
Abstract
Myeloid-derived suppressor cells (MDSCs) expand during inflammation and exhibit immunomodulatory functions on innate and adaptive immunity. However, their impact on trauma-induced immune responses, characterized by an early pro-inflammatory phase and dysregulated adaptive immunity involving lymphocyte apoptosis, exhaustion and unresponsiveness is less clear. Therefore, we adoptively transferred in vitro-generated MDSCs shortly before experimental blunt chest trauma (TxT). MDSCs preferentially homed into spleen and liver, but were undetectable in the injured lung, although pro-inflammatory mediators transiently increased in the bronchoalveolar lavage (BAL). Surprisingly, MDSC treatment strongly increased splenocyte numbers, however, without altering the percentage of splenic leukocyte populations. T cells of MDSC-treated TxT mice exhibited an activated phenotype characterized by expression of activation markers and elevated proliferative capacity in vitro, which was not accompanied by up-regulated exhaustion markers or unresponsiveness towards in vitro activation. Most importantly, also T cell expansion after staphylococcal enterotoxin B (SEB) stimulation in vivo was unchanged between MDSC-treated or untreated mice. After MDSC transfer, T cells preferentially exhibited a Th1 phenotype, a prerequisite to circumvent post-traumatic infectious complications. Our findings reveal a totally unexpected immunostimulatory role of adoptively transferred MDSCs in TxT and might offer options to interfere with post-traumatic malfunction of the adaptive immune response.
Similar content being viewed by others


Introduction
Myeloid-derived suppressor cells (MDSCs) are defined as immature myeloid cells with potent immunoregulatory activity whose differentiation is favoured in cancer and other pathological conditions associated with chronic inflammation. MDSC induction, expansion and activation is induced by various factors produced by tumours or bone marrow (BM) stroma in response to chronic inflammation such as IL-6, GM-CSF, G-CSF or VEGF in combination with pro-inflammatory molecules released by activated T cells and myeloid cells or bacterial and viral danger signals as IFN-γ, IL-1β, IL-13 and TLR-ligands1. Murine MDSCs consist of two major populations: granulocytic MDSCs (gMDSC) identified by a CD11b+Ly-6G+Ly-6Clow phenotype and monocytic MDSCs (mMDSCs), which are CD11b+Ly-6G−Ly-6Chigh. Although T cells are the preferred and major targets of MDSC action they modulate the function of other immune cells such as B-, NK- and dendritic cells. Immunosuppressive factors produced by MDSCs are versatile and comprise enzymes catabolizing amino acids essential for T cell activation, proliferation and function such as arginase-1, iNOS and indoleamine 2,3-dioxygenase (IDO), immunosuppressive cytokines like IL-10 and TGF-β, the production of ROS as well as the expression of membrane molecules leading to T cell exhaustion and apoptosis2,3,4. Beside the ability to inhibit expansion and functions of T cells, MDSCs influence the type of immune response by modulating the Th1/Th2 balance depending on the disease entity. While a shift towards type 2 immunity is reported after bone marrow and solid organ transplantation, viral infections, pregnancy or certain cancers, MDSCs promote Th1 responses in airway inflammation5,6,7,8,9,10,11.
Most of the knowledge regarding the immunosuppressive role of MDSCs has come from studies in the context of cancer. MDSCs in cancer patients and tumour bearing mice strongly suppress the adaptive immune response and favour tumour growth12,13. However, their role in other pathological conditions is less well understood. After traumatic insults danger signals activate the innate immune system leading to a local and systemic pro-inflammatory response, which is counterbalanced by a systemic posttraumatic immune suppression characterized by an impaired adaptive immunity leading to a strongly increased risk for opportunistic infections and multi organ failure14,15. Due to the post-traumatic inflammatory environment, MDSC accumulation occurs after various traumatic injuries such as spinal cord injury, blunt chest trauma (TxT), sepsis or peripheral tissue trauma. Isolated MDSCs from traumatized animals up-regulate immunosuppressive enzymes such as arginase-1 and iNOS and decrease T cell proliferation in vitro16,17,18,19,20,21. The in vivo action of trauma-induced MDSCs is less clear and apparently model dependent. Blocking MDSC expansion by anti-Gr-1 antibody in experimental sepsis prevents Th2 polarization of the immune response20, while MDSC depletion by anti-Gr-1 inhibits Th1-specific cytokine production without affecting Th2 cytokines in a blunt chest trauma model19.
Studying the role of MDSCs in vivo is limited by the lack of antibodies specifically deleting MDSCs. The Gr-1 antibody fails to deplete all MDSC subsets in all organs and also eliminates mature neutrophils19,22,23,24. Alternatively, MDSCs at high numbers, quality and purity can be generated in vitro and utilized for adoptive transfer experiments. In vitro-generated MDSCs are mostly generated from bone marrow (BM) cells, which are cultured for a short period in GM-CSF/G-CSF or M-CSF, often combined with additional factors such as IL-6, TNF α or dexamethasone5,25,26,27. In vitro-generated MDSCs exhibit an increased expression of immunosuppressive molecules and efficiently suppress T cell proliferation in vitro. Adoptive transfer of in vitro-generated MDSCs in experimental models of allogeneic bone marrow or solid organ transplantation promote immune tolerance by preventing lethal graft-versus-host disease (GVHD) and allograft rejection5,25,26,27. Of note, transplantation of in vitro-generated MDSCs in a model of spinal cord injury at the side of injury reduce local inflammation and promote tissue regeneration17, while the effects of systemically applied MDSCs on the trauma immune response are largely unknown. Therefore, we adoptively transferred in vitro-generated MDSCs and analyzed the immune modulating effects on the innate and adaptive immune system in a clinically relevant model of blunt chest trauma.
Results
In vitro-generated MDSCs home preferentially in the spleen of TxT mice
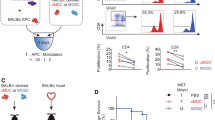
MDSCs were generated from BM cells of B6.SJL mice in the presence of GM-CSF. After four days more than 90% of the cells expressed CD11b and Gr-1, of which granulocytic MDSCs (CD11b+Ly-6G+Ly-6Clow) were induced at higher percentages as monocytic MDSCs (CD11b+Ly-6G-Ly-6Chigh) (Fig. 1A). Compared to CD11b+ cells isolated from BM, in vitro-generated MDSCs exhibited increased expression of arginase-1, iNOS, HO-1, TGF-β and TNF α, while IDO and IL-10 levels were unchanged and Cox-2 expression was even lower than in BM-isolated CD11b+ cells (Fig. 1B). MDSCs efficiently inhibited the proliferation of CD4+ and CD8+ T cells and suppression slightly varied with each preparation but usually ranged between 60–80% at a MDSC: T cell ratio of 1:1 (Suppl. Fig. S1).

Generation, characterization and homing of adoptively transferred in vitro-generated MDSCs. MDSCs were generated in vitro from BM cells of B6.SJL mice (H-2b, CD45.1+) by incubation with GM-CSF. (A) After four days cells were stained for CD11b and Gr-1 expression and the expression of Ly-6C and Ly-6G on the CD11b positive cells indicated the distribution of the MDSC subpopulations. (B) MDSCs were analyzed for the expression of immunosuppressive mediators by qRT-PCR and relative expression to AIP was calculated. (C) MDSCs were injected one hour before TxT in B6 mice (H-2b, CD45.2+) and homing was analyzed in spleen (S), liver (L) and bone marrow (BM) by analyzing the percentage of CD45.1 expressing cells and subsequently numbers of MDSCs were calculated. Data show one representative FACS staining from 5 independent experiments performed (A). (B) shows the mean value ± SD of 4 different MDSCs preparations analyzed and (C) of 5–11 mice/group analyzed. *P≤0.05; **P≤0.01; ***P≤0.001. Significance was calculated by Student’s t test.
To clarify in vivo survival and homing, MDSCs were injected one hour before B6 mice underwent TxT. Due to the congenic marker CD45.1 expressed by B6.SJL mice, transplanted CD45.1+ MDSCs could be identified in syngeneic CD45.2+ B6 mice. 6 hours after TxT, adoptively transferred MDSCs are predominantly found in the liver, accounting for 12% of liver leukocytes and to a lesser extent in spleen and bone marrow. Two days after injection, MDSCs account for less than 0.1% of total bone marrow and splenocytes and 0.3% of liver leukocytes, while at day seven MDSCs compose 1.4% of splenocytes accounting for 1.3 × 105 MDSCs (Fig. 1C). Most interestingly, MDSCs were not attracted into the injured lung and not detectable in lymph nodes at any time analyzed. These results show, that low numbers of MDSCs preferentially populate the spleen and survive in vivo at least seven days after transfer.
The early TxT-induced local and systemic pro-inflammatory immune response is not substantially modulated by adoptively transferred in vitro-generated MDSCs
Since MDSCs are attracted and activated by pro-inflammatory cytokines and chemokines and likewise MDSCs release soluble factors to exert their immunosuppressive function, we analyzed the expression of cytokines and chemokines in the bronchoalveolar lavage (BAL) (Fig. 2A) and the serum (Fig. 2B) of TxT mice treated or untreated with MDSCs 6, 24 and 48 hours after TxT. Pro-inflammatory factors described to be induced shortly after TxT such as IL-6, G-CSF or MCP-1 were slightly increased 6 hours after TxT in MDSC-treated mice in the BAL. Possibly, these factors are derived from the transplanted MDSCs, since in vitro-generated MDSCs produce elevated levels of IL-6, G-CSF or MCP-1 compared to CD11b cells isolated from BM (Suppl. Fig. S2), although no invading MDSCs could be detected in the lungs 6 hours after injection by flow cytometry. Already 24 h after TxT, concentrations decreased to levels measured in untreated TxT mice. No additional systemic increase of IL-6, G-CSF and MCP-1 was detectable by MDSCs after TxT. Since MDSCs influence the type of immune response, we determined the concentration of Th1/Th2 specific cytokines. In BAL and serum the concentration of the Th2-specific cytokine IL-5 was increased in MDSC-treated mice 6 hours after TxT, but unaltered compared to untreated animals at later time points. In vitro-generated MDSCs, however, do not produce IL-5 (Suppl. Fig. S2). Concentrations of all other Th1- and Th2-specific cytokines analyzed (IFN-γ, IL-10, -13) were not affected by MDSC treatment. All together, these data show that adoptively transferred MDSCs transiently effect cytokine and chemokine levels especially in BAL fluid but do not exhibit long-term effects neither locally in the injured organ nor systemically.

Adoptive transfer of in vitro-generated MDSCs in TxT mice does not substantially modulate the expression of cytokines in BAL and serum. TxT mice were treated with MDSCs or left untreated and BAL fluids (A) and serum (B) were analyzed for cytokine concentrations 6, 24 and 48 hours after TxT. Data present the mean value ± SD of 6–13 mice/group analyzed. *P≤0.05; **P≤0.01; ***P≤0.001. Significance was calculated by Mann-Whitney-U-Test comparing TxT w/o MDSC with TxT + MDSC at each time point.
Adoptive transfer of in vitro-generated MDSCs increases splenic leukocyte numbers
Although cytokine and chemokine levels were hardly influenced by MDSC transfer, a massive increase in spleen cell numbers was observed. Two days after adoptive MDSC transfer and TxT, splenocyte numbers were 1.4 fold elevated and increase was maintained after seven days (Fig. 3A). Transplanted MDSCs did not significantly contribute to elevated spleen cell numbers as indicated in Fig. 1C. Although splenic cell numbers increased, the distribution of the different leukocyte subsets (CD3+, CD4+, CD8+, Treg (CD4+ CD25+ FoxP3+), CD19+, Breg (CD19+ CD1dhigh CD5+) and CD11b+) analyzed was unchanged two and seven days after MDSC-treatment (Fig. 3B). Due to elevated splenocyte numbers, total cell numbers of the different cell populations were increased in MDSC-treated mice compared to untreated controls (Suppl. Fig. S3A). To clarify, whether MDSC-induced expansion of splenocytes is specific for the “traumatic environment”, we injected MDSC in untreated B6 mice. Two and seven days after MDSC injection no increase in splenocyte numbers was detected (Suppl. Fig. S3B). In summary, these results show that treatment of TxT mice with in vitro-generated MDSCs induces splenic leukocyte expansion.

MDSC treatment increases the number of splenic leukocytes, but does not alter the distribution of the different leukocyte subpopulations. MDSC-treated or untreated mice were subjected to TxT. (A) Numbers of splenocytes were determined at different time points. (B) The distribution of the different leukocyte subsets was analyzed by flow cytometry. Data represent the mean value ± SD for 5–15 mice/group analyzed. **P≤0.01; ***P≤0.001. Significance was calculated by Student’s t-test comparing TxT w/o MDSC with TxT + MDSC for each time point.
Adoptive transfer of in vitro-generated MDSCs induces T cell activation without induction of exhaustion
Since adoptive transfer of MDSCs induced expansion of T cells, which represent the most prominent targets of MDSC action, we defined the impact of MDSCs on T cell activation. Seven days after MDSC injection and TxT, spleen cells were stained after 24 hours culture for activation markers. Most of the T cells independent whether they were derived from MDSC-treated or untreated TxT mice were naïve T cells (CD44low CD122- CCR7+ (CD197) CD62L+), although a decrease from 74% to 56% for CD4+ and from 55% to 32% for CD8+ T cells was observed in MDSC-treated mice. Likewise about 7% of the CD4+ T cells and 4% of the CD8+ T cells co-expressed the activation markers CD25 and CD69 and T cells showed diminished IL-7 R (CD127) expression indicating that the presence of MDSCs apparently sensitized T cells towards an activated state (Fig. 4A). This was further confirmed by the finding that CFSE-labelled splenic T cells isolated from MDSC-treated TxT mice after 7 days start to proliferate when cultured in medium for 4 days (Fig. 4B). While CD3+ T cells from untreated TxT mice showed a spontaneous proliferation of 11%, 63% of T cells from MDSC-treated TxT mice divided in culture. Since persistent activation is often associated with T cell exhaustion, expression of exhaustion markers was defined 24 hours after culture. Only BTLA (CD272) was expressed on CD4+ and CD8+ T cells, however, independent of MDSC-treatment. All other analyzed exhaustion markers PD-1 (CD279), CTLA-4 (CD152), LAG-3 (CD223) and CD160 were not up-regulated (Fig. 4C). In summary, the presence of adoptively transferred MDSCs activates T cells without driving them into exhaustion.

Adoptive transfer of in vitro-generated MDSCs activates TxT-induced T cells without inducing exhaustion. Mice were untreated or adoptively transferred with MDSCs and subsequently received TxT. (A) 7 days later, splenocytes were cultured for 24 h and CD4+ and CD8+ T cells were stained for markers defining naïve and activated cells. (B) At day 7 after TxT, CFSE-labelled splenocytes were cultured in medium and 4 days later proliferation of CD3+ T cells was analyzed. (C) 7 days after TxT, splenocytes were cultured for 24 h and expression of exhaustion markers was defined on CD4+ and CD8+ T cells. **P≤0.01, ***P≤0.001 Data represent the mean value ± SD of 8–10 animals/group (A) and 13–14 mice/group (B). (C) FACS staining’s show the expression of markers for 3 mice/group and is representative for one experiment out of 2 experiments performed. Significance was calculated by Student’s t-test.
Adoptive transfer of in vitro-generated MDSCs in TxT mice does not impair the proliferative capacity of T cells in vitro and in vivo after activation
Since T cells from TxT mice received an activation signal by adoptively transferred MDSCs, we next clarified whether T cells maintained their ability to proliferate after activation. Seven days after TxT, spleen cells from MDSC-treated or untreated mice were activated by PHA, ConA or anti-CD3/CD28 antibodies. Surprisingly, the presence of MDSCs in TxT mice did not strongly modulate the proliferative capacity of CD3+ T cells. While PHA-induced proliferation was slightly increased, the response towards ConA was faintly attenuated and no difference was detected for anti-CD3/CD28 activation (Fig. 5A). To clarify, whether MDSC-treated TxT mice can respond to T cell activation in vivo, staphylococcal enterotoxin B (SEB), which specifically induces expansion of T cells bearing vβ8 TCRs28, was injected 24 hours after TxT. MDSC-treatment in the absence of SEB had no impact on the percentage of vβ8 expressing T cells, and SEB injection induced the proliferation of CD4+ and CD8+ vβ8+ T cells, however, to the same extend in MDSC-treated and untreated animals (Fig. 5B). All together, these results show that adoptively transferred in vitro-generated MDSCs in the context of TxT do not inhibit the proliferative capacity of T cells in vitro and in vivo.

T cells from MDSC-treated mice maintain their proliferative capacity in vitro and in vivo. MDSC-treated or untreated mice were subjected to TxT. (A) 7 days after TxT, spleen cells of MDSC-treated or untreated TxT mice were CFSE-labelled and stimulated with medium, PHA, ConA or anti-CD3/28 antibodies and after 4 days proliferation of CD3+ T cells was analyzed and percentage of specific proliferation was calculated. (B) 24 h after TxT, mice were injected with SEB and the percentage of proliferating vß8+ CD4+ and vß8+ CD8+ T cells was defined 48 hours later by flow cytometry. *P≤0.05; ***P≤0.001. Data represent the mean value ± SD of 13–14 mice/group (A) and 3–4 mice/group (B). Significance was calculated by Student’s t-test comparing TxT w/o MDSC with TxT + MDSC for each stimulus (A) and by one-way ANOVA with Sidak as post test (B).
T cells from TxT mice maintain a Th1 phenotype in the presence of in-vitro generated MDSCs
Since post-traumatic immunosuppression is associated with a shift in the immune response towards type 2 immunity29,30 and MDSCs are modulators of the Th1/Th2 balance, we analyzed the polarization of T cells after adoptive transfer of MDSCs. MDSC-treated or untreated mice received TxT and seven days later spleen cells were restimulated with PMA/ionomycin, and cytokine expression of CD3+ T cells was analyzed by intracellular flow cytometry (Fig. 6A). Most of the T cells exhibit a Th1 phenotype. Less than 2% of T cells expressed IL-10 and IL-13, while IL-13 expressing cells were slightly increased in MDSC-treated mice. Since IL-4 and IL-5 expressing cells were not detectable by flow cytometry, mRNA expression in isolated splenic T cells of MDSC-treated and untreated mice after PMA/ionomycin stimulation was analyzed. Independent of the presence or absence of MDSCs, Th1-associated cytokines were strongly expressed, while Th2-associated cytokine expression was low. However, T cells from MDSC-treated mice exhibited higher expression of IL-4, -5, -10 and -13 (Fig. 6B). In summary, adoptive transfer of MDSCs in TxT mice does not significantly shift the Th1/Th2 balance but slightly promote the induction of Th2 cells.

The adoptive transfer of in vitro-generated MDSCs maintains the Th1/Th2 balance in TxT mice. Mice were untreated or adoptively transferred with MDSCs and subsequently received TxT. (A) 7 days after TxT, splenocytes were restimulated with PMA/Iono and the intracellular expression of Th1- and Th2-associated cytokines was determined in the CD3+ T cell population by flow cytometry. (B) Alternatively, after PMA/Iono stimulation, CD3+ T cells were isolated and qRT-PCRs for the expression of Th1-and Th2-associated cytokines were performed and relative expression to AIP was calculated. *P≤0.05; **P≤0.01; ***P≤0.001. Data represent the mean value ± SD for 3–4 mice/group in (A) and (B) and significance was calculated by Student’s t-test comparing TxT w/o MDSC with TxT + MDSC for each cytokine.
Discussion
Severe trauma does not only affect the function of the injured organ but is associated with profound changes in immunohomeostasis characterized by an imbalanced and overwhelming innate immune response and a severe maladaptive immunity. MDSCs are induced after various experimental traumatic injuries due to systemic inflammation; however, their immunomodulatory effects on the subsequent immune response are less well defined. To our knowledge, we show here for the first time that the adoptive transfer of in vitro-generated MDSCs shortly before experimental TxT induces the expansion and activation of T cells without dampening their proliferative capacity, inducing exhaustion or influencing the Th1/Th2 balance. These results indicate that in the context of TxT, adoptively transferred MDSCs do not exhibit classical immunosuppressive functions but rather act immunostimulatory.
Recently, we showed that MDSCs are induced locally in the lung and systemically in the spleen shortly after TxT. TxT-induced MDSCs inhibited T cell proliferation when tested for their immunosuppressive capacity in vitro. By treating mice with the anti-Gr-1 antibody, which is commonly used for MDSC depletion, we found that the presence of MDSCs dampened the early systemic pro-inflammatory response and guaranteed the induction of Th1 cells19. Since, anti-Gr-1 treatment does not exclusively and entirely deplete all MDSCs19,23,24, adoptive transfer of in vitro-generated MDSCs offers an attractive alternative to define their influence on disease-induced immune responses and finally their therapeutic potential. In contrast to experimental models of autoimmunity or solid and BM transplantations, not much is known about the effect of in vitro-generated MDSCs on the course of traumatic injuries and their influence on the trauma-induced immune response. In vitro-generated MDSCs applied intraspinally after spinal cord injury reduce inflammation and promote tissue generation17. Furthermore, the adoptive transfer of MDSCs exhibit a cardioprotective role in heart failure31 and immature myeloid cells resembling MDSCs enhance bone fracture healing32.
Therefore, we adoptively transferred in vitro-generated MDSCs one hour before mice received TxT and analyzed changes in innate and adaptive immune responses. GM-CSF-induced MDSCs derived from BM cells exhibited increased expression of arginase-1, iNOS, HO-1, TGF-β and TNF-α and efficiently suppressed allogeneic-induced T cell proliferation in vitro. Adoptively transferred MDSCs homed preferentially in the spleen, but low numbers of MDSCs were also found in the liver and bone marrow. Although no MDSC infiltration into the injured lung was detected, pro-inflammatory mediators typically elevated in the BAL after TxT were further increased six hours after MDSC treatment while at later time points MDSC did not affect cytokine and chemokine expression. Likewise, no systemic effect was detectable indicating that the post-traumatic early inflammatory response cannot be efficiently modulated by adoptively transferred MDSCs.
Interestingly, the adoptive transfer of MDSCs strongly increased total splenocyte numbers particular in TxT mice, since non-traumatized mice did not exhibit elevated cell numbers pointing to an influence of the TxT microenvironment on MDSC functions. Since the ratio of the different splenic leukocyte subpopulations was unaltered, total cell numbers of all subpopulations analyzed were increased. B cells with a regulatory phenotype are expanded after adoptive transfer of in vitro-generated MDSCs in a murine model of systemic lupus erythematosus (SLE) and experimental autoimmune encephalomyelitis, however, MDSC treatment in this model simultaneously decreases the numbers of activated T cells33,34. Similarly, Tregs are reported to be induced after MDSC transfer dependent on the model used. While adoptive transfer of MDSCs in mice with collagen-induced arthritis or SLE show no Treg induction35,36, Treg frequency is increased for instance in experimental asthma or viral myocarditis models37,38.
Increase of all splenic leucocyte populations after MDSC transfer is to our knowledge not described yet, however, several studies indicate that MDSCs exhibit alternative functions in place of immunosuppression. MDSCs induced by inflammatory bowel disease in mice and patients not only fail to suppress autologous T cell responses but even enhance T cell proliferation in vitro39. Adoptive transfer of MDSCs in skin-transplanted mice strongly increases the numbers of infiltrating T cells in the transplant and induces the expansion of APCs and activated CD25+ CD69+ T cells in the spleen pointing to a state of systemic activation, which, surprisingly, does not lead to transplant rejection but correlates with prolonged skin graft survival40. Immunostimulatory capacities are also described for ascites-derived CD11b+ Gr-1+ leading to enhanced T cell proliferation in vitro and in vivo41. Likewise, T cells conditioned in vitro with MDSCs show an increased anti-tumour activity after adoptive T cell based immunotherapy, which is associated with increased IFN-γ expression and diminished mTOR signalling42.
The above reported immunoactivating functions of MDSCs in models of autoimmunity, cancer and transplantation correspond to our findings in the TxT model. After MDSC-treatment T cells from TxT mice showed elevated expression of activation markers and increased proliferative capacity in the absence of activation signals. However, these intrinsic potential to proliferate does not impair the ability to respond to appropriate activation signals in vitro and in vivo and most interestingly was not associated with T cell exhaustion, although in vitro-generated MDSCs exhibit high expression of PD-L1.
Beside their impact on T cell activation and expansion, MDSCs strongly modulate the type of T cell response induced. Dysregulation of the Th1/Th2 balance frequently occurs after severe traumatic injuries and suppression of Th1 responses associated with enhanced Th2 immunity contribute to the predisposition of injured individuals for infections, sepsis and impaired pathogen defence29,30,43,44. Although adoptively transferred MDSCs exhibited a strong effect on T cell expansion and activation, their influence on T cell polarization was marginal. Th2-associated cytokines were expressed at low levels after TxT and slightly increased in the presence of MDSCs, but the predominance of Th1 immunity required to resolve post-traumatic infections was maintained.
Our studies clearly indicate a discrepancy between the in vitro and in vivo action of MDSCs. While in vitro-generated MDSCs strongly prevent T cell proliferation in vitro, they induce T cell expansion in vivo. Studies by Schmidt et al. could also show that tumour-induced MDSCs efficiently suppress CTLs in vitro, but not in vivo following adoptive transfer45. This strongly implies that the microenvironment, immune response and MDSC functions form an intricately interwoven network of mutual influences. MDSCs isolated from late septic mice and subsequently transferred to septic mice decrease pro-inflammatory cytokines, increase bacterial clearance and dramatically improve survival rates. If MDSCs, however, are isolated from early septic mice representing an altered inflammatory environment and subsequently transferred into septic mice they than support disease progression46.
Adoptive transfer of in vitro-generated MDSCs in allogeneic BM transplantation models inhibit GVHD by inducing type 2 immunity without affecting T cell numbers5, while MDSCs generated in the exactly same manner induced lymphocyte expansion and T cell activation without influencing the Th1/Th2 balance in the TxT model. Likewise, the transfer of MDSCs in models of asthma-related airway inflammation diminished the inflammatory injury by shifting the balance towards a Th1 response11,47. Most interestingly, this is independent, whether MDSCs were derived from mice treated with LPS or isolated from BM of tumour bearing mice, although MDSCs in the context of cancer are known to promote Th2 responses9,10,48. Depending on the disease model in which MDSCs are induced or on the time point they are isolated, MDSCs exhibit versatile mechanisms of immunomodulation comprising the release of soluble factors, the induction of other regulatory cell types and the direct interaction with their target cells.
How MDSCs induce the induction and activation of lymphocytes in TxT is currently unclear. Comparing the transcriptome, proteome and secretome of MDSCs isolated from models such as BM transplantation and TxT, in which MDSCs exhibit totally opposite immunomodulatory effects, might depict candidates responsible for immunoactivating or immunosuppressing functions of MDSCs.
Further studies have to elucidate whether the immunostimulatory effect of adoptively transferred MDSCs is found also in other trauma patterns and most importantly, whether similar changes in the immune response are observed when MDSCs are injected after the traumatic injury, especially in order to evaluate their therapeutic potential to improve the post-traumatic immunohomeostasis. This might be of particular relevance for diseases such as sepsis, often manifested by severe, long-term maladaptive immune responses and immunosuppression associated with lymphocyte exhaustion and apoptosis, a failure to return to normal immunohomeostasis and inefficient response to secondary infections49.
Taken together, we suppose that in vitro-generated MDSCs in the context of traumatic injury do not exhibit immunosuppressive activity but activate the T cell response, which might be beneficial to dampen post-traumatic T cell malfunctions. Furthermore, we could show that MDSCs are immunoregulators with a high plasticity which after adoptive transfer acquire regulatory functions ranging from T cell inhibition to T cell activation according to the disease entity and that it is worthwhile and absolutely necessary to define the effect of adoptive MDSC transfer in each individual disease model.
Material and Methods
Animals and blunt chest trauma (TxT)
All animal experiments were performed according to the international regulations for the care and use of laboratory animals and were approved by the local Ethical Committee (No. 1196 and 1297, Regierungspräsidium Tübingen, Germany). Male C57BL/6 mice (B6, H-2b, CD45.2) (Janvier, France) were used for TxT at an age between 11 to 15 weeks. Male B6 and B6.SJL-PtprcaPepcb/BoyJ (B6.SJL, H-2b, CD45.1) mice (breeding pairs obtained from The Jackson Laboratory and bred at University of Ulm) were between 6–14 weeks for MDSC generation from bone marrow (BM). TxT was induced by a single blast wave centered on the thorax under sevoflurane anesthesisa as described previously19. In brief, compressed air was delivered in the upper chamber of the blast wave generator, which is divided by the lower chamber by a Mylar polyester film. As soon as the pressure in the upper part exceeded the defined resistance of the membrane, the film ruptured towards the nozzle and released a reproducible single blast wave and contusion of the lung, which is not associated with histological alterations in liver or abdomen. The sternum cylinder distance was 1.5 cm. Mice received buprenorphin 0.03 mg/kg 30 minutes before TxT and every 8 hours during the first 24 hours after TxT as analgetic treatment.
Bronchoalveolar lavage (BAL) and blood serum
BAL samples were obtained as described previously19. Briefly, trachea was exposed, cannulated and afterwards, 0.5 ml ice-cold PBS was injected and recovered. 1 µl proteinase inhibitor cocktail was added to 100 µl BAL and samples were centrifuged at 13,000 rpm for 1 min. Supernatant fluid was stored at −80 °C. Blood was collected after a puncture of the mandibular vein, incubated at room temperature for 30 min and centrifuged at 13,000 rpm at 4 °C for 15 min. Cytokine stabilization buffer (U-CyTech biosciences) was added to the serum and stored at −80 °C. Cytokine concentrations were determined by Procartaplex Multiplex Immunoassays (Thermo Fisher Scientific) and analyzed on a BIO RAD-Bio-Plex 200 System (Bio-Rad).
MDSC generation and adoptive transfer of MDSCs
3 × 105 BM cells/ml extracted from the femur and tibia were cultured with granulocyte-macrophage colony-stimulating factor (GM-CSF) (250U/ml) (Peprotech) for 4 days. If more than 90% of the cells expressed CD11b and Gr-1, 2 × 107 MDSCs were adoptively transferred into tail veins of B6 mice one hour before TxT.
Isolation of cells
Magnetic bead isolation
CD3+ T cells were isolated from spleens by CD3ε MicroBead Kit and CD11b+ cells were isolated from BM by using CD11b MicroBead Kit (Miltenyi). Cells were positively selected by MACS technology according to manufacture’s protocol. Purity of all isolated cells ranged between 85–99%.
Leukocytes of different organs were isolated according to following protocols:
Bone marrow
Under sterile conditions, BM was isolated from the femurs and tibias of mice and a single cell suspension was prepared by dissociating cell clumps with a syringe followed by the lysis of erythrocytes (0.15 M NH4Cl, 1 mM KHCO3, 0.1 mM Na2EDTA).
Spleen
The spleen was extracted from mice and a single cell suspension was prepared by gently pressing the spleen through a cell strainer (∅ 70 μm) and subsequently, the erythrocytes were lysed.
Liver
The liver was perfused with liver perfusion medium (Invitrogen Life Technologies) followed by liver digest medium (Invitrogen Life Technologies) afterwards removed and digested for 30 min at 37 °C. Liver cells were gently pressed trough a cell strainer (∅ 70 µm) and lymphoid cells were separated by centrifugation 60 × g for 5 min. Lymphoid cells in the supernatant were collected, washed and resuspended in PBS supplemented with 1% FCS and diluted in 70% Easycoll (Biochrom) in a 1:1 ratio (≙ 35% Easycoll) and then overlaid onto 70% Easycoll and centrifuged for 20 min at 950 × g. Lymphoid cells were collected from the interface, washed and subsequently erythrocytes were lysed.
CFSE-labelling and T cell activation in vitro and in vivo
2 × 106 spleen cells were labelled with 5 µM CFSE (Thermo Fisher Scientific) at 37 °C for 10 min, immediately washed with ice-cold PBS-5%FCS, and subsequently used for proliferation assays. 2.5 × 106/ml CFSE-labelled spleen cells of TxT mice were activated with anti-CD3 (cl. 145–2C11, 0.005 µg/ml) and anti-CD28 (cl. 37.51, 0.005 µg/ml) (CD3/28) antibodies (BD Bioscience), phytohaemagglutinin (PHA, 1.25 µg/ml, Sigma), concanavalin A (Con A, 1.25 µg/ml, Sigma) or incubated with medium and proliferation of CD3+ T cells was determined on day 4 by flow cytometry. Percentage of specific proliferation = (% stimulus-induced proliferating T cells - % proliferating T cells in medium alone)/(100 - % proliferating T cells in medium alone) x100. 24 hours after TxT 50 µg staphylococcal enterotoxin B (SEB, Sigma-Aldrich)/mouse or PBS was injected i.v. in the tail vein. Expansion of vß8+ T cells was determined 48 h later by flow cytometry.
Flow cytometry
A total of 5 × 105 cells were stained in FACS-medium (PBS-10% FCS-0.2% NaN3). Dead cells were gated out by analyzing 7-amino-actinomycin-D (7-AAD, Sigma-Aldrich) negative cells. For intracellular cytokine detection, spleen cells were restimulated with phorbol myristate acetate (PMA) (20 ng/ml) plus ionomycin (1 µM) (Calbiochem) in the presence of brefeldin A (10 µg/ml) (Sigma-Aldrich). After 5 hours, cells were stained for CD3 expression, fixed with 4% paraformaldehyde, subsequently lysed with 0.1% saponin (Sigma-Aldrich) and stained for cytokine expression. Staining of regulatory T cells (T reg) was performed according to the manufacturer’s instructions by using the Foxp3 Transcription Factor Staining Buffer Kit (Life Technologies). Antibodies used are specified in Supplementary Table 1. Flow cytometry samples were measured on LSR II flow cytometer (BD Bioscience).
RNA preparation and quantitative reverse-transcription polymerase chain reaction (qRT-PCR)
RNA was isolated and complementary DNA was synthesized as previously described19 and qRT-PCR was performed with a CFX Connect™ Real-Time PCR Detection System (Bio-Rad) using a LightCyler FastStart DNA Master PLUS SYBR Green I Kit (Roche Diagnostics). The qRT-PCR results were normalized using mouse aryl hydrocarbon receptor-interacting protein (AIP) as house keeping gene. Primer sets (Thermo Fisher Scientific) used are listed in Supplementary Table 2.
Statistics
Data were analyzed by either using a Student’s t test, Mann-Whitney-U-test or one-way ANOVA followed by a Sidak test as a post hoc test for multiple comparisons. Results were considered as significant if P≤ 0.05.
Data Availability
The data sets generated during the current study are available from the corresponding author on reasonable request.
References
Condamine, T. & Gabrilovich, D. I. Molecular mechanisms regulating myeloid-derived suppressor cell differentiation and function. Trends Immunol 32, 19–25, https://doi.org/10.1016/j.it.2010.10.002 (2011).
Veglia, F., Perego, M. & Gabrilovich, D. Myeloid-derived suppressor cells coming of age. Nat Immunol 19, 108–119, https://doi.org/10.1038/s41590-017-0022-x (2018).
Zhao, Y., Wu, T., Shao, S., Shi, B. & Zhao, Y. Phenotype, development, and biological function of myeloid-derived suppressor cells. Oncoimmunology 5, e1004983, https://doi.org/10.1080/2162402X.2015.1004983 (2016).
Chen, J. et al. Suppression of T cells by myeloid-derived suppressor cells in cancer. Hum Immunol 78, 113–119, https://doi.org/10.1016/j.humimm.2016.12.001 (2017).
Messmann, J. J. et al. In vitro-generated MDSCs prevent murine GVHD by inducing type 2 T cells without disabling antitumor cytotoxicity. Blood 126, 1138–1148, https://doi.org/10.1182/blood-2015-01-624163 (2015).
Liao, J. et al. Dexamethasone potentiates myeloid-derived suppressor cell function in prolonging allograft survival through nitric oxide. J Leukoc Biol 96, 675–684, https://doi.org/10.1189/jlb.2HI1113-611RR (2014).
Jeisy-Scott, V. et al. Increased MDSC accumulation and Th2 biased response to influenza A virus infection in the absence of TLR7 in mice. PLoS One 6, e25242, https://doi.org/10.1371/journal.pone.0025242 (2011).
Kostlin, N. et al. Granulocytic Myeloid-Derived Suppressor Cells Accumulate in Human Placenta and Polarize toward a Th2 Phenotype. J Immunol 196, 1132–1145, https://doi.org/10.4049/jimmunol.1500340 (2016).
Gabitass, R. F., Annels, N. E., Stocken, D. D., Pandha, H. A. & Middleton, G. W. Elevated myeloid-derived suppressor cells in pancreatic, esophageal and gastric cancer are an independent prognostic factor and are associated with significant elevation of the Th2 cytokine interleukin-13. Cancer Immunol Immunother 60, 1419–1430, https://doi.org/10.1007/s00262-011-1028-0 (2011).
Gonda, K. et al. Myeloid-derived suppressor cells are increased and correlated with type 2 immune responses, malnutrition, inflammation, and poor prognosis in patients with breast cancer. Oncol Lett 14, 1766–1774, https://doi.org/10.3892/ol.2017.6305 (2017).
Arora, M. et al. TLR4/MyD88-induced CD11b+Gr-1 int F4/80+ non-migratory myeloid cells suppress Th2 effector function in the lung. Mucosal Immunol 3, 578–593, https://doi.org/10.1038/mi.2010.41 (2010).
Chesney, J. A., Mitchell, R. A. & Yaddanapudi, K. Myeloid-derived suppressor cells-a new therapeutic target to overcome resistance to cancer immunotherapy. J Leukoc Biol 102, 727–740, https://doi.org/10.1189/jlb.5VMR1116-458RRR (2017).
Ostrand-Rosenberg, S. & Fenselau, C. Myeloid-Derived Suppressor Cells: Immune-Suppressive Cells That Impair Antitumor Immunity and Are Sculpted by Their Environment. J Immunol 200, 422–431, https://doi.org/10.4049/jimmunol.1701019 (2018).
Huber-Lang, M., Lambris, J. D. & Ward, P. A. Innate immune responses to trauma. Nat Immunol 19, 327–341, https://doi.org/10.1038/s41590-018-0064-8 (2018).
Islam, M. N., Bradley, B. A. & Ceredig, R. Sterile post-traumatic immunosuppression. Clin Transl. Immunology 5, e77, https://doi.org/10.1038/cti.2016.13 (2016).
Wang, L., Yu, W. B., Tao, L. Y. & Xu, Q. Myeloid-derived suppressor cells mediate immune suppression in spinal cord injury. J Neuroimmunol 290, 96–102, https://doi.org/10.1016/j.jneuroim.2015.11.023 (2016).
Saiwai, H. et al. Ly6C+Ly6G- Myeloid-derived suppressor cells play a critical role in the resolution of acute inflammation and the subsequent tissue repair process after spinal cord injury. J Neurochem 125, 74–88, https://doi.org/10.1111/jnc.12135 (2013).
Ruan, X. et al. Anti-HMGB1 monoclonal antibody ameliorates immunosuppression after peripheral tissue trauma: attenuated T-lymphocyte response and increased splenic CD11b (+) Gr-1 (+) myeloid-derived suppressor cells require HMGB1. Mediators Inflamm 2015, 458626, https://doi.org/10.1155/2015/458626 (2015).
Husecken, Y. et al. MDSCs are induced after experimental blunt chest trauma and subsequently alter antigen-specific T cell responses. Sci Rep 7, 12808, https://doi.org/10.1038/s41598-017-13019-6 (2017).
Delano, M. J. et al. MyD88-dependent expansion of an immature GR-1(+)CD11b(+) population induces T cell suppression and Th2 polarization in sepsis. J Exp Med 204, 1463–1474, https://doi.org/10.1084/jem.20062602 (2007).
Makarenkova, V. P., Bansal, V., Matta, B. M., Perez, L. A. & Ochoa, J. B. CD11b+/Gr-1+ myeloid suppressor cells cause T cell dysfunction after traumatic stress. J Immunol 176, 2085–2094 (2006).
Rossner, S. et al. Myeloid dendritic cell precursors generated from bone marrow suppress T cell responses via cell contact and nitric oxide production in vitro. Eur J Immunol 35, 3533–3544, https://doi.org/10.1002/eji.200526172 (2005).
Ribechini, E., Leenen, P. J. & Lutz, M. B. Gr-1 antibody induces STAT signaling, macrophage marker expression and abrogation of myeloid-derived suppressor cell activity in BM cells. Eur J Immunol 39, 3538–3551, https://doi.org/10.1002/eji.200939530 (2009).
Ma, C. et al. Anti-Gr-1 antibody depletion fails to eliminate hepatic myeloid-derived suppressor cells in tumor-bearing mice. J Leukoc Biol 92, 1199–1206, https://doi.org/10.1189/jlb.0212059 (2012).
Marigo, I. et al. Tumor-induced tolerance and immune suppression depend on the C/EBPbeta transcription factor. Immunity 32, 790–802, https://doi.org/10.1016/j.immuni.2010.05.010 (2010).
Yang, F. et al. TNFalpha-induced M-MDSCs promote transplant immune tolerance via nitric oxide. J Mol Med (Berl) 94, 911–920, https://doi.org/10.1007/s00109-016-1398-z (2016).
Zhao, Y. et al. Dexamethasone-Induced Myeloid-Derived Suppressor Cells Prolong Allo Cardiac Graft Survival through iNOS- and Glucocorticoid Receptor-Dependent Mechanism. Front Immunol 9, 282, https://doi.org/10.3389/fimmu.2018.00282 (2018).
Newell, K. A., Ellenhorn, J. D., Bruce, D. S. & Bluestone, J. A. In vivo T-cell activation by staphylococcal enterotoxin B prevents outgrowth of a malignant tumor. Proc Natl Acad Sci USA 88, 1074–1078 (1991).
O’Sullivan, S. T. et al. Major injury leads to predominance of the T helper-2 lymphocyte phenotype and diminished interleukin-12 production associated with decreased resistance to infection. Ann Surg 222, 482–490; discussion 490–482 (1995).
Miller, A. C., Rashid, R. M. & Elamin, E. M. The “T” in trauma: the helper T-cell response and the role of immunomodulation in trauma and burn patients. J Trauma 63, 1407–1417, https://doi.org/10.1097/TA.0b013e31815b839e (2007).
Zhou, L. et al. Cardioprotective Role of Myeloid-Derived Suppressor Cells in Heart Failure. Circulation 138, 181–197, https://doi.org/10.1161/CIRCULATIONAHA.117.030811 (2018).
Levy, S. et al. Immature myeloid cells are critical for enhancing bone fracture healing through angiogenic cascade. Bone 93, 113–124, https://doi.org/10.1016/j.bone.2016.09.018 (2016).
Park, M. J. et al. Myeloid-Derived Suppressor Cells Induce the Expansion of Regulatory B Cells and Ameliorate Autoimmunity in the Sanroque Mouse Model of Systemic Lupus Erythematosus. Arthritis Rheumatol 68, 2717–2727, https://doi.org/10.1002/art.39767 (2016).
Casacuberta-Serra, S. et al. Myeloid-derived suppressor cells expressing a self-antigen ameliorate experimental autoimmune encephalomyelitis. Exp Neurol 286, 50–60, https://doi.org/10.1016/j.expneurol.2016.09.012 (2016).
Fujii, W. et al. Myeloid-derived suppressor cells play crucial roles in the regulation of mouse collagen-induced arthritis. J Immunol 191, 1073–1081, https://doi.org/10.4049/jimmunol.1203535 (2013).
Ji, J. et al. Myeloid-derived suppressor cells contribute to systemic lupus erythaematosus by regulating differentiation of Th17 cells and Tregs. Clin Sci (Lond) 130, 1453–1467, https://doi.org/10.1042/CS20160311 (2016).
Fan, H. Z. et al. Passive transfer of lipopolysaccharide-derived myeloid-derived suppressor cells inhibits asthma-related airway inflammation. Eur Rev Med Pharmacol Sci 19, 4171–4181 (2015).
Su, N., Yue, Y. & Xiong, S. Monocytic myeloid-derived suppressor cells from females, but not males, alleviate CVB3-induced myocarditis by increasing regulatory and CD4(+)IL-10(+) T cells. Sci Rep 6, 22658, https://doi.org/10.1038/srep22658 (2016).
Kontaki, E. et al. Aberrant function of myeloid-derived suppressor cells (MDSCs) in experimental colitis and in inflammatory bowel disease (IBD) immune responses. Autoimmunity 50, 170–181, https://doi.org/10.1080/08916934.2017.1283405 (2017).
Drujont, L. et al. Evaluation of the therapeutic potential of bone marrow-derived myeloid suppressor cell (MDSC) adoptive transfer in mouse models of autoimmunity and allograft rejection. PLoS One 9, e100013, https://doi.org/10.1371/journal.pone.0100013 (2014).
Tomihara, K. et al. Antigen-specific immunity and cross-priming by epithelial ovarian carcinoma-induced CD11b(+)Gr-1(+) cells. J Immunol 184, 6151–6160, https://doi.org/10.4049/jimmunol.0903519 (2010).
Raber, P. L. et al. T cells conditioned with MDSC show an increased anti-tumor activity after adoptive T cell based immunotherapy. Oncotarget 7, 17565–17578, https://doi.org/10.18632/oncotarget.8197 (2016).
Held, K. S., Steward, O., Blanc, C. & Lane, T. E. Impaired immune responses following spinal cord injury lead to reduced ability to control viral infection. Exp Neurol 226, 242–253, https://doi.org/10.1016/j.expneurol.2010.08.036 (2010).
Gupta, D. L., Bhoi, S., Mohan, T., Galwnkar, S. & Rao, D. N. Coexistence of Th1/Th2 and Th17/Treg imbalances in patients with post traumatic sepsis. Cytokine 88, 214–221, https://doi.org/10.1016/j.cyto.2016.09.010 (2016).
Schmidt, K. et al. Differently immunogenic cancers in mice induce immature myeloid cells that suppress CTL in vitro but not in vivo following transfer. Blood 121, 1740–1748, https://doi.org/10.1182/blood-2012-06-436568 (2013).
Brudecki, L., Ferguson, D. A., McCall, C. E. & El Gazzar, M. Myeloid-derived suppressor cells evolve during sepsis and can enhance or attenuate the systemic inflammatory response. Infect Immun 80, 2026–2034, https://doi.org/10.1128/IAI.00239-12 (2012).
Song, C. et al. Passive transfer of tumour-derived MDSCs inhibits asthma-related airway inflammation. Scand J Immunol 79, 98–104, https://doi.org/10.1111/sji.12140 (2014).
Sinha, P., Clements, V. K., Bunt, S. K., Albelda, S. M. & Ostrand-Rosenberg, S. Cross-talk between myeloid-derived suppressor cells and macrophages subverts tumor immunity toward a type 2 response. J Immunol 179, 977–983 (2007).
van der Poll, T., van de Veerdonk, F. L., Scicluna, B. P. & Netea, M. G. The immunopathology of sepsis and potential therapeutic targets. Nat Rev Immunol 17, 407–420, https://doi.org/10.1038/nri.2017.36 (2017).
Acknowledgements
The authors thank Sylvia Muche and Ingrid Knape (both from the Department of Pediatrics and Adolescent Medicine) and Annette Palmer and Sonja Braumüller (both from the Institute of Clinical and Experimental Trauma-Immunology) for excellent technical assistance. This study was supported by the grant CRC1149 “Danger Response, Disturbance Factors and Regenerative Potential after Acute Trauma” (Project A07) from the Deutsche Forschungsgemeinschaft (DFG) and TPI2 from Boehringer Ingelheim Ulm University BioCenter (BIU). M. Klingspor was supported by the “Promotionsprogramm Experimentelle Medizin” University Medical Center Ulm.
Author information
Authors and Affiliations
Contributions
M. Kustermann performed experiments, analyzed and interpreted the data, created the figures and reviewed the manuscript. M. Klingspor performed experiments, analyzed and interpreted the data and reviewed the manuscript. MHL helped in establishing the TxT model and critically reviewed the manuscript. KMD performed data interpretation and critically reviewed the manuscript. GS created the study concept and design, performed analysis and interpretation of the data and wrote the manuscript. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kustermann, M., Klingspor, M., Huber-Lang, M. et al. Immunostimulatory functions of adoptively transferred MDSCs in experimental blunt chest trauma. Sci Rep 9, 7992 (2019). https://doi.org/10.1038/s41598-019-44419-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-44419-5
This article is cited by
-
Reprogramming hematopoietic stem cell metabolism in lung cancer: glycolysis, oxidative phosphorylation, and the role of 2-DG
Biology Direct (2024)
-
Exportin 1 governs the immunosuppressive functions of myeloid-derived suppressor cells in tumors through ERK1/2 nuclear export
Cellular & Molecular Immunology (2024)
-
RETRACTED ARTICLE: Expansion of monocytic myeloid-derived suppressor cells ameliorated intestinal inflammatory response by radiation through SOCS3 expression
Cell Death & Disease (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
