
Abstract
Identification of adipose-specific genes has contributed to an understanding of mechanisms underlying adipocyte development and obesity. Herein, our analyses of the recent Genotype-Tissue Expression (GTEx) database revealed 38 adipose-specific/enhanced protein coding genes, among which 3 genes were novel adipose-specific, and 414 highly differentially expressed genes (DEGs) between subcutaneous and omental adipose depots. By integrative analyses of genome-wide association studies (GWASs), 14 adipose-specific/enhanced genes and 60 DEGs were found to be associated with obesity-related traits and diseases, consolidating evidence for contribution of these genes to the regional fat distribution and obesity phenotypes. In addition, expression of HOXC cluster was up-regulated in subcutaneous adipose tissue, and the majority of the HOXB cluster was expressed highly in omental adipose tissue, indicating differential expression patterns of HOX clusters in adipose depots. Our findings on the distinct gene expression profiles in adipose tissue and their relation to obesity provide an important foundation for future functional biological studies and therapeutic targets in obesity and associated diseases.
Similar content being viewed by others


Introduction
Obesity is one of the major global health issues because of its relation to various metabolic complications including type 2 diabetes, coronary heart disease, hypertension, dyslipidemia and a number of cancers1,2,3,4. Previous studies regarding obesity identified a significant contribution of genetic factors to obesity traits5,6. Among genetic factors, genes prominently expressed in adipose tissue are involved in various metabolic and endocrine functions of adipose tissue such as adipocyte development, lipid metabolism, glucose homeostasis and immune/inflammatory responses7,8,9. Altered expression of these adipose-specific genes leads to an increased release of fatty acids, hormones, and pro-inflammatory cytokines that contribute to obesity-related metabolic diseases10.
Functional studies about adipose-specific genes have increased our understanding of adipocyte biology and their etiological significance for the obesity and related diseases. These adipose-specific genes include genes encoding LEP/leptin (adipokine)11,12, ADIPOQ/adiponectin (adipokine)13,14, peroxisome proliferator-activated receptor gamma (PPARγ; adipose-specific transcription factor)15,16,17, and fatty acid binding protein 4 (FABP4; adipocyte fatty acid binding protein)18,19. In the early 2000s, high throughput screening methods including gene filter and gene chip arrays became available. Several groups, including ours, identified adipose-specific genes including ADSF/resistin20 and ATGL/desnutrin21. Our group has also identified several adipose-specific genes (e.g., ISG12b1 and ACVR1C) based on the relative level of gene expression through mining bioinformatics databases9,22. To rigorously screen additional novel adipose-specific genes in humans, we have used the most recent Genotype-Tissue Expression (GTEx) data that provide up-to-date RNA-Seq transcriptomic profiling for various human tissues from hundreds of postmortem donors which is a suitable resource for identifying tissue-specific genes across multiple tissues23,24.
A large number of candidate genes for obesity have been documented by genome-wide association studies (GWAS) to determine genetic factors associated with obesity25,26,27,28,29. Despite findings from these studies, evidence linking adipose-specific genes and obesity in humans is still unclear. The primary objectives of this study were to identify novel adipose-specific genes and consolidate candidate genes for obesity-susceptibility by integrating GWAS data. In addition, depot-related expression of HOX genes in subcutaneous and visceral (omental) adipose tissues was examined to comprehensively evaluate developmental gene expression patterns for regional fat distribution.
Herein, 3 novel common adipose-specific genes and 414 differentially expressed genes (DEGs) between subcutaneous and omental adipose depots were identified. By integrating data of GWAS, evidence of interrelationships between those genes and major obesity-related traits or diseases including adiposity, type 2 diabetes, blood lipids, inflammation, and waist-to-hip ratio, were solidified. Furthermore, differential expression patterns of HOX genes in different adipose tissue depots were identified. Overall, our analysis of diverse databases have identified novel adipose-specific genes and consolidated evidence for their genetic relationship with obesity, providing a basis for further elucidation of therapeutic targets for obesity and related diseases.
Results
Identification of adipose-specific genes
Prior to initiating our workflow (Fig. 1), the GTEx dataset was downloaded from the GTEx portal (www.gtexportal.org), and then adipose-specific genes under the category of adipose-enhanced genes were explored. Distribution of medians in the GTEx dataset was first examined by plotting the number of genes against their relative median values, defined as a median expression value of subcutaneous or omental adipose tissue divided by an average of other medians (Fig. 2a). Most of the data were centered around the value 1 (indicating no difference), and fewer values on the right side of the value 1 represent adipose-enhanced expression (for example, expression of 64 subcutaneous adipose genes and 85 omental adipose genes were more than 10-fold). After the above initial evaluation of the dataset, adipose-specific genes were investigated under rigorous criteria of more than median-5-fold in all pairwise comparisons and an FDR-corrected P value < 0.01. As a result, 14 subcutaneous adipose- and 11 omental adipose-specific protein coding genes were identified (Fig. 2b; Supplementary Table 1). There were 9 genes that were overlapped between subcutaneous and omental tissues, and SLC19A3 has not been reported in terms of its function in adipose tissue (Fig. 2c; Supplementary Table 1). Regarding subcutaneous exclusive expression, the functions of the CSN1S1 gene and LVRN (also named as AQPEP) gene have not been reported, respectively. Heat maps and boxplots display expression levels of the adipose-specific genes in various tissues with extreme specificity to adipose tissue (Fig. 3a; Supplementary Figs 1–6; Supplementary Table 1). Semi-quantitative RT-PCR and/or Western blot analysis confirmed adipose-specific expression of CSN1S1, SLC19A3, and LVRN (AQPEP) (Fig. 3b; Supplementary Fig. 7). Adipose-enhanced genes with at least median-5-folds, with the exception of one or two pairwise comparisons, are also found and listed (Figs 2b and 3a; Supplementary Table 1).

Work flow to identify adipose-related genes in humans, which were used to consolidate obesity-associated genes. Gene TPMs across 46 human tissues were collected from the GTEx v7 study, followed by data processing in three ways: (1) newly identified adipose-specific genes were investigated using GEO database and literature; (2) adipose depot-related DEGs of developmental genes were analyzed to construct a depot-based HOX expression map; and, (3) both adipose-enhanced genes and depot DEGs were located to mapped obesity-related loci published in GWAS.

Classification of genes highly expressed in in human adipose tissues. (a) Distribution of adipose-enhanced genes, in which a median expression value in adipose tissue is more than 10-fold higher compared to an average of median expression values from all other tissues. (b) Adipose-specific genes whose median expression is more than 5-fold higher in adipose tissue compared to all other tissues (All 5 folds), and other adipose-enhanced genes whose median expression is more than 5-fold higher in adipose tissue compared to all other tissues, except one tissue (5 folds, except 1) and two tissues (5 folds, except 2). The significance of differences was set at a threshold of FDR-adjusted P value < 0.01. (c) Venn diagram of subcutaneous- and omental-specific genes. Genes whose expression is specific to both subcutaneous and omental adipose tissues are indicated in the intersection (n = 9), among which one gene, SLC19A3, is functionally unreported in adipose tissue.

Expression of adipose-specific genes and adipose-enhanced genes. (a) Heat map of the expression of adipose-specific genes (all 5 folds) and adipose-enhanced genes (5 folds, except 1 and 5 folds, except 2) in various tissues [A: subcutaneous adipose tissue, A2: omental adipose tissue, and B through AS: 44 non-adipose tissues (B: Adrenal Gland, C: Artery - Aorta, D: Artery - Coronary, E: Artery - Tibial, F: Brain - Amygdala, G: Brain - Anterior cingulate cortex (BA24), H: Brain - Caudate (basal ganglia), I: Brain - Cerebellar Hemisphere, J: Brain - Cerebellum, K: Brain - Cortex, L: Brain - Frontal Cortex (BA9), M: Brain - Hippocampus, N: Brain - Hypothalamus, O: Brain - Nucleus accumbens (basal ganglia), P: Brain - Putamen (basal ganglia), Q: Brain - Spinal cord (cervical c-1), R: Brain - Substantia nigra, S: Colon - Sigmoid, T: Colon - Transverse, U: Esophagus - Gastroesophageal Junction, V: Esophagus - Mucosa, W: Esophagus - Muscularis, X2: Heart - Atrial Appendage, Y: Heart - Left Ventricle, Z: Kidney - Cortex, AA: Liver, AB: Lung, AC: Minor Salivary Gland, AD: Muscle - Skeletal, AE: Nerve - Tibial, AF: Ovary, AG: Pancreas, AH: Pituitary, AI: Prostate, AJ: Skin - Not Sun Exposed (Suprapubic), AK: Skin - Sun Exposed (Lower leg), AL: Small Intestine - Terminal Ileum, AM: Spleen, AN: Stomach, AO: Testis, AP: Thyroid, AQ: Uterus, AR: Vagina, AS: Whole Blood)]. Functionally unreported adipose-specific genes are indicated with red filled circles. Tissues with median fold changes less than five compared to adipose tissue are marked with black unfilled squares. (b) Semi-quantitative RT-PCR and/or Western blot analysis (WB) of CSN1S1, SLC19A3, and LVRN (AQPEP) in selected tissues: subcutaneous adipose tissue (Ad), brain (Br), heart (He), kidney (Ki), lung (Lu), liver (Li), and skeletal muscle (Mu). Target sizes for RT-PCR: 213 bp (CSN1S1), 170 bp (SLC19A3) and 193 bp (LVRN (AQPEP)). Expected sizes for full-length proteins were 21.7 kDa and 55.7 kDa for CSN1S1 and SLC19A3, respectively. Cyclophilin (CYC) (325 bp) and GAPDH (38 kDa) were used as a reference gene and protein, respectively.
Comparative profiling of unreported adipose-specific genes
To obtain insight into roles of unreported adipose-specific genes in adipose tissue, functional annotations of the genes and information from literature describing their potential functions in other tissues and results from analysis of GSE data were summarized (Table 1). Among these adipose-specific genes, CSN1S1 is involved in immune/inflammatory responses30, SLC19A3 is responsible for thiamine transport and subsequent energy production31, and LVRN (AQPEP) is a member of the aminopeptidase family that catalyzes peptide catabolic processes32. Of particular interest, GSE data (GSE9624 and GSE12050) showed that SLC19A3 and CSN1S1 genes were expressed significantly higher in omental and subcutaneous adipose tissue, respectively, of an obese population than a lean population (Table 1). Given that those genes have more significant expression in fat cell fraction containing mature adipocytes than in stromal vascular fraction mainly containing preadipocytes and tendency of increasing expression during differentiation of human adipose-derived stem/stromal cells (Table 1), these three unreported adipose-specific genes may play important roles in adipogenensis and lipid metabolism and remain to be investigated.
GWAS associated phenotypes of adipose-specific/enhanced genes
The recent GWAS catalog of 71,423 records for 2,688 diseases or traits was used to select and classify 34 diseases or traits into four obesity-associated categories: obesity, type 2 diabetes, blood lipids, and inflammation (Supplementary Table 2). In order to identify obesity candidate genes among adipose-specific/enhanced genes, GWAS mapped and reported genes in those four categories were integrated with adipose-specific/enhanced genes. In total, 14 adipose-specific/enhanced genes were matched with those mapped and reported genes: obesity-related (4 genes), blood lipids-related (7 genes), type 2 diabetes-related (2 genes), and inflammation-related (4 genes) groups (Table 2). Among the 14 genes, 2, 5, and 7 genes were subcutaneous, omental, and common adipose-specific genes, respectively (Table 2; Supplementary Table 2). 13 genes have one-related GWAS catagories. Specifically, PPARG gene belongs to all four GWAS catagories (Table 2; Supplementary Table 2). Genes functionally non-reported in adipose tissue showed a link with a GWAS obesity trait (SLC19A3 and UPK3B genes) and a GWAS blood lipids trait (LVRN, CD300LG, and HAS1 genes) (Table 2). Linking of these adipose-specific/enhanced genes to those GWAS categories solidified evidence of their interrelationships with obesity and obesity-related traits.
DEGs between adipose depots and associated obesity-related traits
DEGs between subcutaneous and omental adipose tissues were identified and then subjected to GWAS integration and KEGG pathway analyses to understand functional characteristics of those genes. A total of 414 DEGs (log2 fold change ≥3) were found, among which 86 and 328 DEGs showed up-regulated expression in subcutaneous and omental adipose tissue, respectively, and the protein-coding DEGs were 48 and 214 in subcutaneous and omental adipose tissues, respectively (Fig. 4a; Supplementary Table 3; Supplementary Fig. 8).

Classification and whole genome visualization of differentially expressed genes between two different adipose depots. (a) Classification of DEGs between subcutaneous and omental adipose tissues. DEGs in the two adipose depots were categorized into five groups including protein-coding, lincRNA, pseudogene, antisense RNA, and others. (b) Plotting DEGs between two different adipose tissues. The graphics illustrate association between DEGs in two different adipose depots and previously reported genes from GWAS studies plotted on ideograms of all chromosomes. Lines are plotted on chromosomal regions corresponding to the matched locations between DEGs and GWAS loci and then connected to different shapes representing different tissue abundancy: diamond, highly expressed genes in subcutaneous adipose tissue; circle, highly expressed genes in omental adipose tissue. GWAS phenotypes are color-coded to represent different categories. A detailed list of phenotypes and associations is presented in Supplementary Table 4.
Using the recent GWAS catalog, 26 diseases or traits were grouped into seven relevant adipose depot-related categories (Supplementary Table 4). In order to find the evidence of a link between the adipose depot-related DEGs and GWAS locus, mapped and reported genes in those seven categories were integrated with DEGs listed in Supplementary Table 4. Among DEGs, a total of 60 genes were matched with GWAS mapped and reported genes in those seven categories with some overlaps: waist circumferences-related (3 genes), hip circumferences-related (3 genes), waist-to-hip ratio-related (5 genes), adiposity-related (25 genes), blood lipids-related (7 genes), inflammation-related (27 genes) and visceral fat-related (5 genes) (Fig. 4b; Supplementary Table 4). Among the 60 genes, 10 and 50 genes were up-regulated in subcutaneous and omental adipose tissue, respectively (Fig. 4b; Supplementary Table 4). Most inflammation-related GWAS genes (26 out of 27) were up-regulated in omental adipose tissue compared with subcutaneous adipose tissue, indicating omental adipose tissue plays a more important role in inflammation than subcutaneous adipose tissue. 48 and 8 DEGs have one and two related GWAS categories, respectively; and TBX15 and CNTNAP2 genes belong to three GWAS categories; and FAM101A gene is a candidate gene for five GWAS categories (Fig. 4b; Supplementary Table 4).
KEGG pathways analysis showed that up-regulated DEGs in subcutaneous adipose tissue were enriched in nicotine addiction, GABAergic synapse, morphine addiction, tissue remodeling, relaxin signaling pathway, retrograde endocannabinoid signaling, and neuroactive ligand-receptor interaction (Fig. 5: category 1 through 7; Supplementary Table 5). Further KEGG pathway analysis showed that significantly up-regulated DEGs in omental adipose tissue were highly related to categories including JAK-STAT signaling pathway, IL-17 signaling pathway, TNF signaling pathway, salmonella infection, amoebiasis, inflammatory bowel disease (IBD) and cytokine-cytokine receptor interaction, indicating those DEGs in omental adipose tissue might be involved in immune/inflammatory responses, pathological infection, and cell signaling. (Fig. 5: category 8 through 19; Supplementary Table 5).

KEGG pathway analysis of up-regulated DEGs in subcutaneous and omental adipose tissue. Chords represent a detailed relationship between the expression levels of up-regulated DEGs (left semicircle perimeter) and their enriched KEGG pathways (right semicircle perimeter). For each gene, the expression value (TPM) of up-regulated DEGs in omental adipose tissue is shown by colored rectangles. Details in Supplementary Tables 5 and 6.
Overall, many DEGs which showed distinct expression patterns between subcutaneous and omental adipose tissues were associated with adipose depot-related parameters such as waist-to-hip ratio and different enriched pathways, strengthening the evidence that those DEGs may be potential indicators of regional adipose tissue distribution.
Dissecting expression patterns of HOX clusters in adipose tissues
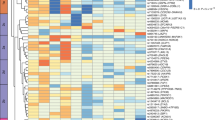
Considering differential expression of some development-related genes between adipose depots, expression pattern of homeotic genes in subcutaneous and omental adipose tissues were investigated. Interestingly, many homeobox (HOX) genes, a subfamily of homeotic genes, showed a distinct expression pattern in subcutaneous and omental adipose tissues (Fig. 6, Supplementary Table 6). First, the HOXA cluster displayed an anterior-to-posterior expression pattern. Specifically, expression levels of 3′ HOXA4 gene controlling anterior regions and a central group gene, HOXA5, had a significantly higher expression in omental adipose tissue compared to subcutaneous adipose tissue; whereas, 5′ HOXA genes (HOXA9, HOXA10, HOXA11, and HOXA13) controlling posterior regions had a significantly higher expression in leg subcutaneous compared to omental adipose tissue. Second, HOXB and HOXC clusters showed an almost opposite expression pattern regardless of the group. The expression level of the HOXB cluster was significantly higher in omental adipose tissue than subcutaneous adipose tissue except for HOXB13; whereas, the HOXC cluster had a significantly greater expression in subcutaneous adipose tissue compared to omental adipose tissue. Last, the HOXD cluster showed a similar expression pattern as the HOXC cluster except for HOXD9, HOXD10, and HOXD12. In summary, the unique expression pattern of these four HOX clusters in subcutaneous and omental adipose tissue appears to be required for the development of human adipose depots.

Heat map representation of expression patterns of human HOX clusters in subcutaneous and omental adipose tissues. The 39 HOX genes are arranged into four distinct chromosomal clusters and designated as HOXA, HOXB, HOXC, and HOXD. The locations of HOX gene expression in the human body are indicated with apricot, violet, blue, and green. The GTEx biospecimen source sites for collection of the following adipose tissues are highlighted with yellow: subcutaneous adipose tissues are derived from beneath the skin of the leg; and omental adipose tissues are collected from the large fold of parietal peritoneum. The scale bar depicts the degrees of log2 expression value (white, low expression; blue, high expression). The yellow stars indicate differential expression (DE) between adipose tissues (FDR < 0.01).
Discussion
Transcriptomic data from the GTEx project served as a valuable asset and have been used for studies on genetic variation and gene regulatory networks in multiple human tissues24,33,34,35. Due to large-scale sampling, these data enabled us to perform comprehensive profiling of tissue-specific genes. In this study, novel adipose-specific genes were identified using large-scale analysis of the human transcriptome, and their relation to obesity-related phenotypes was investigated through integrative analysis. Our approaches present effective identification of adipose-specific genes and an evidence base for a causal association between expression of adipose-specific genes with human obesity and related metabolic disorders.
In our study, 3 novel adipose-specific protein-coding genes (CSN1S1, SLC19A3, and LVRN) were identified through a comprehensive assessment with 44 non-adipose tissues. Their high expressions in either or both subcutaneous and omental adipose tissues suggest an unknown genetic mechanism underlying adipose tissue development and functions. Besides its nutritional role as a milk protein, CSN1S1 protein is also expressed in human blood monocytes where it induces the expression of proinflammatory cytokines such as IL-1β, IL-8, and IL-6 via toll-like receptor 4 (TLR4). Given these cytokines stimulate various innate and adaptive immune responses and play a key role in inflammatory and autoimmune disorders30 and an exclusive expression of CSN1S1 in subcutaneous adipose tissue, a proinflammatory role of CSN1S1 protein in subcutaneous adipose tissue needs to be investigated. SLC19A3, also known as thiamine transporter 2 (THTR2), is one of the transporters of thiamine which is a member of the B family of vitamins, and upon uptake into intestinal epithelial cells, thiamine is converted into a coenzyme required for energy production from glucose metabolism36. As a growing amount of evidence highlights the role of deficiency of essential micronutrients such as thiamine and its derivatives in the progress of obesity31,37, this adipose-specific thiamine transporter gains special interest as a future therapeutic target. The LVRN gene encodes aminopeptidase Q (APQ/AQPEP) which is a cell surface protein expressed on human extravillous trophoblasts (EVTs) in the placenta. It hydrolyzes N-terminal amino acid of multiple peptide substrates expressed abundantly in the placenta such as angiotension III, kisspeptin-10, and endokinin C32. During placentation, LVRN plays a regulatory role in EVT migration to the uterus and its similarity in enzyme activity to an adipocyte-derived aminopeptidase was reported32, but its expression and functions in adipocytes have yet to be investigated.
In addition, genes showing adipose-enhanced expression, that have not been reported regarding their function in adipose tissue, included beta-defensin 132 (DEFB132), CD300 molecule like family member g (CD300LG), and glycogenin 2 (GYG2). The human DEFB132, also named HEL-75, is a secretory protein that is previously reported to be expressed highly in the human epididymis at both the transcription and translation levels38. DEFB132 showed dose- and time-dependent antibacterial activity without affecting fertilization indicating its main role in host defense38. CD300LG, also referred to as nepmucin, CLM-9, and TREM-4, is a type I membrane glycoprotein containing a mucin-like domain, and a single immunoglobulin (Ig) V-like domain. It is expressed in vascular endothelial cells, mediates lymphocyte rolling via its mucin-like domain, and promotes lymphocyte adhesion and migration across endothelial cells through the Ig domain. It has been reported that CD300LG binds various polar lipids39. GYG2 is one of the two self-glucosylating glycogenins that initiate the synthesis of glycogen, a storage form of a large amount of glucose as an energy source40. Unlike GYG1 which is ubiquitously expressed, GYG2 was highly expressed in adipose tissue with a lesser degree in the brain and a very low degree in skeletal muscle which is consistent with a previous report40. Although a dispensable role of GYG2 in liver glycogen synthesis has been reported because of low expression of GYG2 in the liver, its function in adipose tissue remains to be investigated, focusing on the regulatory role of GYG2 in glucose and lipid metabolism in adipose tissue and an extended role in whole-body glucose homeostasis. Also, expression patterns of well-known adipose-specific genes, such as FABP4 and ITLN1 (omentin), were confirmed by this study as previously reported elsewhere41,42. Combined analysis of the GEO database43, a functional annotation and literature search, revealed novel adipose-specific/enhanced genes, and highlight their biological functions and pathways where dysregulation may lead to changes in adult and child adiposity, as shown in the case of expression of SLC19A3 gene which is found to be significantly elevated in the child obese population.
Over the last decade, GWAS have documented numerous single nucleotide polymorphisms (SNPs) or variations at genetic loci associated with obesity-related phenotypes25,26,27,28,29. However, most GWAS loci should be interpreted with caution because of potential issues, such as difficulty in defining specific functional genes that are phenotypically- or pathologically-related, due to broad candidate chromosome regions for a particular genetic variation. In our study, adipose-specific genes identified using the GTEx RNA-Seq data were used to facilitate a comprehensive understanding of their association with GWAS phenotypes. Our integrative analysis showed that these adipose-specific genes are associated with obesity and related diseases including obesity, type 2 diabetes, blood lipid concentration, and inflammation. A robust association of inflammation-related GWAS candidate genes with omental adipose DEGs indicates omental adipose tissue plays a significantly more important role in inflammatory responses than subcutaneous adipose tissue. Further combined analyses on DEGs with GWAS revealed that some DEGs are perfectly matched with GWAS candidate genes for hip circumferences, waist circumferences, and waist-to-hip ratio. Since these body attributes are derived from regional fat deposition, our observation through the analyses is consistent with the notion that distribution of fat is regulated by genetic factors including differentially expressed genes in subcutaneous or visceral adipose tissue and genetic variants are linked to altered distribution of fat44. In this regard, the observed different expression levels of these genes between subcutaneous and omental adipose tissues may explain the positive relationship between these DEGs and distribution of adipose tissue depots in our study.
KEGG pathway analysis revealed that ligand-receptor interaction is one of the pathways that are enriched in relation to DEGs in subcutaneous adipose tissue. In this enriched pathway, genes encoding γ-aminobutyric acid type A receptor alpha 4 subunit (GABRA4), γ-aminobutyric acid type A receptor Pi subunit (GABRP), neuropeptide Y receptor Y2 (NPY2R), relaxin family peptide receptor 2 (RXFP2), and opioid receptor kappa 1 (OPRK1) are involved. In the brain, the γ-aminobutyric acid type A receptors (GABAARs), such as GABRA4 and GABRP are responsible for neurotransmitter signalling between NPY/AgRP neurons and POMC/CART neurons45,46, regulating food intake and body weight47,48. In adipocytes, the presence of γ-aminobutyric acid (GABA), GABA-synthesizing enzyme, GABA transporters, and GABA receptors suggests a functional role of GABA in adipose tissue49. Also, a very recent rodent study mentions the potential link of peripheral GABA signaling to the development of systemic metabolic dysfunction in obesity50. In this regard, those differentially expressed receptors in subcutaneous adipose tissue may play a significant role in peripheral adipose metabolism via systemic/paracrine signalling and needs to be further investigated. In addition, matrix metalloproteinases (MMPs)-mediated tissue remodelling was enriched with subcutaneous adipose DEGs encoding matrix metalloproteinase-3 (MMP-3) and -13 (MMP-13). This suggests that modification of structural integrity through these MMPs may be essential for distinct features of subcutaneous adipose tissue. MMPs are capable of cleaving protein components of extracellular matrix (ECM) and ensuing ECM remodeling plays important roles in maintaining adequate tissue function51,52. It has recently been reported that inhibition of MMP-13 led to reduced body adiposity in mice and decreased adipocyte differentiation of 3T3-L1 preadipocytes, suggesting an involvement of MMPs in adipocyte development53. Thus, these MMPs may play an important role in providing morphological and functional characteristics of subcutaneous adipose tissue as a protective tissue and further investigation is needed. On the other hand, most omental adipose DEGs were enriched in immune- and inflammation-related pathways including the JAK-STAT signaling pathway, IL-17 signaling pathway and TNF signaling pathway, implicating the role of their gene product in the regulation of immune/inflammatory responses as proinflammatory cytokines and chemokines. These data are consistent with previous evidence that adaptive immunity might be the most significant characteristic for omental adipose tissue54. In particular, some genes involved in immune/inflammatory responses and infection (e.g., IL19, MAPK15, and SERPINB10) and components of a complement system (e.g., C4B, VTN, BDKRB1, and C4BPA) have not been previously reported regarding their expression and functions in human omental adipose tissue. Therefore, these inflammation- or complement-related DEGs may need to be studied to fully characterize the immune system of omental adipose tissue.
HOX genes encode transcription factors that dominate the expression of developmental genes responsible for determining the specific anatomical structures, and regulate regional patterning of the body55. In humans, a total of 39 HOX gene family members are organized in four paralogous genomic clusters on 4 different chromosomes56. During body axis elongation, expression of the HOX genes controls a regional identity in a temporal and spatial collinear manner. In particular, 3′ HOX genes in group 1–4 (cervical) control the development of the branchial area, central HOX genes in groups 5–8 control the thoracic portion of the body, and 5′ HOX genes in groups 9–13 control the lumbo-sacral region57. Given these roles of HOX genes, our transcriptional profiling data suggest that the HOXA cluster displays a unique expression in regional fat depots following the anterior–posterior axis. Also, as shown in Fig. 5, different expression patterns among the HOXA, HOXB, HOXC, and HOXD clusters were observed between subcutaneous and omental adipose tissues. The significantly greater expression of HOXC cluster and some of HOXD genes in subcutaneous adipose tissue suggests that these genes may be important for controlling the development of subcutaneous adipose tissue. On the contrary, the majority of HOXB genes had a significantly greater expression in omental adipose tissue, suggesting that HOXB cluster genes may play critical roles in characterization of omental adipose tissue. Taken together, it appears that different HOX codes, which are combinatorial expression patterns of HOX genes for the specification of regional identity58, exist between subcutaneous and omental adipose tissues. These depot-specific HOX codes might partially contribute in differentiating the anatomical structure, regulating distribution, and determining functional characteristics of the two adipose depots.
Comprehensive analysis of the relationship between adipose-specific genes and genetic loci associated with obesity traits is essential to elucidating the genetic architecture underlying human obesity and related diseases. Thus, our findings on novel adipose-specific genes provide new insights into the susceptibility genes of obesity and related diseases. Continued attempts in characterization of adipose-specific genes and DEGs will be needed to increase our understanding of the etiology of obesity. In conclusion, our analysis identified adipose-specific genes and revealed evidence for their relationship with genetic causality of obesity, providing an important foundation for further elucidation of genetic factors and therapeutic target genes of obesity and associated diseases.
Methods
Data Collection
The most recent publicly available RNA-seq data reported in the GTEx Analysis V7 (dbGaP Accession: phs000424.v7.p2, release date: June 30, 2017) release was downloaded from the GTEx portal (www.gtexportal.org). This comprehensive data set includes gene expression values, which were normalized using transcripts per million (TPM), from non-diseased normal tissues of postmortem human donors (n = 752). For the current study, samples with RNA integrity number (RIN) of 6.0 or higher were initially chosen. Mammary tissue was excluded from the data due to its high fat content, as well as non-tissue samples such as cultured transformed fibroblasts and EBV-transformed lymphocytes. Our analysis was then confined to tissues from at least 30 donors, resulting in a total of 10,098 sampled tissues from 46 tissues including subcutaneous and omental adipose tissues, 13 brain subregions, whole blood, and other organ tissues as listed in Supplementary Table 1. The sample size for each tissue ranged from 35 to 560.
Data Analysis
Initially, 56,202 human protein-coding and noncoding genes were filtered by criteria based on expression values (i.e., in a given tissue, at least 20% of TPM values is more than 0.1 and median TPM > 0.5). After filtering, adipose-specific genes and adipose-enhanced genes were identified using a median fold change with a concomitant one-way ANOVA-based test and the Benjamini–Hochberg false discovery rate (FDR) adjustment for multiple testing using the Bioconductor limma package59. Adipose-specific genes were defined as genes with a median fold change higher than 5.0 in adipose tissue versus all other tissues and FDR-adjusted P value < 0.01 in all pair-wise comparisons between adipose tissue and the rest of tissues. In detail, genes with at least a median 5-fold change and whose expression is significantly different between tissues based on ANOVA based test, formulated as [FDR of i versus j pairwise comparison] < 0.01, where i = A1 or A2 (adipose) and j = B, C, D, ···, or AS (non-adipose), were selected. Adipose-enhanced genes were defined as genes having a median fold change higher than 5.0 in adipose tissue versus all other tissues, with an exception of 1 tissue or 2 other tissues (for those 1 or 2 tissues, higher median expression in adipose tissue was selected) and FDR-adjusted P value < 0.01. For example, an adipose-enhanced CD300LG gene showed 26% median expression level in testis compared to subcutaneous adipose tissue, which is less than 5-fold higher, but 3.8-fold higher, in subcutaneous adipose tissue (Supplementary Table 1). Genes showing higher expression in those 1 or 2 tissues than in adipose tissue were excluded from the category of adipose-enhanced. For grouping protein-coding and noncoding genes [long intergenic non-coding RNA (linc RNA), pseudogene, small nuclear RNA (snRNA), and antisense RNA], the Ensembl Gene IDs were aligned to the human reference genome using the Ensembl gene GRCh38 release 91 annotation file (https://www.ensembl.org/info/data/ftp/index.html). Heat maps were generated using the R package heatmap360.
Semi-quantitative RT-PCR
Total RNAs from the adult human brain, heart, kidney, lung, liver and skeletal muscle were purchased from Agilent Technologies (Santa Clara, CA) and adult human RNA from adipose tissue was purchased from Clontech Laboratories (Mountain View, CA). To measure the quantity of RNA, a Nanodrop spectrophotometer (Thermo Scientific, Wilmington, DE) was used. Approximately 1 µg of RNA was reverse-transcribed in a 20 µL total reaction to cDNA using Moloney murine leukemia virus (M-MLV) reverse transcriptase (Invitrogen). The thermal cycle of reverse transcription (RT) was 65 °C for 5 min, 37 °C for 52 min, and 70 °C for 15 min. Exactly 1 µL of cDNA samples was used as a template for PCR in a 25 µL total reaction with AmpliTaq Gold DNA polymerase (Applied Biosystems, Carlsbad, CA). The conditions for this reaction were 95 °C for 10 min, appropriate cycles with linear amplification ranges of 94 °C for 30 s, 55 °C for 30 s, 72 °C for 30 s, with an additional extension step at 72 °C for 10 min. PCR products were separated by using 1% agarose gel electrophoresis. Following forward and reverse primers for humans were designed on different exons for multi-exon genes to avoid genomic DNA contamination: CSN1S1 (forward: 5′-CCTACCCCTAT GCTGTTT-3′, reverse: 5′-TCCTTGAGAGGAGAAATTCA-3′), SLC19A3 (forward: 5′-ACTTGCCA GTCAGCATT-3′, reverse: 5′-GTTTGTTGCGATGAGGTTA-3′), and LVRN (AQPEP) (forward: 5′-GGAGAACCGTAACTACAGATTT-3′, reverse: 5′-TGAAAGATAGCCACAAGCTAT-3′). Human cyclophilin (CYC; forward: 5′-CTCCTTTGAGCTGTTTGCAG-3′, reverse: 5′-CACCACATGCTTG CCATCC-3′) was used as a reference gene.
Western blot analysis
Human tissue lysates from subcutaneous adipose tissue, brain, heart, kidney, lung, liver and skeletal muscle were purchased from Protein Biotechnologies Inc (Ramona, CA). Western blot analysis with tissue protein lysates was performed as described in our previous report61. In detail, equal amounts of protein lysates were loaded onto gels before wet-transfer to PVDF membranes (Bio-Rad, Hercules, CA). The membranes were blocked for 30 min and then incubated with CSN1S1 (H00001446-D01P; 1:750; Novus Biologicals, LLC, Centennial, CO) or SLC19A3 (NBP1–69703; 1:500; Novus Biologicals, LLC) antibodies at 4 °C overnight. The next day, after washing, an appropriate secondary antibody (HRP-linked anti-rabbit IgG (HAF008); 1:5000; R&D systems Inc., Minneapolis, MN) was applied to the membrane before washing and developing with ECL plus reagents and X-ray films (both materials from GE Healthcare Biosciences, Pittsburgh, PA). GAPDH (antibody: 10494-1-AP; 1:1000; Proteintech Group, Chicago, IL) was used as a reference protein.
Profiling of adipose-specific genes
To investigate biological functions and subcellular localization of three unreported common adipose-specific genes, the GeneCards database of human genes (http://www.genecards.org/) and literature were explored. The PubMed database was searched for previous studies using the keywords ‘gene name AND adipose’ or ‘gene name AND adipocyte’ or ‘gene name AND obesity’ and functionally unreported genes in adipose tissue were determined. For further identification of obesity relatedness of each of those genes, data were retrieved from the Gene Expression Omnibus (GEO) Series (GSE) through the GEO2R interface (http://www.ncbi.nlm.nih.gov/geo/geo2r/).
Comprehensive comparison between adipose depots
Read count matrix from RNA-Seq of subcutaneous adipose tissue and omental adipose tissue was downloaded from GTEx portal. The Bioconductor DESeq. 2 package62 was used to normalize all samples for sequencing depth and calculate the DEGs between subcutaneous adipose tissue and omental adipose tissue. To obtain significant DEGs, the combined criteria of FDR-adjusted P value < 0.01 and the absolute log2 fold change >3 was used, where a fold change is defined as the expression in samples of subcutaneous adipose tissue divided by the expression in samples of omental adipose tissue.
Association analyses with GWAS phenotypes
The GWAS data used for our analyses were obtained from the public GWAS Catalog (https://www.ebi.ac.uk/gwas/) on 06/25/2018. Reported and mapped genes of GWAS shared with adipose-specific/enhanced genes and DEGs were used to analyze the relationships between gene expression and phenotypic traits in adipose tissues. Whole genome visualization was performed by PhenoGram63 to plot DEGs with associated traits using categories of GWAS phenotypes.
GO functional and pathway enrichment analyses
For the sets of DEGs, the Gene Ontology (GO) function and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analyses were performed using R package, clusterProfiler (version 3.6.0)64. The enriched GO terms and KEGG pathways were displayed with the significance of the enrichment of each gene set under the criteria of FDR-corrected P value < 0.05. The visualization of relevant KEGG pathways was generated using GOPlot (version 1.0.2)65.
References
Smyth, S. & Heron, A. Diabetes and obesity: the twin epidemics. Nat Med 12, 75–80, https://doi.org/10.1038/nm0106-75 (2006).
Artham, S. M., Lavie, C. J., Milani, R. V. & Ventura, H. O. Obesity and hypertension, heart failure, and coronary heart disease-risk factor, paradox, and recommendations for weight loss. Ochsner J 9, 124–132 (2009).
Lauby-Secretan, B. et al. Body Fatness and Cancer–Viewpoint of the IARC Working Group. N Engl J Med 375, 794–798, https://doi.org/10.1056/NEJMsr1606602 (2016).
Ford, N. D., Patel, S. A. & Narayan, K. M. Obesity in Low- and Middle-Income Countries: Burden, Drivers, and Emerging Challenges. Annu Rev Public Health 38, 145–164, https://doi.org/10.1146/annurev-publhealth-031816-044604 (2017).
Loos, R. J. Recent progress in the genetics of common obesity. Br J Clin Pharmacol 68, 811–829, https://doi.org/10.1111/j.1365-2125.2009.03523.x (2009).
Herrera, B. M. & Lindgren, C. M. The genetics of obesity. Curr Diab Rep 10, 498–505, https://doi.org/10.1007/s11892-010-0153-z (2010).
Dandona, P., Aljada, A. & Bandyopadhyay, A. Inflammation: the link between insulin resistance, obesity and diabetes. Trends Immunol 25, 4–7 (2004).
Peterfy, M., Phan, J. & Reue, K. Alternatively spliced lipin isoforms exhibit distinct expression pattern, subcellular localization, and role in adipogenesis. J Biol Chem 280, 32883–32889, https://doi.org/10.1074/jbc.M503885200 (2005).
Li, B., Shin, J. & Lee, K. Interferon-stimulated gene ISG12b1 inhibits adipogenic differentiation and mitochondrial biogenesis in 3T3-L1 cells. Endocrinology 150, 1217–1224, https://doi.org/10.1210/en.2008-0727 (2009).
Hotamisligil, G. S. Inflammation, metaflammation and immunometabolic disorders. Nature 542, 177–185, https://doi.org/10.1038/nature21363 (2017).
Zhang, Y. et al. Positional cloning of the mouse obese gene and its human homologue. Nature 372, 425–432, https://doi.org/10.1038/372425a0 (1994).
Pan, W. W. & Myers, M. G. Jr. Leptin and the maintenance of elevated body weight. Nat Rev Neurosci 19, 95–105, https://doi.org/10.1038/nrn.2017.168 (2018).
Hu, E., Liang, P. & Spiegelman, B. M. AdipoQ is a novel adipose-specific gene dysregulated in obesity. J Biol Chem 271, 10697–10703 (1996).
Achari, A. E. & Jain, S. K. Adiponectin, a Therapeutic Target for Obesity, Diabetes, and Endothelial Dysfunction. Int J Mol Sci 18, https://doi.org/10.3390/ijms18061321 (2017).
Tontonoz, P., Hu, E., Graves, R. A., Budavari, A. I. & Spiegelman, B. M. mPPAR gamma 2: tissue-specific regulator of an adipocyte enhancer. Genes Dev 8, 1224–1234 (1994).
Tsai, Y. S. & Maeda, N. PPARgamma: a critical determinant of body fat distribution in humans and mice. Trends Cardiovasc Med 15, 81–85, https://doi.org/10.1016/j.tcm.2005.04.002 (2005).
Lefterova, M. I. & Lazar, M. A. New developments in adipogenesis. Trends Endocrinol Metab 20, 107–114, https://doi.org/10.1016/j.tem.2008.11.005 (2009).
Hunt, C. R., Ro, J. H., Dobson, D. E., Min, H. Y. & Spiegelman, B. M. Adipocyte P2 gene: developmental expression and homology of 5′-flanking sequences among fat cell-specific genes. Proc Natl Acad Sci USA 83, 3786–3790 (1986).
Furuhashi, M., Saitoh, S., Shimamoto, K. & Miura, T. Fatty Acid-Binding Protein 4 (FABP4): Pathophysiological Insights and Potent Clinical Biomarker of Metabolic and Cardiovascular Diseases. Clin Med Insights Cardiol 8, 23–33, https://doi.org/10.4137/CMC.S17067 (2014).
Kim, K. H., Lee, K., Moon, Y. S. & Sul, H. S. A cysteine-rich adipose tissue-specific secretory factor inhibits adipocyte differentiation. J Biol Chem 276, 11252–11256, https://doi.org/10.1074/jbc.C100028200 (2001).
Villena, J. A., Roy, S., Sarkadi-Nagy, E., Kim, K. H. & Sul, H. S. Desnutrin, an adipocyte gene encoding a novel patatin domain-containing protein, is induced by fasting and glucocorticoids: ectopic expression of desnutrin increases triglyceride hydrolysis. J Biol Chem 279, 47066–47075, https://doi.org/10.1074/jbc.M403855200 (2004).
Song, Y., Ahn, J., Suh, Y., Davis, M. E. & Lee, K. Identification of novel tissue-specific genes by analysis of microarray databases: a human and mouse model. PLoS One 8, e64483, https://doi.org/10.1371/journal.pone.0064483 (2013).
Consortium, G. T. The Genotype-Tissue Expression (GTEx) project. Nat Genet 45, 580–585, https://doi.org/10.1038/ng.2653 (2013).
Consortium, G. T. et al. Genetic effects on gene expression across human tissues. Nature 550, 204–213, https://doi.org/10.1038/nature24277 (2017).
Speliotes, E. K. et al. Association analyses of 249,796 individuals reveal 18 new loci associated with body mass index. Nat Genet 42, 937–948, https://doi.org/10.1038/ng.686 (2010).
Bradfield, J. P. et al. A genome-wide association meta-analysis identifies new childhood obesity loci. Nat Genet 44, 526–531, https://doi.org/10.1038/ng.2247 (2012).
Sung, Y. J. et al. Genome-wide association studies suggest sex-specific loci associated with abdominal and visceral fat. Int J Obes (Lond) 40, 662–674, https://doi.org/10.1038/ijo.2015.217 (2016).
Lu, Y. et al. New loci for body fat percentage reveal link between adiposity and cardiometabolic disease risk. Nat Commun 7, 10495, https://doi.org/10.1038/ncomms10495 (2016).
Akiyama, M. et al. Genome-wide association study identifies 112 new loci for body mass index in the Japanese population. Nat Genet 49, 1458–1467, https://doi.org/10.1038/ng.3951 (2017).
Vordenbaumen, S. et al. Human casein alpha s1 induces proinflammatory cytokine expression in monocytic cells by TLR4 signaling. Mol Nutr Food Res 60, 1079–1089, https://doi.org/10.1002/mnfr.201500792 (2016).
Maguire, D., Talwar, D., Shiels, P. G. & McMillan, D. The role of thiamine dependent enzymes in obesity and obesity related chronic disease states: A systematic review. Clin Nutr ESPEN 25, 8–17, https://doi.org/10.1016/j.clnesp.2018.02.007 (2018).
Maruyama, M. et al. Laeverin/aminopeptidase Q, a novel bestatin-sensitive leucine aminopeptidase belonging to the M1 family of aminopeptidases. J Biol Chem 282, 20088–20096, https://doi.org/10.1074/jbc.M702650200 (2007).
Li, X. et al. The impact of rare variation on gene expression across tissues. Nature 550, 239–243, https://doi.org/10.1038/nature24267 (2017).
Mohammadi, P., Castel, S. E., Brown, A. A. & Lappalainen, T. Quantifying the regulatory effect size of cis-acting genetic variation using allelic fold change. Genome Res 27, 1872–1884, https://doi.org/10.1101/gr.216747.116 (2017).
Saha, A. et al. Co-expression networks reveal the tissue-specific regulation of transcription and splicing. Genome Res 27, 1843–1858, https://doi.org/10.1101/gr.216721.116 (2017).
Zhao, R. & Goldman, I. D. Folate and thiamine transporters mediated by facilitative carriers (SLC19A1-3 and SLC46A1) and folate receptors. Mol Aspects Med 34, 373–385, https://doi.org/10.1016/j.mam.2012.07.006 (2013).
Gangolf, M. et al. Thiamine status in humans and content of phosphorylated thiamine derivatives in biopsies and cultured cells. PLoS One 5, e13616, https://doi.org/10.1371/journal.pone.0013616 (2010).
Lin, Y. Q. et al. Cloning and identification of a novel sperm binding protein, HEL-75, with antibacterial activity and expressed in the human epididymis. Hum Reprod 23, 2086–2094, https://doi.org/10.1093/humrep/den084 (2008).
Cannon, J. P., O’Driscoll, M. & Litman, G. W. Specific lipid recognition is a general feature of CD300 and TREM molecules. Immunogenetics 64, 39–47, https://doi.org/10.1007/s00251-011-0562-4 (2012).
Nilsson, J. et al. LC-MS/MS characterization of combined glycogenin-1 and glycogenin-2 enzymatic activities reveals their self-glucosylation preferences. Biochim Biophys Acta 1844, 398–405, https://doi.org/10.1016/j.bbapap.2013.11.002 (2014).
Terra, X. et al. FABP 4 is associated with inflammatory markers and metabolic syndrome in morbidly obese women. Eur J Endocrinol 164, 539–547, https://doi.org/10.1530/EJE-10-1195 (2011).
Yang, R. Z. et al. Identification of omentin as a novel depot-specific adipokine in human adipose tissue: possible role in modulating insulin action. Am J Physiol Endocrinol Metab 290, E1253–1261, https://doi.org/10.1152/ajpendo.00572.2004 (2006).
Barrett, T. et al. NCBI GEO: archive for functional genomics data sets–10 years on. Nucleic Acids Res 39, D1005–1010, https://doi.org/10.1093/nar/gkq1184 (2011).
Schleinitz, D., Bottcher, Y., Bluher, M. & Kovacs, P. The genetics of fat distribution. Diabetologia 57, 1276–1286, https://doi.org/10.1007/s00125-014-3214-z (2014).
Ohlson, J., Pedersen, J. S., Haussler, D. & Ohman, M. Editing modifies the GABA(A) receptor subunit alpha3. RNA 13, 698–703, https://doi.org/10.1261/rna.349107 (2007).
Zagmutt, S., Mera, P., Soler-Vazquez, M. C., Herrero, L. & Serra, D. Targeting AgRP neurons to maintain energy balance: Lessons from animal models. Biochem Pharmacol 155, 224–232, https://doi.org/10.1016/j.bcp.2018.07.008 (2018).
Wu, Q., Boyle, M. P. & Palmiter, R. D. Loss of GABAergic signaling by AgRP neurons to the parabrachial nucleus leads to starvation. Cell 137, 1225–1234, https://doi.org/10.1016/j.cell.2009.04.022 (2009).
Vasconcelos, L. H., Souza, I. L., Pinheiro, L. S. & Silva, B. A. Ion Channels in Obesity: Pathophysiology and Potential Therapeutic Targets. Front Pharmacol 7, 58, https://doi.org/10.3389/fphar.2016.00058 (2016).
Nicolaysen, A., Gammelsaeter, R., Storm-Mathisen, J., Gundersen, V. & Iversen, P. O. The components required for amino acid neurotransmitter signaling are present in adipose tissues. J Lipid Res 48, 2123–2132, https://doi.org/10.1194/jlr.M700021-JLR200 (2007).
Denis, C. J. & Lambeir, A. M. The potential of carboxypeptidase M as a therapeutic target in cancer. Expert Opin Ther Targets 17, 265–279, https://doi.org/10.1517/14728222.2012.741122 (2013).
Jones, C. B., Sane, D. C. & Herrington, D. M. Matrix metalloproteinases: a review of their structure and role in acute coronary syndrome. Cardiovasc Res 59, 812–823 (2003).
Traurig, M. T. et al. Differential expression of matrix metalloproteinase 3 (MMP3) in preadipocytes/stromal vascular cells from nonobese nondiabetic versus obese nondiabetic Pima Indians. Diabetes 55, 3160–3165, https://doi.org/10.2337/db06-0373 (2006).
Shih, C. L. & Ajuwon, K. M. Inhibition of MMP-13 prevents diet-induced obesity in mice and suppresses adipogenesis in 3T3-L1 preadipocytes. Mol Biol Rep 42, 1225–1232, https://doi.org/10.1007/s11033-015-3861-2 (2015).
Palming, J. et al. Plasma cells and Fc receptors in human adipose tissue–lipogenic and anti-inflammatory effects of immunoglobulins on adipocytes. Biochem Biophys Res Commun 343, 43–48, https://doi.org/10.1016/j.bbrc.2006.02.114 (2006).
Mark, M., Rijli, F. M. & Chambon, P. Homeobox genes in embryogenesis and pathogenesis. Pediatr Res 42, 421–429, https://doi.org/10.1203/00006450-199710000-00001 (1997).
Apiou, F. et al. Fine mapping of human HOX gene clusters. Cytogenet Cell Genet 73, 114–115, https://doi.org/10.1159/000134320 (1996).
Cantile, M., Procino, A., D’Armiento, M., Cindolo, L. & Cillo, C. HOX gene network is involved in the transcriptional regulation of in vivo human adipogenesis. J Cell Physiol 194, 225–236, https://doi.org/10.1002/jcp.10210 (2003).
Hunt, P. & Krumlauf, R. Deciphering the Hox code: clues to patterning branchial regions of the head. Cell 66, 1075–1078 (1991).
Ritchie, M. E. et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res 43, e47, https://doi.org/10.1093/nar/gkv007 (2015).
Zhao, S., Guo, Y., Sheng, Q. & Shyr, Y. Advanced heat map and clustering analysis using heatmap3. Biomed Res Int 2014, 986048, https://doi.org/10.1155/2014/986048 (2014).
Ahn, J. et al. Identification of the avian RBP7 gene as a new adipose-specific gene and RBP7 promoter-driven GFP expression in adipose tissue of transgenic quail. PLoS One 10, e0124768, https://doi.org/10.1371/journal.pone.0124768 (2015).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq. 2. Genome Biol 15, 550, https://doi.org/10.1186/s13059-014-0550-8 (2014).
Wolfe, D., Dudek, S., Ritchie, M. D. & Pendergrass, S. A. Visualizing genomic information across chromosomes with PhenoGram. BioData Min 6, 18, https://doi.org/10.1186/1756-0381-6-18 (2013).
Yu, G., Wang, L. G., Han, Y. & He, Q. Y. Clusterprofiler: an R package for comparing biological themes among gene clusters. OMICS 16, 284–287, https://doi.org/10.1089/omi.2011.0118 (2012).
Walter, W., Sanchez-Cabo, F. & Ricote, M. GOplot: an R package for visually combining expression data with functional analysis. Bioinformatics 31, 2912–2914, https://doi.org/10.1093/bioinformatics/btv300 (2015).
Acknowledgements
The data used for the analyses described in this manuscript were obtained from the GTEx Portal on 11/22/2017. This work was partially supported by the United States Department of Agriculture National Institute of Food and Agriculture Grant (Project No. 2016-08413), the Ohio Agricultural Research and Development Center Research SEEDS funding (Project No. 2017-069), and the National Nature Science Foundation of China (Project No. 31660648).
Author information
Authors and Affiliations
Contributions
K.L. conceived the study. J.A., H.W. and K.L. developed the study design. J.A. and H.W. performed data processing and statistical analysis. H.W. conducted GWAS integration analysis. J.A., H.W. and K.L. analysed and interpreted the data. J.A. and H.W. drafted the manuscript. All authors read and approved the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ahn, J., Wu, H. & Lee, K. Integrative Analysis Revealing Human Adipose-Specific Genes and Consolidating Obesity Loci. Sci Rep 9, 3087 (2019). https://doi.org/10.1038/s41598-019-39582-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-39582-8
This article is cited by
-
Reduced plasma GDF10 levels are positively associated with cholesterol impairment and childhood obesity
Scientific Reports (2024)
-
The temporal relationship between parental concern of overeating and childhood obesity considering genetic susceptibility: longitudinal results from the IDEFICS/I.Family study
International Journal of Behavioral Nutrition and Physical Activity (2021)
-
Chronic stress, epigenetics, and adipose tissue metabolism in the obese state
Nutrition & Metabolism (2020)
-
Distinct abdominal and gluteal adipose tissue transcriptome signatures are altered by exercise training in African women with obesity
Scientific Reports (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
