
Abstract
Ticks are notorious vectors for various pathogens that cause infections in animals and humans worldwide. Rickettsia spp., a zoonotic tick-borne pathogen that could be used as a weapon agent, is widely spread in China. In the present study, ticks were collected for species identification and Rickettsia screening. PCR amplification targeting the tick 18s rRNA gene was first conducted for species validation, and then, amplification was conducted for the Rickettsia housekeeping gene for the infection rate and phylogenetic analysis. The collected ticks were identified as Haemaphysalis longicornis, 7.36% of which were Rickettsia-positive. The phylogenetic analysis showed that the Rickettsia in the parasitic ticks belonged to a novel genotype, whose closest genetic relationship was with Rickettsia heilongjiangenesis. The samples were collected in Dandong, a city on the border between China and North Korea. Considering the geographical and biological situations of the sampling sites, more extensive surveillance and risk evaluation of the tick species and tick-borne diseases are required.
Similar content being viewed by others


Introduction
As hematophagous arthropods, ticks are notorious vectors for various pathogens. Approximately 900 species of ticks have been detected around the world, 36 of which were found in China1,2,3. Ticks can act as vectors, reservoirs, or amplifiers for various pathogens, including viruses, bacteria, Rickettsia species, spirochetes, protozoa, mycoplasma, chlamydia, Bartonella, and nematodes, to cause infections in animals and humans worldwide4. Among these, tick-borne rickettsia, the Xinjiang haemorrhagic fever, Lyme disease caused by spirochetes, and Q fever have been detected in China5. Rickettsia spp., a pathogen of the spotted fever group, represents an important biological weapon agent, which could cause a great public health crisis4. The life cycles of most tick-borne rickettsia are largely unknown. In natural vertebrate hosts, which are mammals, but not normally humans, infections may result in rickettsemia, which allows non-infected ticks to become infected and permits the perpetuation of the natural cycle6,7,8.
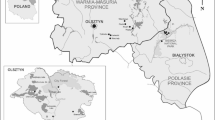
Northeast China possesses mountainous terrain and contains abundant biological resources, providing an ecological and biological basis for ticks to survive and reproduce. However, the distribution and infection incidence of these tick species and tick-borne diseases in this area remain unclear. Kuandian Manchu Autonomous County, a large county that is affiliated with Dandong City, southeast of the Liaoning Province, is close to the Democratic People’s Republic of Korea, where the Yalu River acts as the border line. In the present study, ticks were collected from Kuandian County to examine the species and Rickettsia harboured.
Results
Morphological characteristics and molecular identification of parasitic ticks
Ticks were collected in the Kuandian Manchu Autonomous County, which is located in northeast China and is close to North Korea, along the Yalu River (Fig. 1). About 6000 ticks were collected in 2017 from June 5th to 12th. All of these ticks were morphologically identified as Haemaphysalis longicornis (Fig. 2), the representative, marked features of which were as follows. (1) The body was broadly oval or subcircular and cinnamomeous. (2) The capitulum was short and wedge-shaped. The basis capituli was rectangular, with a strong posterior and triangular cornu. The palpal exterior margin was slightly convex, with a coniform present. 5/5 dentition of hypostome. (3) The scutum was subcircular and covered the body anteroposteriorly, with clear and slender festoons. Medium sized punctuations were uniformly and densely distributed. The cervical grooves were short and curved, while the lateral grooves were clear and narrow. (4) On the venter, the anal opening was medioventral with the anal groove. The genital opening was level with the coxa II. The anterior part between the groove was a depressed lunula, while the posterior part was a convex lunula. The spiracular plate was broadly oval or subcircular. (5) The legs were of moderate length with numerous setae. The coxa I was located posteriorly with a strong cone-shaped spur, and the coxa II had a short but clear ventral spur.

Tick sampling sites in Kuandian County, China-North Korea. The red region represents Kuandian County. The GPS locations of the sampling sites are labelled with their coordinates.

Ticks were observed with a stereomicroscope, and the morphological features of the scutum, venter, and capitulum were recorded. (a) The scutum was subcircular, covering the body anteroposteriorly, with clear and slender festoons. Medium-sized punctuations were uniformly and densely distributed. Cervical grooves were short and curved, while the lateral grooves were clear and narrow. The legs were of moderate length with numerous setae. The coxa II had a short but clear ventral spur. (b) On the venter, the anal opening was about medioventral with the anal groove. The genital opening was level with the coxa II. The anterior part was a depressed lunula. The spiracular plate was broadly oval and subcircular. On the legs, the coxa I was posterior with a strong cone-shaped spur. (c,d) The capitulum was short and wedge-shaped. The basis capituli was rectangular was a strong posterior and a triangular cornu. The palpal exterior margin was slightly convex, with corniform present. Five out of five met the definition of hypostome.
The tick DNA was extracted and subjected to PCR amplification. Forty-two of the 489 positive samples (8.5%, selected according to the gender, host, and stage of the tick life cycle) were sent for sequencing of the rrs gene, and 40 of which were successfully sequenced. The sequences from all of the samples were completely identical. The phylogenetic tree for the rrs gene showed that these ticks were closest to Haemaphysalis longicornis (Fig. 3). Combined with the morphological characteristics and molecular evidence, these ticks were classified as H. longicornis.

Phylogenetic tree for the representative tick 16 s rDNA gene. The tree was constructed using the maximum likelihood algorithm implemented in the Molecular Evolutionary Genetics Analysis (MEGA) 7 software. The black dot represents the sequence acquired from this study.
Molecular identification and genotypes of Rickettsia
Genomic DNA was extracted from representative tick samples and subjected to PCR amplification of the gltA gene. Among the 489 tested samples, 36 were gltA positive, generating an average positive rate of 7.36%. The Rickettsia-positive rate for each sampling site was calculated. The positive rates for these sites ranged from 4.10% to 9.22%, with the village of Erdaogou (9.22%) having the highest, followed by Kuandian (7.92%) and Shandian (7.79%); whereas, the positive rates for Lazigou (6.19%) and Penghai (4.10%) were relatively low for all of the collection sites (Table 1). The 36 Rickettsia-positive DNA samples were then amplified and sequenced for both ompA and ompB, which are two commonly used genotyping genes. The PCR amplification results showed that 27 of the 36 samples were ompA positive, and 32 were ompB positive; 24 (66.7%) were positive for both genes (Fig. 4). All of the PCR products from the positive samples (35 in total) were sent for sequencing. The sequences for each gene from all of the samples were completely identical. The phylogenetic tree showed that the Rickettsia detected herein belongs to a novel genotype (Fig. 5). According to the criteria for novel Rickettsia determination based on gene sequences, an isolate can be classified as a new Rickettsia genotype when it exhibits no more than one of the following degrees of nucleotide similarity with the most homologous validated species: ≥99.8% and ≥99.9% for the rrs and gltA genes, respectively, and, when amplifiable, ≥98.8%, ≥99.2%, and ≥99.3% for ompA, ompB, and gene D, respectively9. Thus, we made a sequence alignment based on our sequenced genes, gltA, ompA and ompB, between the Rickettsia detected in this study and R. heilongjiangensis or R. japonica. The results of the alignment with R. heilongjiangensis, the most homologous validated species, are shown in Fig. 6, for which the percentages for the nucleotide identities were 99.6% for the gltA gene (Fig. 6a), 94.66% for the ompA gene (Fig. 6b), and 98.63% for the ompB gene (Fig. 6c). The sequence alignment with R. japonica was showed in Figure S1. Even though the sequence alignment results were promising, the evidence to prove that the newly detected Rickettsia is a novel Rickettsia species was insufficient. Therefore, we temporarily determined that the detected rickettsiae is a novel genotype.

PCR amplification results for the Rickettsia-positive samples. Gene amplification of ompA (a) and ompB (b). For both figures, Lanes 1 and 26: Markers; Lanes 2–14: gltA-positive samples from the village of Erdaogou; Lanes 15–22: gltA-positive samples from the village of Kuandian; Lanes 23–25 and 27–29: gltA-positive samples from the village of Lazigou; Lanes 30–35: gltA-positive samples from the village of Shangdian; Lanes 36–38: gltA-positive samples from the village of Penghai; Lane 39: negative control; and Lane 40: positive control. All positive samples were sent for sequencing.

Phylogenetic trees for the ompA and ompB genes. The trees were constructed using the sequences of ompA (a) and ompB (b), using the maximum likelihood (ML) algorithm implemented in the Molecular Evolutionary Genetics Analysis (MEGA) 7 software. The black dots represent the sequences acquired from this study.

Sequence alignment between the Rickettsia detected in this study and R. heilongjiangensis. The sequence alignments for gltA, ompA, and ompB were conducted using the Multiple Sequence Alignment in the DNAMAN software.
Discussion
In the present study, ticks, collected from the Liaoning province, were morphologically and phylogenetically shown to be Haemaphysalis longicornis, which has been detected in both animals and humans and as a host for various kinds of emerging pathogens. For instance, in China, during the past 15 years, H. longicornis has been found in: (1) the Heilongjiang and Jilin provinces, along with Haemaphysalis concinna, Dermacentor nuttalli, Dermacentor silvarum, and Ixodes persulcatus, conveying Rickettsia spp.10; (2) in the Hebei province, conveying the spotted fever group Rickettsia11; (3) in the Henan province, along with Rhipicephalus microplus, conveying Anaplasma spp., Rickettsia spp., Babesia spp., Theileria spp., Ehrlichia spp., and severe fever with thrombocytopenia syndrome bunyavirus (SFTSV)12,13; and (4) in the Zhejiang province, along with Amblyomma testudinarium and Ixodes sinensis, conveying Rickettsia spp.14. Other studies from South Korea, Japan, New Calenonia, and New Zealand have also illustrated the extensive distribution and high incidence of ticks and tick-borne diseases15,16,17,18,19,20,21. Close attention has been paid to the geographic distribution of ticks, as tick-borne diseases put the public health at risk. According to the review by Chen1, at least 104 species from 7 genera of ticks have been circulated in China, but the ticks collected in the present study were homogenous. Therefore, the distribution of ticks in the Liaoning province remains unclear and needs further investigation.
The Rickettsia-positive rate among all the specimens was 7.36% (36/489). The Rickettsia we detected have the closest genetic relationship with R. heilongjiangenesis, but they are located on a separate branch, implying that they might belong to a novel genotype. The qualification of new a Rickettsia genotype remains to be further defined. As a zoonotic disease, at least five species of spotted fever group Rickettsia have been detected in China, including R. heilongjiangenesis, Rickettsia sibirica, Rickettsia raoultii, Rickettsia slovaca, and Rickettsia felis4,22. In previous studies, the Rickettsia-positive rate in Xinyang in the Henan province (6.25%, 9/144) and in the Jilin and Heilongjiang provinces (6.12%) was about 6%10,12,23. Compared to these regions in China, the Rickettsia-positive rate at our sampling site, the national border line in the Liaoning province, was relatively high. This high positive rate implies a high infection risk and a potential public health problem. Rickettsia is mutually transmitted through arthropods and humans, representing an important pathogen that is circulated by ticks. With their high prevalence and species diversity, as well as the geographical spectrum, ticks in northeast China may be able to transmit numerous pathogens, causing potential severe infections in both animals and humans. In the sampling regions, livestock are mainly bred by free range. This sharply increases the possibility for human-tick-livestock contact and transmission. However, neither tick species, nor tick-borne diseases, have been thoroughly investigated in these regions.
Although our current investigation on ticks and tick-borne diseases at the China-North Korea border is not completely comprehensive, the results we obtained do have several implications. First, unlike the research in other provinces in north-eastern China (the Heilongjiang and Jilin provinces)10, all 489 ticks collected in the Liaoning province were H. longicornis, indicating that H. longicornis is the predominant tick species in the sampling region. Furthermore, the identified Rickettsia genotype is also different from those observed during previous research studies in northeast China. Second, because the sampling site was only located along the border area, the distribution of both the ticks and the Rickettsia in other regions of the Liaoning province remains unknown. Likewise, it is of great interest to determine whether H. longicornis and the novel Rickettsia genotype also circulate. Third, further investigation into the distribution of Rickettsia and other vector-borne pathogens at the border line is important for risk evaluation for emerging infectious diseases. In our ongoing work, we are trying to widen the sampling spectrum for other tick species and tick-borne pathogens and to investigate the distribution of the novel Rickettsia genotype in different arthropods in this region. These studies will greatly contribute to our understanding of ticks and tick-borne diseases and will allow us to evaluate the circulating risk.
Methods
Tick sampling
The sampling cites were located in the Kuandian Manchu Autonomous County. The altitudes and latitudes of sampling sites are shown in Table 2. Ticks were manually collected from animal skin without damage and then carefully placed into 70% ethanol. Each sample was labelled with the area name, latitude, longitude, host type, and the collection date and site. All specimens were then sent to the laboratory at the Shenyang Agricultural University and stored for further examination.
Morphological identification
The morphological features of the ticks were individually observed by a stereomicroscope. The back, abdomen, shield plate, gas door plate, false head base, lateral furrow, and genital orifice were carefully examined for species identification.
Molecular identification
The protection solution was removed, and the specimens were vortexed and rinsed with 75% ethanol for 1 h. Then, they were rinsed with distilled water three times. The DNA was extracted using a DNA extraction kit (Tiangen Biochemical Technology (Beijing) Co., Ltd.), according to the manufacturer’s protocols, and stored at −20 °C. PCR amplification was first conducted with paired universal primers that targeted the 18S rRNA gene (rrs) for tick species and then the Rickettsia citrate synthase gene (gltA) to calculate the positive rate24. The Rickettsia-positive samples were then passaged for the detection of two more genes: outer membrane protein A (ompA) and B (ompB)3,9. Double distilled water and the recombined plasmids of each gene were used as the negative and positive controls, respectively. The primer sequences are shown in Table 3 24. All PCR products were sequenced by Sanger sequencing (Sangon Biotech Co., Ltd, Shanghai, China). Related Rickettsia sequences were extracted from the Genbank database and edited along with the generated sequences. The phylogenetic tree was made based on the sequence distance method using the neighbor-joining (NJ) and maximum likelihood (ML) algorithms implemented in the Molecular Evolutionary Genetics Analysis (MEGA) 7 software25.
All animal sampling operations were performed properly according to the protocols proposed by NEON and AfiVIP: TOS Protocol and Procedure: Tick and Tick-borne Pathogen Sampling and Ticks: Tick surveillance. The morphological and molecular identification was performed in the Biosafe Laboratories of Shenyang Agricultural University using the relevant equipment with formal approval.
References
Chen, Z. et al. Ticks (acari: ixodoidea: argasidae, ixodidae) of China. Experimental & applied acarology 51, 393–404, https://doi.org/10.1007/s10493-010-9335-2 (2010).
Yu, Z. et al. Tick-borne pathogens and the vector potential of ticks in China. Parasites & vectors 8, 24, https://doi.org/10.1186/s13071-014-0628-x (2015).
Adjou Moumouni, P. F. et al. Molecular detection of spotted fever group rickettsiae in Amblyomma variegatum ticks from Benin. Ticks and tick-borne diseases 7, 828–833, https://doi.org/10.1016/j.ttbdis.2016.03.016 (2016).
Wei, Q. Q. et al. The first detection of Rickettsia aeschlimannii and Rickettsia massiliae in Rhipicephalus turanicus ticks, in northwest China. Parasites & vectors 8, 631, https://doi.org/10.1186/s13071-015-1242-2 (2015).
Chinikar, S. et al. Surveillance and laboratory detection system of Crimean-Congo haemorrhagic fever in Iran. Transboundary and emerging diseases 55, 200–204, https://doi.org/10.1111/j.1865-1682.2008.01028.x (2008).
Parola, P., Paddock, C. D. & Raoult, D. Tick-borne rickettsioses around the world: emerging diseases challenging old concepts. Clinical microbiology reviews 18, 719–756, https://doi.org/10.1128/CMR.18.4.719-756.2005 (2005).
Burgdorfer, W., Friedhoff, K. T. & Lancaster, J. L. Jr. Natural history of tick-borne spotted fever in the USA Susceptibility of small mammals to virulent Rickettsia rickettsii. Bulletin of the World Health Organization 35, 149–153 (1966).
Norment, B. R. & Burgdorfer, W. Susceptibility and reservoir potential of the dog to spotted fever-group rickettsiae. American journal of veterinary research 45, 1706–1710 (1984).
Zhang, J. Z. et al. Genetic classification of “Rickettsia heilongjiangii” and “Rickettsia hulinii”, two Chinese spotted fever group rickettsiae. Journal of clinical microbiology 38, 3498–3501 (2000).
Liu, H. et al. Characterization of rickettsiae in ticks in northeastern China. Parasites & vectors 9, 498, https://doi.org/10.1186/s13071-016-1764-2 (2016).
Zou, Y. et al. Detection of spotted fever group Rickettsia in Haemaphysalis longicornis from Hebei Province, China. The Journal of parasitology 97, 960–962, https://doi.org/10.1645/GE-2751.1 (2011).
Zhuang, L. et al. Identification of tick-borne pathogen diversity by metagenomic analysis in Haemaphysalis longicornis from Xinyang, China. Infectious diseases of poverty 7, 45, https://doi.org/10.1186/s40249-018-0417-4 (2018).
Chen, Z. et al. Tick-borne pathogens and associated co-infections in ticks collected from domestic animals in central China. Parasites & vectors 7, 237, https://doi.org/10.1186/1756-3305-7-237 (2014).
Sun, J. et al. Detection of spotted fever group Rickettsiae in ticks from Zhejiang Province, China. Experimental & applied acarology 65, 403–411, https://doi.org/10.1007/s10493-015-9880-9 (2015).
Heath, A. Biology, ecology and distribution of the tick, Haemaphysalis longicornis Neumann (Acari: Ixodidae) in New Zealand. New Zealand veterinary journal 64, 10–20, https://doi.org/10.1080/00480169.2015.1035769 (2016).
Park, S. W. et al. Prevalence of severe fever with thrombocytopenia syndrome virus in Haemaphysalis longicornis ticks in South Korea. Ticks and tick-borne diseases 5, 975–977, https://doi.org/10.1016/j.ttbdis.2014.07.020 (2014).
Kim, C. M. et al. Tick-borne rickettsial pathogens in ticks and small mammals in Korea. Applied and environmental microbiology 72, 5766–5776, https://doi.org/10.1128/AEM.00431-06 (2006).
Noh, Y. et al. Molecular detection of Rickettsia species in ticks collected from the southwestern provinces of the Republic of Korea. Parasites & vectors 10, 20, https://doi.org/10.1186/s13071-016-1955-x (2017).
Lee, J. H. et al. Identification of the spotted fever group rickettsiae detected from Haemaphysalis longicornis in Korea. Microbiology and immunology 47, 301–304 (2003).
Tateno, M. et al. Molecular survey of arthropod-borne pathogens in ticks obtained from Japanese wildcats. Ticks and tick-borne diseases 6, 281–289, https://doi.org/10.1016/j.ttbdis.2015.01.009 (2015).
Mediannikov, O., Davoust, B., Cabre, O., Rolain, J. M. & Raoult, D. Bartonellae in animals and vectors in New Caledonia. Comparative immunology, microbiology and infectious diseases 34, 497–501, https://doi.org/10.1016/j.cimid.2011.09.002 (2011).
Anderson, B. E., McDonald, G. A., Jones, D. C. & Regnery, R. L. A protective protein antigen of Rickettsia rickettsii has tandemly repeated, near-identical sequences. Infection and immunity 58, 2760–2769 (1990).
Guo, L. The distribution and molecular evidence of ticks and tick-borne diseases in the boundry area of Xinjiang province Master thesis, Shihezi University (2014).
Labruna, M. B. et al. Molecular evidence for a spotted fever group Rickettsia species in the tick Amblyomma longirostre in Brazil. Journal of medical entomology 41, 533–537 (2004).
Kumar, S., Stecher, G. & Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Molecular biology and evolution 33, 1870–1874, https://doi.org/10.1093/molbev/msw054 (2016).
Acknowledgements
This work was supported by National Special Project on Research and Development of Key Biosafety Technology (2016YFC1200100, 2017YFD0500305, 2017YFD0500901), Shenyang Key Research and Development Plan (17-161-3-00) and construction project of Liaoning Engineering Laboratory of zoonosis (01072917001).
Author information
Authors and Affiliations
Contributions
Z. Chen and X. Han conceived and designed the study, H. Guan, Y. Zhong, Q. Zhang and X. Han performed the experiments and analyzed the data, F. Jiang participate in sample collection, Qi Zhang, H. Xu and Z. Chen draft and revised the manuscript.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Xu, H., Zhang, Q., Guan, H. et al. High Incidence of a Novel Rickettsia Genotype in Parasitic Haemaphysalis longicornis from China-North Korea Border. Sci Rep 9, 5373 (2019). https://doi.org/10.1038/s41598-019-41879-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-41879-7
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
