRC9,10. However, their ability to detect pre-cancerous or small lesions is limited.
Contributors to the pathogenesis of CRC include chronic inflammation and the accumulation of genetic, epigenetic, diet, and environmental factors11,12. As the well-described carcinogenic potential of infectious agents contributes to more than 18% of the global cancer burden (e.g., gastric cancer, which can be caused by Helicobacter pylori)13, emerging evidence suggests that a dysbiosis of human gut microbiota is associated with CRC13,14,15,16. It has been hypothesized that certain pathogens interact with the colon epithelium by influencing the host’s immune system, increasing its mutagenic potential through chronic inflammation, possessing bacteria-derived virulence factors, and creating DNA-damaging and non-DNA-damaging metabolites17. For example, Fusobacterium nucleatum (Fn) is prevalent in CRC and pre-malignant colorectal lesions18,19 and has been associated with a poor prognosis20. Alternations in the composition of the gut microbiome have also been observed along the adenoma-carcinoma sequence16. An altered microenvironment that leads to a different gut microbe composition is thought to be a biomarker that can differentiate healthy subjects from those with colonic neoplasms16,21,22. To analyse these specific dysbiotic features, the human faecal microbiome may be a new detection tool for CRC14,16,21,22. Furthermore, manipulating the gut microbiome may affect the progression of colonic neoplasms.
However, previous studies that used differing clustering and grouping strategies produced heterogeneous results14,15,16,21,22. Given that the human gut microbiome can be characterized by changes in the level of one of three robust genera—Bacteroides, Prevotella, and Ruminococcus—these categories have been defined as “enterotypes”23. Enterotypes are stable and are strongly associated with long-term diets; a protein and animal fat-rich diet has been associated with the Bacteroides-dominated enterotype, while a carbohydrate-rich diet has been linked to the Prevotella-dominated enterotype24. We hypothesized that changes in the gut microbiome in patients with colorectal neoplasms are different among enterotypes.
Here, we systemically investigated the microbial composition of human stool samples at various points along the conventional adenoma to carcinoma sequence using enterotype-based and co-abundance group (CAG) analysis.
Results
Sample collection and NGS OTU mapping
We analysed stool samples from 283 individuals, including 104 from normal controls, 117 from patients with adenomatous polyps, and 62 from patients with CRC (Table 1). One-hundred seventy-three of the subjects were males, and 110 were females. Their ages ranged from 40 to 86, with a mean of 60.96 ± 10.11 years old. We generated 13,671,987 quality-filtered sequence reads, with 48,311 average reads per sample. Sequence reads were mapped to the bacteria in the SILVA database. We mapped all sequences into 277 genera. The most dominant bacterial phyla were Bacteroidetes, Proteobacteria, and Firmicutes, which covered more than 95% of our sequenced reads. These phyla were present in all individuals, with minor variations between groups (Table S1).
Enterotypes and biodiversity analysis
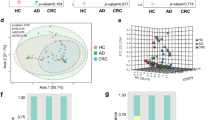
At the genus level, Bacteroides, Escherichia, and Prevotella contributed to the majority of the human gut microbiota, with an average prevalence of 36.52%, 16.03%, and 9.84%, respectively (Fig. 1, Table S2 and Fig. S2A). The weighted principal coordinates analysis (PCoA) of all stool samples demonstrated strong clustering into three enterotypes that were dominated by the 3 genera: enterotype 1 contained a high proportion of Bacteroides (≥40% of all genera, with more Bacteroides than Prevotella); enterotype 2 contained a high proportion of Prevotella (≥30% of all genera, with more Prevotella than Bacteroides); and enterotype 3 contained a higher proportion of Escherichia mixed with other genera (Figs 2 and S2B–D). The PCoA plot represents the microbiota of all faecal samples, which were significantly different and clearly separated into the 3 enterotypes. However, there was no difference in the incidence of colorectal neoplasms between enterotypes, although enterotypes 1 and 3 contained most of the cases (Supplementary Table S3).

Overall microbial flora at the genus level The average of the top 25 genera of each group, which occupied more than 90% of the relative abundance of the three groups. These groups shared the same top 16 genera, arranged in a slightly different order.

PCoA plot of enterotypes using weighted PCoA Enterotypes were defined as microbial flora dominated by genus Bacteroides, Prevotella, or Escherichia. Samples with a relative abundance of Bacteroides over 40% with levels greater than Prevotella were assigned to enterotype 1. Samples with a relative abundance of Prevotella of over 30% with levels greater than or equal to Bacteroides were assigned to enterotype 2. All others were assigned to enterotype 3, which was found to be dominated by Escherichia.
We then calculated the richness and Shannon diversity index between the normal, adenoma, and CRC groups, which were not significantly different between groups (Fig. 3A). When we took the enterotype into consideration, CRC group members that are in enterotype 3 are significantly richer than their adenoma and normal counterparts in the same enterotype (p < 0.01, Fig. 3B). However, the Shannon diversity and richness measurements were not different between the groups within the 3 enterotypes. Subgroup analysis also showed a trend towards increasing richness from stage 0 to late-stage CRC, although the Shannon diversity index remained equivocal (Supplementary Fig. S1A–C).

(A) Richness and Shannon diversity indices by group in all faecal samples The Shannon diversity index and the binary logarithm of the genus richness of each sample were calculated in all three groups. Each group had a similar richness and Shannon diversity index, with only the cancer group having a slightly higher variation in richness that was not significant. Each dot represents one sample, and outlier samples are marked as red dots. (B) Richness index by group across enterotypes The binary logarithm of the genus richness of each sample in the three enterotypes. Enterotype 1 and 2 had a similar richness in each group. In enterotype 3, the cancer group had a significantly higher and more varied richness. (*p < 0.01).
Faecal microbiota differs between the CRC, adenoma and normal control groups in different enterotypes
Overall, the abundance of sixteen of the genera was significantly different between the normal control, adenoma, and CRC groups. In particular, the relative abundance of Fusobacterium, Enterococcus, and Morganella was significantly greater in CRC patients relative to those with adenomas (all p < 0.01, Fig. 4A, Table S4A).

(A) Relative overall sample abundance in the 3 groups Binary logarithms of the relative abundance of a single genus in the normal, adenoma, and cancer groups. Each genus was present in more than 50% of the samples in the cancer group. Most of the significant differences were between the cancer group and the other 2 groups. (B) Relative abundance between the 3 groups in enterotype I The binary logarithm of the relative abundance of a single genus in enterotype I. Both genera were present in more than 50% of the cancer group samples. The significance observed in the Citrobacter levels was between the cancer group and the other 2 groups, while Bacteroides levels were significantly different between cancer and adenoma groups. (C) Relative abundance between the 3 groups in enterotype II A binary logarithm of the relative abundance of a single genus in enterotype II. All genera were present in more than 50% of the cancer group samples. Fusobacterium levels were significantly different between cancer and normal groups, Coprococcus levels were different between cancer and adenoma groups, and Clostridium differences were between the normal and adenoma groups. (D) The relative abundance between 3 groups in enterotype III Binary logarithm of relative abundance of single genus in enterotype III, and all genera present in more than 50% of samples in cancer group. Most of the significances are between cancer group and other 2 groups. Each dot represents one sample, and outlier samples are marked as red dots. (*p < 0.01).
Given that the enterotype was influenced by long-term diet, we assumed that dysbiotic features might be different within enterotypes. Further, we analysed the abundance of different groups within the 3 enterotypes. In enterotype 1, Bacteroides and Citrobacter were less common in individuals with CRC. In enterotype II, Fusobacterium was more abundant in individuals with CRC, while the Coprococcus levels were lower. The abundance of twelve genera were significantly different in enterotype III. In particular, we observed an overexpression of pathogenic bacteria, including Aeromonas, Enterococcus, Fusobacterium, and Porphyromonas (all p < 0.01, Fig. 4B–D, Supplementary Table S4B–D).
“Key bacteria” in the transition from a pre-cancerous polyp to CRC
We further performed a subgroup analysis to observe changes in the abundance of “key bacteria” during the transition from an advanced polyp to early-stage (stage 0, 1, or 2) CRC. We found a significantly decreased abundance of four butyrate-producing bacteria during this progression: Eubacterium, Roseburia, Faecalibacterium, and Oscillospira (all p < 0.01, Fig. S3). Of note, less Oscillospira was found in stage 0 CRC relative to advanced polyps, while reduced Haemophilus was observed in stage 1 and 2 CRC relative to stage 0 CRC (all p < 0.01, Fig. 5).

Relative abundance in the transition from an advanced adenoma to stage 0 CRC to early-stage CRC A binary logarithm of the relative abundance of a single genus between the two sub-groups. The relative abundance of Oscillospira in the CRC stage 0 group was significantly lower than in the advanced adenoma group and was also present in approximately 35% fewer samples in the CRC stage 0 group. The relative abundance of Haemophilus in the CRC stage 0 group was significantly higher than in the CRC stage 1, 2 group. Each dot represents one sample, and outlier samples are marked as red dots. (*p < 0.01).
CRC correlation clustering and classifiers for CRC
We performed a Spearman’s correlation analysis to identify CAGs between the normal, adenomatous polyp and CRC groups. Only the genera whose appearance were greater than 50% in cancer group were selected. Using different combinations of samples from the normal (N), adenomatous polyp (A), and CRC (C) groups (Supplementary Figs S4–S7 and Tables S5–S12), we found that the CAG created from the combination of N and C faecal samples had the best ability to classify adenoma and CRC samples (Table S6, Fig. 6). The abundance of cluster 3 significantly increased in the CRC group, whereas that of cluster 5 decreased. In enterotype 3 (Escherichia-predominated enterotype), the CRC group contained decreased levels of clusters 5 and 7 (Fig. 6).

The relative abundance of clustered CAGs, with significant differences between groups Normal and cancer samples were selected for Pearson’s correlation analysis, and 7 CAGs were combined. The abundance of cluster 3 was significantly increased in the CRC group, whereas cluster 5 was decreased. In enterotype 3 (the Escherichia-predominated enterotype), the CRC group had decreased levels of clusters 5 and 7.
Network of bacteria
The composition of each CAG varied between different sample combinations. However, cluster 2, which contained 10 genera—Brenneria, Cronobacter, Erwinia, Escherichia, Nitrobacter, Paracoccus, Pectobacterium, Photorhabdus, Shigella, and Sporosarcina—stayed the same in all groups. Furthermore, the genera also clustered together in all enterotypes. These 10 genera clustered in every group, with a high correlation coefficient (Table S13, in the cancer group). This steady CAG provided a satisfactory standard for identifying specific genera that differed between groups. We identified two genera, Clostridium and Coprococcus, whose correlation coefficient with this CAG varied between the normal, adenoma, and cancer groups in enterotype 1 (Figs 7A and S8A). In enterotype 2, 9 genera, including Citrobacter, Clostridium, Fusobacterium, Klebsiella, Lactobacillus, Leclercia, Peptostreptococcus, Synergistes, and Veillonella, had a high variation between groups in their correlation with this CAG (Figs S8B and S9). We also found that the Blautia, Clostridium, Klebsiella, Leclercia, Oscillospira, Veillonella, and Xenorhabdus genera had substantially different correlation coefficients within this CAG in the normal, adenoma, and cancer groups in enterotype 3 (Figs 7B and S8C).

(A) Network analysis of stool microbiota using Pearson’s correlation coefficients (Enterotype I) Correlation coefficients between 10 genera of CAG 2, Clostridium and Coprococcus. (B) Enterotype III Correlation coefficients network between 10 genera of CAG and 2 and 7 other genera.
Discussion
The study confirmed that faecal microbiota differ along the adenoma-to-carcinoma sequence and across enterotypes. A previous metagenome-wide association study reported a greater prevalence of CRC faecal microbiota, suggesting an overgrowth of potential pathogenic taxa16. Identical findings were observed in the present study in CRC from enterotype 3 and in late-stage CRC. The increased abundance of Fusobacterium, Enterococcus, and Aeromonas in the CRC group was consistent with previous reports14,15,25. The high abundance of Porphyromonas that was previously reported was observed only in enterotype 314,15. Beyond Bacteroides-dominated and Prevotella-dominated enterotypes, we observed an Escherichia-dominated enterotype 3 in the Taiwanese population that was different from the Ruminococcus enterotype26.
Consensus is that no single bacteria is representative of the dysbiosis of CRC and that increased levels of potentially pathogenic bacteria are not the only biomarkers of CRC15,16,27. The loss of potentially beneficial taxa may be more predictive of colorectal neoplasms27. In this study, we identified that a decreased abundance of CAG cluster 5 and cluster 7, composed primarily of butyrate-producing bacteria, is a suitable marker of CRC. In previous study of Flemer, B. et al.15, they mentioned that “no single OTU tested being increased in all individuals with CRC” and “community structure can be more informative than abundance differences of individual taxa”. Although we identified several significant genera in different enterotypes, not a single genus showed significance in all groups. Here, we not only tried to identify significant markers in groups, but also found a highly correlated group of 10 genera—Brenneria, Cronobacter, Erwinia, Escherichia, Nitrobacter, Paracoccus, Pectobacterium, Photorhabdus, Shigella, and Sporosarcina—stayed the same in all groups and enterotypes. This might suggest a least part of the gut bacteria function as groups.
Clinically, CRCs occurred in patients with relatively healthy diets or in vegetarians, who were considered to be at a decreased risk of CRC28,29. Genetic and environmental factors may play a role in this situation. Given that enterotypes are associated with long-term diet, we assumed that the “key bacteria” contributing to CRC may be different between enterotypes23,26. The Prevotella enterotype is dominated by fibre-using bacteria that ferment dietary fibre into short chain fatty acids (SCFAs)14. Subjects with the Prevotella enterotype have been reported to have a lower serum low-density lipoprotein level, which is associated with a lower cardiometabolic risk30. Metabolic syndrome is a risk factor for the incidence and recurrence of CRC and is a poor prognostic factor after radical resection31,32. In our Prevotella-enterotype cohort, enriched Fusobacterium and depleted Coprococcus levels were consistent with the results of a previous study that analysed stool and mucosa samples from CRC patients15. Coprococcus is a butyrate-producing anaerobe with immunomodulatory and anti-inflammatory properties33. Coprococcus comes is associated with a healthy gut and is particularly common in healthy Mongolians34. This finding may play a key role in the pathogenesis of CRC in this enterotype.
To date, no study has reported on microbiota changes during the transition sequence from advanced adenoma to carcinoma in situ to early CRC. For the first time, we found that Oscillospira levels were significantly reduced in stage 0 CRC, whereas Haemophilus was reduced in early-stage CRC. Oscillospira are under-studied anaerobes and butyrate-producing bacteria associated with leanness that have been found to be reduced in humans in the setting of inflammation35,36. The two genera may act as competitors in the healthy gut. The increasing richness of these organisms during the transition from pre-cancerous lesions to late-stage CRC may arise from the overgrowth of harmful bacteria as sequela of the two depleted taxa.
A bacterial driver-passenger model was previously proposed for CRC to explain individual variations between CRC patients and healthy subjects37. The gut microbiota of CRC patients carries more “driver” bacteria with pro-carcinogenic features that can interact with the intestinal microenvironment but are then outcompeted by “passenger” bacteria. In our study, the abundance of Bacteroides and Citrobacter in enterotype 1 and Bacteroides, Eubacterium, Faecalibacterium, Ruminococcus, Bilophila, and Roseburia in enterotype 3 decreased along the adenoma-carcinoma sequence, suggesting that these bacteria act as “driver” bacteria, a finding consistent with that of a previous report37. In addition, increases in Fusobacterium and Clostridium in enterotype 2 and enterotype 3 and Pseudomonas, Aeromonas, and Porphyromonas in enterotype 3 in the CRC group were consistent with “passenger” bacteria as sequela of CRC.
A strength of our study is that we systemically analysed different stages of colorectal neoplasms along the conventional adenoma-carcinoma sequence and across different enterotypes11. Our findings confirmed that faecal microbiota is a potentially favourable detection tool for CRC. Current screening tools use FIT, which detects human globin and is less influenced by diet or drugs7. However, the sensitivity of FIT studies varies from 65%-81% for CRC and is less than 30% for advanced neoplasms, which need detectable haemoglobin in the stool for increased accuracy7. The multi-target stool DNA test improves the cancer detection rate, with a sensitivity of 92.3% for CRC and 42.4% for pre-cancerous lesions10. The DNA test detects gene mutations presented in a shedding adenoma or tumour that improves its diagnostic accuracy in the setting of CRC. However, this test is still limited in the setting of non-cancerous neoplasms, and its accuracy may also be confounded by tumour size. Combined, the faecal metagenomic test and FIT might improve CRC detection sensitivity dramatically22. It is important to perform further validation tests, and the addition of an enterotype analysis should be considered. Furthermore, to increase the clinical value of such tests, it is necessary to develop affordable stool tests combined with a stool occult blood test. The ultimate goal is to provide a more predictive non-invasive screening tool, which may increase patient interest in receive screening tests and reduce clinical load and medical resource cost.
Our study was limited by the small number of subjects in enterotype 2. Second, this is an observational study, and the CAG classifiers need further validation and comparison with FIT. Third, a more cost-effective method is required for clinical translation.
In conclusion, we performed an enterotype-based analysis of CRC human faecal microbiota along the conventional adenoma-carcinoma sequence. We highlight that the dysbiotic features of CRC luminal gut microbiota are different across enterotypes, implying that our results may be confounded by lifestyle and long-term dietary habits. Although the interaction involving the process of carcinogenesis and CRC progression requires further study on the tissue microbiota, faecal microbiota could be a potential tool for the screening of CRC. To improve their predictive value, metagenomic biomarkers may not be composed of a single gene or taxon. A combination of CAG with known increased or decreased abundance should be evaluated. Future studies should include the validation of biomarkers in a different cohort and a comparison with current screening and diagnostic approaches.
Methods
Ethics approval and consent to participate
This study was reviewed and approved by the Ethics Committee of Chung Shan Medical University Hospital (CSMUH No: CS14047). All of the methods were performed in accordance with relevant guidelines and regulations, including any relevant details. Informed consents were obtained from all patients, as approved by the Institutional Review Board.
Patients and sample collection
From 2014 to 2016, 283 participants underwent a screening or surveillance colonoscopy were enrolled at Chung Shan Medical University Hospital, Taichung, Taiwan. All fresh faecal samples were collected from the patients before colonoscopy using Sigma-Transwab (Medical Wire, Corsham, Wiltshire England) with Liquid Amies Transport Medium before their colon preparation procedure and were stored in their home refrigerators at −20 °C prior to transport to the laboratory, where the samples were stored in a freezer at −80 °C. Subjects who were under the age of forty, pregnant, used antibiotics or probiotics within two months of stool collection, had evidence of infection, had undergone a colectomy, received preoperative chemotherapy or radiotherapy, or were diagnosed with inflammatory bowel disease (e.g., Crohn’s disease or ulcerative colitis) or any malignancy were excluded from the study.
Bowel preparation, colonoscopy, and pathology
All participants underwent a conventional bowel preparation that included polyethylene glycol electrolyte lavage powder (containing sodium chloride 21.36 mg, sodium bicarbonate 24.57 mg, potassium chloride 10.83 mg, sodium sulfate anhydrous 82.9 mg, and polyethylene glycol 4000 860.34 mg). Colonoscopies were performed primarily by 7 experienced endoscopists. Based on the colonoscopy findings and pathology reports, subjects were grouped into normal, small adenoma, advanced adenoma (i.e., size ≥1 cm, villous or tubulovillous features, or high grade dysplasia), carcinoma in situ (stage 0), early-stage carcinoma (stage I and II), and late-stage carcinoma (stage III and IV) groups38. Patients who did not receive a complete colonoscopy or had serrated polyps were also excluded from the study.
DNA extraction
In this study, faeces were obtained from the participants. DNA was extracted directly from the stool samples using a QIAamp Fast DNA Stool Mini Kit (Qiagen, Hilden, Germany). For stool samples, a swab was vortexed vigorously and incubated at room temperature for 1 min. An aliquot of 200 μL of each sample was then transferred a microcentrifuge tube containing 950 μL InhibitEX Buffer and then vortexed until it was thoroughly homogenized. An enzyme solution (50 μL of 4 mg/mL lysozyme; 4 mM Tris·HCl, pH 8.0; 0.4 mM EDTA; 0.4% SDS) was added into the sample, which was then incubated at 37 °C for 30 min and 95 °C for 15 min. Particles were pelleted with a centrifuge, and 600 μL of supernatant was transferred into a new tube that contained 45 μL of proteinase K (20 mg/mL) and 600 μL of Buffer AL. After 10 minutes of incubation at 70 °C, 600 μL of ethanol was added to the lysate. Extractions were then performed with QIAamp spin columns according to the QIAamp Fast DNA Stool Mini Kit protocol. The extracted DNA from the stool was eluted with 50 μL Buffer AE. All samples were centrifuged at 18,000 × g for 1 min. Final concentrations were measured using a NanoPhotometer (Implen, Westlake Village, CA USA) and then stored at −20 °C for further analysis.
Library construction and sequencing for the V3-V4 region of the 16S ribosomal RNA gene
The 16S rRNA gene, a molecular marker for identifying bacterial species, consists of nine hypervariable regions. Using 2-step PCR amplification, we can add adaptor sequences into the V3 and V4 hypervariable regions. This region, which provides ample information on the taxonomic classification of microbial communities from specimens associated with human microbiome studies, was used in the Human Microbiome Project39.
The 1st step of PCR is to amplify the V3 and V4 hypervariable regions. The amplicon primers are designed to contain (1) gene-specific sequences selected from work done by Klindworth et al.40; (2) a sequencing primer binding site that allows amplicons to be sequenced via dual-indexed sequencing with the MiSeq system (Illumina, San Diego, CA USA); and 3) a 0 to 7 bp “heterogeneity spacer” that increases the sequence diversity of the 16S rRNA gene libraries41. PCR amplification was performed using a 25 μL reaction volume that contained 12.5 μL of 2X KAPA HiFi HotStart ReadyMix (KAPA Biosystems, Wilmington, MA USA), 0.2 μM each of forward and reverse primer, and 100 ng of the DNA template. The reaction process was executed by raising the solution temperature to 95 °C for 3 min, then performing 25 cycles of 98 °C for 20 sec, 55 °C for 30 sec, and 72 °C for 30 sec, ending with the temperature held at 72 °C for 5 min. Amplicons were purified using the AMPure XP PCR Purification Kit (Beckman Coulter Life Sciences, Indianapolis, IN USA).
The second step of PCR is to add the index adaptors using a 10-cycle PCR programme. The PCR step adds the index 1 (i7), index 2 (i5), sequencing, and common adapters (P5 and P7) required for cluster generation and sequencing. PCR amplification was performed on a 25 μL reaction volume containing 12.5 μL of 2X KAPA HiFi HotStart ReadyMix (KAPA Biosystems, Wilmington, MA USA), 0.2 μM of each index adaptor (i5 and i7), and 2.5 μL of the first-PCR final product. The reaction process was executed by raising the solution temperature to 95 °C for 3 min, then performing 10 cycles of 98 °C for 20 sec, 55 °C for 30 sec, and 72 °C for 30 sec, ending with a 72 °C hold for 5 min. Amplicons were purified using the AMPure XP PCR Purification Kit (Beckman Coulter Life Sciences, Indianapolis, IN USA).
Amplified products were then checked with 2% agarose gel electrophoresis with Novel Juice (GeneDireX, Taiwan). Amplicons were purified using the AMPure XP PCR Purification Kit (Beckman Coulter Genomics, Danvers, MA, USA) and quantified using the Qubit dsDNA HS Assay Kit and a Qubit 2.0 Fluorimeter (Thermo Fisher Scientific, Waltham, MA USA), and qPCR with the Library Quantification Kit for Illumina (KAPA Biosystems, Wilmington, MA USA), all according to their corresponding manufacturer’s instructions.
The PhiX Control library (v3) (Illumina, San Diego, CA USA) was combined with the amplicon library (expected at 20%). The library was clustered to a density of approximately 800–1000 K/mm2. The libraries were processed for cluster generation and sequencing on 250PE MiSeq runs, and one library was sequenced using the standard Illumina sequencing primers, eliminating the need for an eight-index read. Sequencing data were available within approximately 40 h. Image analysis, base calling and data quality assessment were performed using the MiSeq instrument.
16S rRNA gene V3V4 region amplicon sequencing data quality control
Heterogeneity spacers41 and 5′ end primer sequence were identified and removed by in-house script. FASTX-Toolkit (http://hannonlab.cshl.edu/fastx_toolkit) was applied to control that the read quality in 70% or above of read region of each read is higher than Q20. We also applied fastq_quality_trimmer from FASTX-Toolkit to cut the bad quality 3′ tail of each read, and remain the read which length is higher than 100 nts. Finally, we matched read 1 (forward read) and read 2 (reverse reads) for next taxonomy assignment analysis stage.
Taxonomy assignment and OTU table generation
Bowtie2 (2.2.8)42 was applied to align paired sequencing reads that passed quality control to a 16S rRNA gene sequence to reference, the SILVA database (release SILVA_SSU_Parc_115)43,44. We set the parameters were “–very-sensitive–end-to-end–no-mixed–no-discordant–dovetail -X 1000” to make the alignment results with higher specificity. We assigned the taxonomy when both paired reads are 97% or above similarity to the same taxonomy reference. After this taxonomy assignment step, an operational taxonomic unit (OTU) table was generated.
Downstream analysis
Enterotyping based on the relative abundance of Bacteroides and Prevotella
To create a genus-level OTU table, OTUs with the same genus name were merged into one genus. We then calculated the relative abundance of each genus. We classified three enterotypes based on the following criteria: (i) Enterotype I, RAB ≥ 40% and RAB > RAp; (ii) Enterotype II, RAp ≥ 30% and RAp ≥ RAB; (iii) Enterotype III, Others. If the RAB in one sample was greater than or equal to 40% and the RAB was greater than the RAP, the sample was classified as enterotype I. If the RAP was greater than or equal to 30% and the RAP was greater than or equal to the RAB, the sample was classified as enterotype II. Otherwise, the sample was placed into the enterotype III group. RAB represents the relative abundance of Bacteroides, and RAP represents the relative abundance of Prevotella.
Statistical analysis
The richness and Shannon index was used to calculate alpha-diversity. A two-sided Mann-Whitney rank test (python package SciPy 1.0.0) was used to compare the two groups. A correlation analysis using Spearman’s rank correlation coefficient was performed using the corrplot package in R (R Foundation for Statistical Computing, Vienna, Austria), and the co-abundancegroups (CAGs) were defined by the corrplot created heat plots (Figs S4A, S5A, S6A and S7A) and the hierarchical clustering in plots15,45. Tax4Fun46 was used to predict the function and metabolic capabilities of the microbial communities.
Visualization
R software and the ggplot2 and reshape2 packages were used to create boxplots. A heatmap of our functional analysis was illustrated with MORPHEUS (https://software.broadinstitute.org/morpheus). The correlation network of specific genera was built using Cytoscape47.
Data Availability
Sequence data associated with this project have been deposited at the NCBI under study accession SRP131074 (https://www.ncbi.nlm.nih.gov/sra/SRP131074).
References
Siegel, R., Ma, J., Zou, Z. & Jemal, A. Cancer statistics, 2014. CA: a cancer journal for clinicians 64, 9–29, https://doi.org/10.3322/caac.21208 (2014).
Nishihara, R. et al. Long-term colorectal-cancer incidence and mortality after lower endoscopy. The New England journal of medicine 369, 1095–1105, https://doi.org/10.1056/NEJMoa1301969 (2013).
Loberg, M. et al. Long-term colorectal-cancer mortality after adenoma removal. The New England journal of medicine 371, 799–807, https://doi.org/10.1056/NEJMoa1315870 (2014).
Lieberman, D. A. et al. Guidelines for colonoscopy surveillance after screening and polypectomy: a consensus update by the US Multi-Society Task Force on Colorectal Cancer. Gastroenterology 143, 844–857, https://doi.org/10.1053/j.gastro.2012.06.001 (2012).
Ferlay, J. et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. International journal of cancer. Journal international du cancer 136, E359–386, https://doi.org/10.1002/ijc.29210 (2015).
Quintero, E. et al. Colonoscopy versus fecal immunochemical testing in colorectal-cancer screening. The New England journal of medicine 366, 697–706, https://doi.org/10.1056/NEJMoa1108895 (2012).
Levin, B. et al. Screening and surveillance for the early detection of colorectal cancer and adenomatous polyps, 2008: a joint guideline from the American Cancer Society, the US Multi-Society Task Force on Colorectal Cancer, and the American College of Radiology. Gastroenterology 134, 1570–1595, https://doi.org/10.1053/j.gastro.2008.02.002 (2008).
Ling, B. S., Moskowitz, M. A., Wachs, D., Pearson, B. & Schroy, P. C. Attitudes toward colorectal cancer screening tests. Journal of general internal medicine 16, 822–830 (2001).
van Rossum, L. G. et al. Random comparison of guaiac and immunochemical fecal occult blood tests for colorectal cancer in a screening population. Gastroenterology 135, 82–90, https://doi.org/10.1053/j.gastro.2008.03.040 (2008).
Imperiale, T. F. et al. Multitarget stool DNA testing for colorectal-cancer screening. The New England journal of medicine 370, 1287–1297, https://doi.org/10.1056/NEJMoa1311194 (2014).
Arends, M. J. Pathways of colorectal carcinogenesis. Applied immunohistochemistry & molecular morphology: AIMM/official publication of the Society for Applied Immunohistochemistry 21, 97–102, https://doi.org/10.1097/PAI.0b013e31827ea79e (2013).
Brenner, H., Kloor, M. & Pox, C. P. Colorectal cancer. Lancet 383, 1490–1502, https://doi.org/10.1016/s0140-6736(13)61649-9 (2014).
Schwabe, R. F. & Jobin, C. The microbiome and cancer. Nature reviews. Cancer 13, 800–812, https://doi.org/10.1038/nrc3610 (2013).
Yu, J. et al. Metagenomic analysis of faecal microbiome as a tool towards targeted non-invasive biomarkers for colorectal cancer. Gut 66, 70–78, https://doi.org/10.1136/gutjnl-2015-309800 (2017).
Flemer, B. et al. Tumour-associated and non-tumour-associated microbiota in colorectal cancer. Gut 66, 633–643, https://doi.org/10.1136/gutjnl-2015-309595 (2017).
Feng, Q. et al. Gut microbiome development along the colorectal adenoma-carcinoma sequence. Nature communications 6, 6528, https://doi.org/10.1038/ncomms7528 (2015).
Irrazabal, T., Belcheva, A., Girardin, S. E., Martin, A. & Philpott, D. J. The multifaceted role of the intestinal microbiota in colon cancer. Molecular cell 54, 309–320, https://doi.org/10.1016/j.molcel.2014.03.039 (2014).
Castellarin, M. et al. Fusobacterium nucleatum infection is prevalent in human colorectal carcinoma. Genome research 22, 299–306, https://doi.org/10.1101/gr.126516.111 (2012).
Mima, K. et al. Fusobacterium nucleatum and T Cells in Colorectal Carcinoma. JAMA Oncol 1, 653–661, https://doi.org/10.1001/jamaoncol.2015.1377 (2015).
Flanagan, L. et al. Fusobacterium nucleatum associates with stages of colorectal neoplasia development, colorectal cancer and disease outcome. European journal of clinical microbiology & infectious diseases: official publication of the European Society of Clinical Microbiology 33, 1381–1390, https://doi.org/10.1007/s10096-014-2081-3 (2014).
Zackular, J. P., Rogers, M. A., Ruffin, M. T. T. & Schloss, P. D. The human gut microbiome as a screening tool for colorectal cancer. Cancer prevention research 7, 1112–1121, https://doi.org/10.1158/1940-6207.CAPR-14-0129 (2014).
Zeller, G. et al. Potential of fecal microbiota for early-stage detection of colorectal cancer. Molecular systems biology 10, 766, https://doi.org/10.15252/msb.20145645 (2014).
Arumugam, M. et al. Enterotypes of the human gut microbiome. Nature 473, 174–180, https://doi.org/10.1038/nature09944 (2011).
Hsu, S. D. et al. miRTarBase: a database curates experimentally validated microRNA-target interactions. Nucleic Acids Res 39, D163–169 (2011).
Peters, B. A. et al. The gut microbiota in conventional and serrated precursors of colorectal cancer. Microbiome 4, 69, https://doi.org/10.1186/s40168-016-0218-6 (2016).
Wu, G. D. et al. Linking long-term dietary patterns with gut microbial enterotypes. Science 334, 105–108, https://doi.org/10.1126/science.1208344 (2011).
Baxter, N. T., Ruffin, M. T. T., Rogers, M. A. & Schloss, P. D. Microbiota-based model improves the sensitivity of fecal immunochemical test for detecting colonic lesions. Genome Med 8, 37, https://doi.org/10.1186/s13073-016-0290-3 (2016).
Bhopal, R. S. Diet and Colorectal Cancer Incidence. JAMA Intern Med 175, 1726–1727, https://doi.org/10.1001/jamainternmed.2015.4016 (2015).
Printz, C. Vegetarian diet associated with lower risk of colorectal cancer. Cancer 121, 2667, https://doi.org/10.1002/cncr.29582 (2015).
de Moraes, A. C. et al. Enterotype May Drive the Dietary-Associated Cardiometabolic Risk Factors. Front Cell Infect Microbiol 7, 47, https://doi.org/10.3389/fcimb.2017.00047 (2017).
Bardou, M., Barkun, A. N. & Martel, M. Obesity and colorectal cancer. Gut 62, 933–947, https://doi.org/10.1136/gutjnl-2013-304701 (2013).
You, J. et al. Metabolic syndrome contributes to an increased recurrence risk of non-metastatic colorectal cancer. Oncotarget 6, 19880–19890, https://doi.org/10.18632/oncotarget.4166 (2015).
Riviere, A., Selak, M., Lantin, D., Leroy, F. & De Vuyst, L. Bifidobacteria and Butyrate-Producing Colon Bacteria: Importance and Strategies for Their Stimulation in the Human Gut. Front Microbiol 7, 979, https://doi.org/10.3389/fmicb.2016.00979 (2016).
Liu, W. et al. Unique Features of Ethnic Mongolian Gut Microbiome revealed by metagenomic analysis. Sci Rep 6, 34826, https://doi.org/10.1038/srep34826 (2016).
Konikoff, T. & Gophna, U. Oscillospira: a Central, Enigmatic Component of the Human Gut Microbiota. Trends Microbiol 24, 523–524, https://doi.org/10.1016/j.tim.2016.02.015 (2016).
Gophna, U., Konikoff, T. & Nielsen, H. B. Oscillospira and related bacteria - From metagenomic species to metabolic features. Environ Microbiol 19, 835–841, https://doi.org/10.1111/1462-2920.13658 (2017).
Tjalsma, H., Boleij, A., Marchesi, J. R. & Dutilh, B. E. A bacterial driver-passenger model for colorectal cancer: beyond the usual suspects. Nature reviews. Microbiology 10, 575–582, https://doi.org/10.1038/nrmicro2819 (2012).
Edge, S. B. & Compton, C. C. The American Joint Committee on Cancer: the 7th edition of the AJCC cancer staging manual and the future of TNM. Annals of surgical oncology 17, 1471–1474, https://doi.org/10.1245/s10434-010-0985-4 (2010).
Schoen, R. E. et al. Colorectal-cancer incidence and mortality with screening flexible sigmoidoscopy. The New England journal of medicine 366, 2345–2357, https://doi.org/10.1056/NEJMoa1114635 (2012).
Klindworth, A. et al. Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Research 41, e1–e1, https://doi.org/10.1093/nar/gks808 (2013).
Fadrosh, D. W. et al. An improved dual-indexing approach for multiplexed 16S rRNA gene sequencing on the Illumina MiSeq platform. Microbiome 2, 6, https://doi.org/10.1186/2049-2618-2-6 (2014).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat Methods 9, 357–359 (2012).
Pruesse, E. et al. SILVA: a comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic Acids Research 35, 7188–7196 (2007).
Quast, C. et al. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res 41, D590–596 (2013).
Claesson, M. J. et al. Gut microbiota composition correlates with diet and health in the elderly. Nature 488, 178–184, https://doi.org/10.1038/nature11319 (2012).
Aßhauer, K. P., Wemheuer, B., Daniel, R. & Meinicke, P. Tax4Fun: predicting functional profiles from metagenomic 16S rRNA data. Bioinformatics 31, 2882–2884, https://doi.org/10.1093/bioinformatics/btv287 (2015).
Shannon, P. et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome research 13, 2498–2504, https://doi.org/10.1101/gr.1239303 (2003).
Acknowledgements
This study was funded by Chung Shan Medical University Hospital, Taichung, Taiwan (CSH-2014-D-004-Y2) and was particularly supported by “Aiming for the Top University Program” at the National Chiao Tung University (NCTU-104W976 and 105W976) and the Ministry of Education, Taiwan, R.O.C. In addition, grants from the Health and welfare surcharge of tobacco products, Ministry of Health and Welfare, Taiwan [MOHW 103-TD-B-111-08, MOHW 104-TDU-B-212-124-005, MOHW 105-TDU-B-212-134002, MOHW 106-TDU-B-212-144005]. And grants from Ministry of Science and Technology, Taiwan [MOST 103-2628-B-009-001-MY3, MOST 105-2627-M-009-007, MOST 105-2319-B-400-002, MOST 104-2911-I-009-509, MOST 106-2633-B-009-001, MOST 106-2627-M-009-002, MOST 106-2319-B-400-001]. We express our sincere appreciation to the participants for taking part in this study.
Author information
Authors and Affiliations
Contributions
T.W.Y., W.H.L., H.D.H., C.N.H., Y.J.J. and C.C.L. conceived and designed the study; T.W.Y., W.H.L., M.C.T., C.C.W., H.Y.C., C.C.H., B.H.S. and C.C.L. arranged the sample collection and preparation; W.H.L., T.H.S. and H.T.H. extracted DNA and constructed the sequencing libraries; H.D.H., W.H.L., T.L.Y., T.H.S., S.F.Y., F.M.L. and H.M.C. contributed to conception of the research project and coordinated the sequencing; T.W.Y., W.H.L., S.J.T., W.C.H., S.Y.H., W.L.C. contributed to data interpretation. W.H.L., S.J.T., W.C.H., Y.P.C., C.H.C., Y.R.H., Y.R.S. and L.C. analyzed the data and made the figures; T.W.Y. and W.H.L. prepared the draft manuscript. All authors discussed the results and contributed to the preparation of the final manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Yang, TW., Lee, WH., Tu, SJ. et al. Enterotype-based Analysis of Gut Microbiota along the Conventional Adenoma-Carcinoma Colorectal Cancer Pathway. Sci Rep 9, 10923 (2019). https://doi.org/10.1038/s41598-019-45588-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-45588-z
This article is cited by
-
Identification of enterotype and its predictive value for patients with colorectal cancer
Gut Pathogens (2024)
-
Alteration in gut microbial characteristics of patients with acromegaly
Endocrine (2024)
-
Reproducible and opposing gut microbiome signatures distinguish autoimmune diseases and cancers: a systematic review and meta-analysis
Microbiome (2022)
-
A systematic review of microbial markers for risk prediction of colorectal neoplasia
British Journal of Cancer (2022)
-
Acromegaly is associated with a distinct oral and gut microbiota
Pituitary (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
