
Abstract
Clonostachys rosea is a mycoparasitic fungus used for biological control of plant diseases. Its genome contains 31 genes putatively encoding for polyketide synthases (PKSs), 75% of which are arranged in biosynthetic gene clusters. Gene expression analysis during C. rosea interactions with the fungal plant pathogens Botrytis cinerea and Fusarium graminearum showed common and species-specific induction of PKS genes. Our data showed a culture media dependent correlation between PKS gene expression and degree of antagonism in C. rosea. The pks22 and pks29 genes were highly induced during fungal-fungal interactions but not during pigmentation, and gene deletion studies revealed that PKS29 was required for full antagonism against B. cinerea, and for biocontrol of fusarium foot rot on barley. Metabolite analysis revealed that Δpks29 strains has a 50% reduced production (P = 0.001) of an unknown polyketide with molecular formula C15H28O3, while Δpks22 strains lost the ability to produce four previously unknown polyketides named Clonorosein A-D. Clonorosein A and B were purified, their structures determined, and showed strong antifungal activity against B. cinerea and F. graminearum. These results show that PKS22 is required for production of antifungal polyketide Clonorosein A-D, and demonstrate the role of PKS29 in antagonism and biocontrol of fungal plant diseases.
Similar content being viewed by others


Introduction
Polyketides are a structurally diverse group of secondary metabolites with diverse functions important for ecological and evolutionary adaptation of fungi1,2. Polyketides are biosynthesised by large multi-domain enzymes called polyketide synthases (PKSs)1,2. Fungal PKSs are mainly iterative type I enzymes consisting of multiple functional domains: ketoacyl synthase (KS), acyl transferase (AT), dehydratase (DH), enoyl reductase (ER), ketoreductase (KR) and acyl carrier protein (ACP). PKS domains KS, AT and ACP catalyse the biosynthesis of acetyl coenzyme A and malonyl CoA and are essential domains, while KR, DH and ER are optional domains that may or may not be present in a single PKS protein. Based on the presence and absence of these optional domains, type I iterative PKSs can be functionally and phylogenetically grouped into highly reducing (HR) and non-reducing (NR) types, respectively. However, NR-PKSs contain additional unique functional S-acetyltransferase (SAT), product template (PT) and thioesterase (TE) domains1,3,4. Highly reducing PKS enzymes lacking KR, DH or ER are classified as partially-reducing (PR) PKSs. Variability in type, number, and activity of these domains contribute to the vast diversity of polyketide compounds produced by fungal PKSs.
Genes encoding PKSs are often localised in biosynthetic gene clusters (BGC) together with additional biosynthetic genes such as those encoding cytochrome P450 monooxygenases, oxidoreductases, dehydrogenases, acetyltransferases, methyltransferases, and other transferases responsible for modifications of the polyketide backbone to produce the end product. The BGC also includes genes encoding transporters like ATP-binding cassette (ABC) and major facilitator superfamily (MFS) transporters and regulatory proteins, for polyketide efflux and gene expression regulation, respectively3,5.
In fungi, functional characterization of PKSs have mostly been focussed on their role in human and plant pathogenesis and mycotoxin, drug and pigment production1,2,6. A role of polyketides in mycelial growth, and development of asexual and sexual structure has also been demonstrated7,8. However, only limited information is available concerning the role of polyketides in regulating microbial interactions, especially in interactions resulting in biological control of plant diseases. Fungal plant pathogen Fusarium spp. produce the polyketide mycotoxin zearalenone with high antifungal activity, presumably to compete with other fungi present in the same ecological niche9, and it was shown that the ability to detoxify zearalenone is indeed important for the biocontrol ability of the mycoparasitic fungus Clonostachys rosea10,11. Deletion of the PKS gene pks4 in the weak mycoparasitic fungus Trichoderma reesei resulted in loss of pigmentation that made it more sensitive to toxic metabolites produced by the plant pathogenic fungi Alternaria alternata, Botrytis cinerea, Rhizoctonia solani and Sclerotinia sclerotiorum12.
The ascomycete fungus C. rosea (teleomorph name Bionectria ochroleuca)13 is an efficient biocontrol agent (BCA) against numerous plant pathogenic fungi14,15,16, against oomycete17 and plasmodiophorid18 plant pathogens and even against plant parasitic nematodes19. This broad activity against a wide taxonomic range of plant pathogens suggests that interference competition by means of secretion of broad spectrum antibiotic compounds may contribute to the biocontrol activity of C. rosea20. However, only a few peptaibols with antifungal activity21, epipolysulfanyldioxopiperazines with nematicidal activity22, polyterpenoid glisoprenins that inhibit appressorium formation of phytopathogenic fungi23, and a few TMC-151-type polyketide antibiotics with antibacterial properties24 have been identified from C. rosea.
This limited knowledge on production of secondary metabolites in C. rosea is in sharp contrast to the predicted gene content of its genome. The C. rosea strain IK726 genome contains high numbers of genes encoding PKSs (31 genes), non-ribosomal peptide synthetases (NRPSs, 17 genes), ABC (90 genes) and MFS (634 genes) membrane transporters25,26. Functional studies indeed confirmed the role of several ABC and MFS transporters in antagonism, xenobiotic tolerance and in biocontrol by mediating efflux of endogenous or exogenous metabolites in C. rosea10,11,26,27,28. However, the biological roles of PKSs in C. rosea are yet to be studied.
The aim of this study was to investigate the biological functions of PKSs in C. rosea, with emphasis on their role in biotic interactions with relevance for biological control of fungal plant diseases. We hypothesized that the significant copy number expansion of PKS genes is associated with the mycoparasitic lifestyle of C. rosea. To test this hypothesis, we identified PKS BGCs in C. rosea, and studied PKS gene expression patterns under different conditions relevant for mycoparasitic interactions and pigmentation. By generating and analysing gene deletion mutants, we identified previously unknown polyketides and investigated their biological functions. Our data demonstrate that PKSs are important for antagonism and mycoparasitic interactions in C. rosea. Moreover, our results further validate the potential to discover novel compounds by using a genome mining and reverse genetics approach.
Results
Identification and analysis of putative polyketide biosynthetic gene clusters
Analysis of the C. rosea IK726 genome with the antiSMASH software confirmed the presence of 31 PKS genes and one PKS-NRPS hybrid gene25 and predicted 20 putative PKS BGCs (18 BGCs with one PKS gene and two BGCs with two PKS genes; Tables 1 and S1). Conserved domain analysis using translated amino acid (aa) sequences verified the presence of PKSs genes in BGCs (Fig. S1). The predicted PKS BGCs ranged from 36.1 kbp to 59.6 kbp in size, contained 6–14 predicted genes, and were distributed to 20 different scaffolds (Table S1). Seven PKS genes (pks1, pks5, pks11, pks14, pks15, pks17 and pks26) were not predicted to be part of a BGC, sequences of pks30 and pks31 were incomplete and pks32 was a PKS-NRPS hybrid, and were thus excluded from further bioinformatic analyses. Analysis of the predicted BGC genes with the smCOG (secondary metabolite cluster of orthologous groups) and BlastP softwares showed the presence of a core polyketide biosynthetic gene, additional biosynthetic genes, regulatory genes and transport-related genes required for polyketide biosynthesis, backbone modifications, regulation and transport, respectively (Table 1). All PKS BGCs contained one to eight additional biosynthetic genes with the majority encoding putative flavoproteins such as flavin adenine dinucleotide (FAD)-binding domain proteins, flavin-containing monooxygenases and short-chain dehydrogenases/reductases (SDR). All PKS BGCs with the exception of clusters 63, 76, 93, 142 and 186, contained one or two genes encoding transport-related proteins with the majority being MFS transporters. In addition, more than 50% of the BGCs contained one or more genes encoding transcription factors (Table 1). A complete description of genes and their arrangement in the putative BGCs are presented in Table S2 and Fig. S2.
Cluster Blast analysis suggested that C. rosea PKS BGC63 was an ortholog of the characterised citrinin BGC (BGC0001338, Genbank accession number KT781075) of Monascus ruber29. The BGC63 contained seven genes from which a gene encoding a putative serine hydrolase and a gene encoding PKS4 showed ≥49% sequence identity with the serine hydrolase gene CitA (e-value = 1.8 × 10−33) and citrinin PKS gene CitS (e-value = 0) from the citrinin BGC BGC0001338. BGC134 showed similarity with the characterised sorbicillin BGC (BGC0001404, Genbank: AM920436) of Penicillium chrysogenum30. The BGC134 contained 12 genes including genes encoding two transcription factors, a MFS transporter, PKS2 and PKS12 that showed ≥55% sequence identity with genes encoding the transcription factors Orf1 (e-value = 2 × 10−87) and Orf5 (e-value = 1.8 × 10−26), the MFS transporter Orf6 (e-value = 8.4 × 10−176), and the two oppositely transcribed NR-PKS SorB (e-value = 5.6 × 10−244) and HR-PKS SorA (e-value = 0) from the sorbicillin BGC of P. chrysogenum, respectively. Four genes in BGC190 encoding two putative FAD-binding monooxygenases, a MFS transporter and PKS19 showed ≥44% sequence identity with the DEP2 (e-value = 3.4 × 10−115), DEP3 (e-value = 1.2 × 10−68), DEP4 (e-value = 1 × 10−122) and DEP5 (e-value = 0) genes in the depudecin BGC (BGC0000046, Genbank: FJ977165) of Alternaria brassicicola31 (Table S1, Fig. 1).

Schematic representation of C. rosea putative PKS biosynthetic gene clusters (BGCs) showing similarity with genes from characterised BGCs. (A) Citrinin BGC, (B) Sorbicillin BGC, and (C) Depudicin BGC. Type of genes common (≥40% sequence identity; e- value ≥1 × 1025) between two BGC are highlighted with the same colour, white arrows represent genes that are not common between BGC. Genes name are given in parentheses below the arrow, their orientation are indicated, while distance and size are not set to scale. C. rosea protein IDs are given above the arrows. Genbank accession numbers: citA: ALI92654.1; citS: ALI92655.1; orf1: CAP95402.1; sorB: CAP95404.1; sorC: CAP95405.1; orf5: CAP95406.1; orf6: CAP95407.1; dep2: ACZ57545.1; dep3: ACZ57546.1; dep4: ACZ57547.1; dep5: ACZ57548.1.
Antagonistic effect of Clonostachys rosea is culture medium dependent
Antagonistic ability of C. rosea against the plant pathogenic fungi A. alternata, B. cinerea, Fusarium graminearum, and R. solani was determined by measuring their mycelial biomass in C. rosea culture filtrates from culture media differing with regard to carbon source, nitrogen source and pH. The media compositions are provided in Table S3. The mycelial biomass of all plant pathogenic fungi was reduced significantly (P ≤ 0.05) in C. rosea culture filtrates from all culture media, except Czapek-Dox (CZ), in comparison to control medium (Fig. 2). The maximum reduction in biomass was observed in C. rosea culture filtrates from potato dextrose broth (PDB) (83–98%) and malt extract (ME) (73–97%) followed by synthetic minimal salt (SMS) (61%–90%) and synthetic nutrient broth (SNB) (36–72%). Interestingly, A. alternata and R. solani produced significantly more biomass (P ≤ 0.030) in C. rosea culture filtrate from CZ medium, compared with the CZ control (Fig. 2). We observed that C. rosea lowered the pH in CZ medium from 7.2 to 5.5 during incubation, and the result from a follow-up experiment showed that A. alternata and R. solani both produced significantly (P ≤ 0.01) more biomass in CZ medium with pH 5.5 as compared with CZ medium with pH 7.2 (data not shown).

Production of fungal growth-inhibitory compounds by C. rosea in different media. C. rosea was grown in liquid medium broth (czapek-dox, CZ; malt extract, ME; potato dextrose, PDB; synthetic minimal salt, SMS; synthetic nutrient broth, SNB) for 4 days at 25 °C, culture filtrate was collected after removing the mycelium, and was then inoculated with an A. alternata (Aa), B. cinerea (Bc), F. graminearum (Fg) or R. solani (Rs) agar plug. Fungus inoculated into the respective fresh culture broth was used as control. Biomass production in culture filtrates was analysed by determining mycelial dry weight 4 days post-inoculation. Error bars indicate standard deviation based on five biological replicates. Different letters indicate statistically significant differences (P ≤ 0.05) within experiments based on the Fisher’s exact test.
Gene expression analysis
Gene expression analysis of the 31 PKS genes and one PKS-NRPS hybrid gene in C. rosea was carried out in 3 different conditions: (i) during mycelial growth in liquid culture media, (ii) during dual culture interactions with plant pathogenic fungal species B. cinerea or F. graminearum, and (iii) during pigment production on solid agar.
In order to investigate if there was a correlation between degrees of antagonism in different culture media with PKS gene expression, CZ (no significant positive antagonistic effect), PDB (high antagonistic effect) and SNB (medium antagonistic effect) media were selected for PKS gene expression analysis. With the exception for pks25 and pks27, all PKS genes were expressed in the three selected culture media (Fig. 3A). The expression levels of all genes were significantly (P ≤ 0.002) higher in PDB compared with CZ. Similarly, 24 PKS genes showed significantly (P ≤ 0.002) induced expression in SNB compared with CZ. Eighteen genes were induced significantly (P ≤ 0.002) in PDB compared with SNB (Fig. 3A). In PDB, pks29 showed the maximum 3154-fold induction followed by pks8 (926-fold), pks14 (892-fold), pks12 (660-fold), pks30 (640-fold), pks6 (526-fold) and pks19 (506-fold). In SNB, pks29 showed maximum 35-fold induction followed by pks16 (16-fold), pks4, pks9, pks24 (13-fold), and pks12 (12-fold). Interestingly, no induction in gene expression was observed in C. rosea grown in CZ compared with PDB or SNB (Fig. 3A).

Heat map of polyketide synthase gene expression in C. rosea. (A) During mycelial growth in liquid CZ, PDB and SNB culture media. C. rosea mycelia grown in CZ medium were used as control treatment. (B) During interactions with itself (Cr-Cr), B. cinerea (Cr-Bc) or F. graminearum (Cr-Fg). C. rosea interaction with self (Cr–Cr) was used as control treatment. (C) During pigment production. Four days old C. rosea culture plates, incubated at the same conditions were used as control treatment as no pigmentation was observed at this stage. Relative expression level based on RT-qPCR was calculated as the ratio between the target PKS gene and β-tubulin using 2−ΔΔCt method (Livak and Schmittgen, 2001), and compared with the respective control. Statistically significant differences (P ≤ 0.05) in gene expression between treatments were determined using Fisher’s exact test and are indicated by different letters. The scale representing the relative expression intensity values is shown.
During fungal interactions, a common set of 7 PKS genes were significantly (P ≤ 0.020) induced during interactions with both B. cinerea (Cr-Bc) and F. graminearum (Cr-Fg) as compared with the self-interaction control (Cr-Cr) (Fig. 3B), although with moderate fold changes (maximum 4.3-fold induction). Among these, four genes (pks2, pks4, pks16, and pks28) were more highly expressed in Cr-Bc than Cr-Fg, while pks22 showed the opposite pattern with higher expression in Cr-Fg. Another 7 PKS genes (pks3, pks12, pks13, pks14, pks15, pks17 and pks20) were specifically induced 1.4–6.2 fold (P ≤ 0.049) against B. cinerea but not F. graminearum. The expression of pks9 was significantly suppressed (P = 0.001) in both Cr-Bc and Cr-Fg, while pks19 was significantly suppressed (P = 0.001) exclusively during interaction with F. graminearum (Fig. 3B). Seven PKS genes (pks6, pks7, pks21, pks23, pks24, pks26, pks31) were expressed but showed no significant changes in expression during interactions, while transcripts from another 6 PKS genes (pks5, pks11, pks18, pks25, pks27 and pks32) were not detected under these conditions (Fig. 3B).
C. rosea often secretes yellow pigment during cultivation on PDA. To study the role of PKSs in ageing and yellow pigment biosynthesis, PKS gene expression was analysed at the time of occurrence of yellow pigment on agar plates (10 days post-inoculation [dpi]) and compared with an earlier time point (4 dpi) where no pigmentation was evident. Gene expression analysis showed a significant (P ≤ 0.011) induction in expression of 19 PKS genes, significant suppression (P = 0.047) of pks22, and no change in expression of pks29 (P = 0.764) during pigmentation compared with the control (Fig. 3C). The PKS gene pks1 showed the maximum 933-fold induction followed by pks20 (797-fold), pks2 (646-fold), and pks17 (464-fold). Expression of 11 PKS genes (pks3, pks4, pks5, pks11, pks18, pks19, pks23, pks25, pks27, pks31, and pks32) was not detected under these conditions (Fig. 3C).
Sequence analysis of PKS22 and PKS29
Due to their high induction during fungal-fungal interactions but no induction during pigmentation, pks22 and pks29 were selected for functional characterization, as the best candidates for biosynthesis of polyketides involved in microbial interactions. The pks22 and pks29 open reading frames (ORFs) were predicted to encode polypeptides composed of 2490 and 2378 aa residues, respectively. The conserved domain analyses of PKS22 and PKS29 predicted the presence of functional domains KS, AT, DH, ER, KR and ACP similar to the typical HR type polyketide synthase domains, and an additional methyltransferase (MT) domain (Fig. S1), which is in line with previous results from a phylogenetic analysis that placed PKS22 and PKS29 in a HR polyketide group25. The antiSMASH analysis showed that pks22 was a core biosynthetic gene clustered with additional biosynthetic genes putatively encoding an aldo/keto reductase, a tubulin-tyrosine ligase, a FAD-dependent oxidoreductase, an AMP-binding protein and a MFS transporter in the 48.7 kbp long BGC117. Similarly, pks29 was a core biosynthetic gene clustered with additional biosynthetic genes putatively encoding a FAD-dependent oxidoreductase, an AMP-dependent synthetase and ligase, a short-chain dehydrogenase/reductase, a copper type II ascorbate-dependent monooxygenase, a cytochrome b561, a flavin-containing amine oxidoreductase, a tryptophan halogenase, two MFS transporters and a transcription factor in the 54.2 kb long BGC130 (Table 1, Fig. S2). BlastP analysis using the PKS22 aa sequence showed highest similarity (49% identity) with PKSs from Rosellinia necatrix (GAP88125) and Podospora anserina (CDP29160) followed by 44% identity with a lovastatin nonaketide synthase from Escovopsis weberi (KOS22576). The PKS29 aa sequence showed highest similarity (53% identity) with the same PKS from R. necatrix (GAP88125) as PKS22, followed by 50% identity with the lovastatin diketide synthase LovF from Madurella mycetomatis (KXX72797) and 43% identity with a PKS from Glarea lozoyensis (XP_008078275) known to be involved in biosynthesis of the antifungal lipohexapeptide pneumocandin32.
Generation and validation of gene deletion mutants
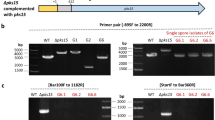
Gene deletion mutants of pks22 and pks29 were generated by replacing the respective orf with the hygB selection cassette by homologous recombination using Agrobacterium tumefaciens-mediated transformation (ATMT). More than 75 hygromycin resistant C. rosea colonies were obtained for each transformation on selection plates containing hygromycin (200 μg/ml) and cefotaxime (300 μg/ml). The individual transformants were sub-cultured on fresh selection plates and were subjected for mutant validation to confirm that the deletion cassette was inserted at the target locus using PCR with primers located within the hygB cassette together with primers located upstream and downstream of the construct (Fig. S3A) as described in our previous studies11,33. The PCR fragments of expected size were amplified in five and four transformants for pks22 and pks29 deletion mutants, respectively, while no amplification was observed in wild type (WT) (Fig. S3B,C), indicating that pks22 or pks29 were replaced correctly by hygB in these transformants. Furthermore, RT-PCR experiments on cDNA using primers specific to the pks22 or pks29 sequence demonstrated the complete loss of pks22 or pks29 transcript in each transformant, whereas an amplification product of desired size was found in WT strain (Fig. S3B,C). In order to confirm that the observed phenotypes were attributed to deletion of pks22 or pks29 and not to ectopic insertions, if any, of the deletion cassette, all 5 independent deletion strains of pks22 (Δpks22A, Δpks22B, Δpks22C, Δpks22D, Δpks22E), and 4 independent deletion strains of pks29 (Δpks29A, Δpks29B, Δpks29C, Δpks29D) were used in phenotypic analyses, unless otherwise specified.
Deletion of PKS genes resulted in phenotypic effects
A significant (P ≤ 0.004) reduction in mycelial growth of Δpks22 strains compared with WT was recorded on PDA and CZ-agar medium (Fig. 4A). However, deletion of pks29 showed no effect on mycelial growth rate (data not shown). Deletion strain Δpks22B showed a significantly lower growth rate compared with the other 4 independent Δpks22 strains, and was thus excluded from further phenotypic analyses. Furthermore, the Δpks22 and Δpks29 strains showed significantly (P ≤ 0.001) increased conidiation on CZ-agar (Fig. 4B). However, no difference in conidiation was observed between WT and deletion strains on PDB medium (data not shown). Dual culture interaction assays showed no significant differences in antagonistic ability between WT and pks22 or pks29 deletion strains against the plant pathogenic fungi A. altarnata, B. cinerea, F. graminearum or R. solani. Dual culture interactions were allowed up to 20 days and no differences in overgrowth on the prey fungi were observed between WT and deletion strains. Furthermore, a culture filtrate test was performed to assess the difference in antagonism by measuring mycelial biomass of B. cinerea or F. graminearum in culture filtrates of C. rosea WT and deletion strains. Similar to the dual culture interaction test on agar plates, no significant differences in biomass of B. cinerea or F. graminearum when grown in WT or Δpks22 liquid culture filtrates were identified (data not shown). However, a significant (P ≤ 0.024) increase in biomass of B. cinerea grown in PDB culture filtrates obtained from Δpks29 strains was found in comparison to biomass produced in WT culture filtrates (Fig. 4C), while the similar experiment against F. graminearum showed no significant differences in biomass production (data not shown). In contrast with the in vitro antagonism tests, a bioassay for F. graminearum foot rot disease on barley showed a significant (P ≤ 0.027) increase of disease severity in barley seedlings previously seed coated with Δpks29 strains as compared with seedlings from seeds coated with WT C. rosea (Fig. 4D). However, disease symptoms on seedlings from seeds coated with Δpks22 strains showed no significant difference compared with WT.

Phenotypic characterizations of C. rosea WT and deletion mutants. (A) Growth rate of WT, and pks22 deletion strains on czapek-dox (CZ) or potato dextrose agar (PDA) medium. Strains were inoculated on solid agar medium, incubated at 25 °C and the growth rate was recorded five days post-inoculation. Error bars represent standard deviation based on 4 biological replicates. (B) Conidiation of WT, pks22 and pks29 deletion strains on CZ medium 12 days post-inoculation. Conidia were harvested in equal volume of water and counted using a Bright-Line Haemocytometer as per instruction of manufacturer. Error bars represent standard deviation based on 4 biological replicates. (C) Culture filtrate test of C. rosea strains. Culture filtrates from WT and deletion strains grown in PDB were collected 10 days post-inoculation and then inoculated with a B. cinerea agar plug. Biomass production in culture filtrates was analysed by determining mycelial dry weight 4 days post-inoculation. Error bars represent standard deviation based on 4 biological replicates. (D) In vivo bioassay to test the biocontrol ability of C. rosea strains against F. graminearum foot rot disease on barley. Barley seeds were coated with C. rosea conidia, and planted in moist sand together with a F. graminearum agar plug. Seedlings were harvested three weeks post-inoculation and disease symptoms were scored on 0–4 scale. The experiment was performed in five biological replicates with 12–15 plants in each replicate. Different letters indicate statistically significant differences (P ≤ 0.05) within the experiments based on the Fisher’s exact test.
Secondary metabolite analyses led to the identification of novel compounds
In order to investigate the involvement of PKS22 or PKS29 in secondary metabolite production, culture filtrates of C. rosea strains were analyzed by ultra-high-performance liquid chromatography mass spectrometry (UHPLC-MS). Our data showed that four metabolites detected at the retention time of 90–98 seconds (s) with m/z values (H+) 256.1907 (compound 1), 270.2063 (compound 2), 258.2057 (compound 3), and 272.2222 (compound 4) in samples from WT and Δpks29 strains were not detected from Δpks22 strains, suggesting that the Δpks22 strains lost the ability to produce compounds 1–4 (Fig. 5A). In addition, UHPLC-MS analysis showed a significant (P = 0.001) 50% reduction of a compound eluted at around 70 s retention time with m/z (Na+) 279.1934 (compound 5) in culture filtrates from Δpks29 strains compared with WT and Δpks22 strains (Fig. 5A,B).

Secondary metabolite analysis of C. rosea WT and deletion strains. (A) Extracted ion chromatograms of compounds 1–5 obtained by UHPLC-MS analysis of WT, and deletion strains culture filtrates. The experiment was performed in three biological replicates. (B) Relative quantification of compounds 5 in culture filtrates of C. rosea WT and deletion strains. Quantification was done by UHPLC-MS using representative extracted-ion chromatograms for the compound. Sample areas are presented as relative area units. Error bars represent standard deviation based on three biological replicates. (C) Proposed structures of compounds 1–4, and key HMBC (solid arrows) and ROESY (dashed arrow) correlations for structure determination of compound 1, along with partial structures of TMC-171A-C, TMC-154, and TMC-151A-F (Kohno et al.34; Kohno et al.35).
According to the mass spectrometry (MS) data, compounds 1–4 had molecular formulae C14H25NO3, C15H27NO3, C14H27NO3, and C15H29NO3, respectively, with unsaturation index three, three, two and two, respectively. Compounds 1 and 2 were isolated by preparative high-performance liquid chromatography (HPLC) (0.25 mg and 0.32 mg, respectively), and their structures were determined by nuclear magnetic resonance (NMR) (Table 2), MS and MS/MS. Using correlation spectroscopy (COSY) and total correlation spectroscopy (TOCSY) NMR experiments, two spin-systems, A and B, were identified in compound 1. Spin-system A comprised two adjacent methylene groups and a heteroatom linked hydrogen, possibly an amide proton, i.e. tentatively -CH2-CH2-NH-CO-. Spin-system B contained signals from three methyl groups (one sp2 linked, two methine linked, and one methylene linked), one sp2 linked hydrogen, two methine groups and two methylene groups, joined to form a CH3-CH2-CH(CH3)-CH2-CH(CH3)-CH=C-CH3 motif. Spin-systems A and B were shown to be connected by heteronuclear multiple bond correlation (HMBC) experiments, by a cross-peak from the sp2 linked methyl group of spin-system B to a carbonyl at δC 169.7, which also had a cross-peak to the methylene group in the proposed amide-end of spin-system A (Fig. 5C). After comparison with the molecular formula C14H25NO3, it was deduced that a COOH group also should be included in the structure, which only could be placed on the other end of spin-system A. This was supported by HMBC cross-peaks from the two methylene groups of spin-system A to a carbonyl carbon at δC 173.8. The C-2/C-3 double bond was determined to have E-configuration by a rotating-frame Overhauser spectroscopy (ROESY) cross-peak between H3-9 and H-4, resulting in the proposed structure of compound 1 (Fig. 5C), for which we propose the name Clonorosein A. The structure for 1 was supported by MS/MS analysis, which yielded one major fragment ion with m/z 167.1426, in agreement with an acylium ion formed by cleavage of the proposed amide bond. The NMR data for compound 2 was very similar to the data for 1, the only major difference was the presence of one extra methylene group in spin-system A, i.e. -CH2-CH2-CH2-NH-CO-, resulting in the proposed structure for the compound 2 (Fig. 5C), which was named Clonorosein B. Compounds 1 and 2 both showed positive specific rotation (+48 and +33, respectively), but the absolute configuration was not determined experimentally for the compounds. Clonorosein A and B are both new structures, but the methylated octenoyl moiety of A and B is present in reduced form in the compounds TMC-171A, B, C, and TMC-15434,35 (Fig. 5C). These compounds were isolated from different strains of C. rosea (formerly Gliocladium roseum/G. catenulatum) TC 1304 and TC 1282 and it is likely that Clonorosein A and B share the configuration at C-4 and C-6 with the configuration at C-16 and C-18 in TMC-171A, B, and C, which was determined to be S34. Thus, the 4S, 6S configuration was proposed for Clonorosein A and B.
Compounds 3 and 4 were proposed, by the respective molecular formulae, to differ from Clonorosein A and B, by the presence of two extra hydrogen atoms (Fig. 5C). Compounds 3 and 4 were not isolated in sufficient amounts or purity for allowing detailed analysis by NMR, but for both compounds the signals from the olefinic H-3 and the sp2 linked methyl group were absent, indicating that the difference between Clonorosein A and compound 3, and between Clonorosein B and compound 4, was the absence of the C-2/C-3 double bond in the fatty acyl part of the molecules. This was also in accordance with MS/MS data. In analogy with the configuration of C-4 and C-6 in compounds 1 and 2, C-2, C-4 and C-6 in compounds 3 and 4 were all tentatively assigned to have S-configuration after comparison with the compounds TMC-151A-F produced by C. rosea TC 128035. Compounds 3 and 4 were given the names Clonorosein C and D, respectively.
The molecular formula of compound 5, C15H28O3, obtained by MS, corresponded to an unsaturation/ring index of two, and could fit many compounds including e.g. a monounsaturated hydroxy substituted fatty acid. Unfortunately, we were not able to isolate compound 5 of sufficient quantity or purity for structure determination by NMR. A similar compound with the same m/z was eluted at 74 s (Fig. 5A), and the mass spectrum and MS/MS data for this compound was very similar to the data for the compound eluting at 70 s. These two compounds are thus presumably of similar structures, and may differ in e.g. relative configuration or perhaps position of substituents.
Clonorosein A and Clonorosein B exhibited antifungal activity
Clonorosein A and B isolated and purified from C. rosea culture filtrates were used to test their antifungal activity by analyzing conidial germination and germ tube development of B. cinerea and F. graminearum. Both compounds significantly inhibited conidial germination (P ≤ 0.028) and germ tube growth (P ≤ 0.042) of B. cinerea at a concentration of 64 µg/ml compared with the control (Fig. 6A,B). The two compounds showed an even more severe effect against F. graminearum with a >50% (P = 0.001) reduction in conidial germ tube length already at 64 µg/ml compared with the control (Fig. 6C). However, no significant difference was found in F. graminearum conidial germination at any tested concentration (data not shown). The antifungal effects of Clonorosein A and B did not appear to be dose-dependent, as no significant differences were observed between the concentrations 64 and 128 µg/ml.

Antifungal activity of compound 1 and compound 2 extracted from C. rosea against plant pathogens B. cinerea and F. graminearum. Conidial suspensions in half-strength potato dextrose broth were inoculated in 96-well microtiter plates containing compound 1 or compound 2. Frequency of germinating conidia was determined 8 hour post-inoculation by counting the number of germinating and non-germinating conidia, while germ tube length was measured using ImageJ software. (A) B. cinerea conidia germination, (B) B. cinerea germ tube length, (C) F. graminearum germ tube length. Abbreviation: 1, compound 1; 2, compound 2. Concentrations of compound are indicated below the bar. Error bars represent standard deviation based on three biological replicates. Different letters indicate statistically significant differences (P ≤ 0.05) within the experiments based on Fisher’s exact test.
Discussion
As an opportunistic, necrotrophic mycoparasite with broad host range, C. rosea has evolved strategies to compete with and antagonise other fungi by producing bioactive secondary metabolites36. This contributes to its usefulness as a biological control agent to protect crop plants against fungal and other diseases in agricultural production systems20. This ability for antibiosis is reflected by the presence of a rich repertoire of genes encoding enzymes associated with secondary metabolite production in the C. rosea genome, including 31 PKSs, one PKS-NRPS hybrid, 17 NRPSs, and 8 terpene synthases25. An evolutionary analysis showed selection for increased PKS gene copy number in C. rosea compared with other hypocrealean fungi such as the mycoparasitic T. atroviride and T. virens (18 putative PKS genes), saprotrophic T. reesei (11 putative PKS genes) and plant pathogenic F. graminearum, F. oxysporum f. sp. lycopersici, F. solani or F. verticillioides (12–16 PKS genes)25. Our work shows that most PKS genes in C. rosea (75%) are located in predicted secondary metabolite biosynthetic clusters, which is a higher proportion compared with the mentioned Trichoderma spp. (50%)37, indicating that the increase of PKS gene copy number in C. rosea has specifically involved genes related with secondary metabolite production.
Our gene expression data further suggest that antagonism is an important driving force behind the selection for increased PKS gene copy number in C. rosea. First, this is supported by the positive correlation between fungal growth inhibitory activity of different culture filtrates and the number and fold-change of expressed PKS genes, with PDB exhibiting the highest growth inhibiting activity and the highest number of expressed PKS genes. The fact that all tested fungal plant pathogens, A. alternata, B. cinerea, F. graminearum and R. solani, displayed reduced growth in C. rosea PDB, ME, SMS and SNB culture filtrates suggests production of compounds with broad spectrum activity, or production of a mix of compounds with different biotic activities. The exact identity of the growth media constituents that trigger PKS gene expression in C. rosea is not known, but it is reported that the source and level of carbon and nitrogen, and pH can influence production of secondary metabolites in fungi38. In contrast, lack of growth inhibitory activity towards any fungus in CZ culture filtrate may suggest that the higher pH (7.3 ± 0.2) and nitrogen (2% NaNO3), and sucrose as the sole carbon source (3% sucrose) in CZ may not represent optimal conditions for PKS gene expression and consequently secondary metabolite production in C. rosea. In fact, C. rosea lowered the pH in CZ medium from 7.2 to 5.5 during incubation, which may explain the increased growth of A. alternata and R. solani in the culture filtrate compared with the CZ control. This result is in line with previous data from F. graminearum where higher number of PKS genes were expressed in PDB medium, rice agar and corn meal medium compared with CZ medium7. Similar results are reported previously from the functional analysis of bik genes responsible for the biosynthesis of the red pigment bikaverin in F. fujikuroi39,40. Expression of bik genes was highly suppressed at neutral and alkaline pH, and at high nitrogen concentration, while their expression was increased at acidic pH and lower nitrogen concentration. The nitrogen and pH-mediated regulation of gene expression was consistent with the production of the bikaverin39,40.
Secondly, 45% of all PKS genes in C. rosea are induced in response to antagonistic interactions with B. cinerea or F. graminearum. PKS genes are also reported to be induced in T. atroviride, T. reesei and T. harzianum during interaction with R. solani and Sclerotinia sclerotiorum, respectively41,42. Data from C. rosea interacting with B. cinerea or F. graminearum revealed both common and species-specific responses in PKS gene expression. This is in line with a recent transcriptome analysis of C. rosea during interactions with B. cinerea or F. graminearum where genes predicted to encode proteins particularly involved in membrane transport and secondary metabolite biosynthesis were differentially expressed towards the two fungi26. The pks9 gene was reported by Nygren et al.26 to be induced specifically against B. cinerea, while it is repressed during interactions with both fungi in the current work. One important difference between these studies that may explain this contradiction is the fact that Nygren et al.26 sampled the hyphal front of C. rosea 24 hours after contact with the antagonists, while samples were taken at contact (24 hours earlier) in the current work.
It is possible that specificity in expression of PKS genes towards different fungal prey species represents an adaptation of the mycoparasitic attack driven by intrinsic differences between the fungal preys. Alternatively, specificity in C. rosea PKS gene expression during fungal-fungal interactions may be influenced by the fungal prey. The mycotoxin deoxynivalenol produced by Fusarium spp. was reported to suppress NAGase gene expression in T. atroviride43, indicating that fungal prey species may have evolved effectors that interfere with the activity of their parasites. In this study, the pks19 gene is specifically suppressed in C. rosea interacting with F. graminearum, but not with B. cinerea. This is interesting, as pks19 is the core gene in a putative depudicin gene cluster in C. rosea, reported to be conserved among distantly related groups of fungi44. Depudicin was shown to be an inhibitor of histone deacetylases (HDACs)31,45 that together with histone acetyltransferases (HATs) influence expression of genes involved in various processes, including secondary metabolite production46,47, through chromatin modifications. Chromatin modification-mediated gene expression has previously been suggested in mycoparasitic Trichoderma spp. during in vitro interaction with the plant pathogenic fungus R. solani41.
However, the fact that all PKS genes in C. rosea are expressed under the standard laboratory conditions used in this study indicates a role for polyketide compounds in normal growth and development in C. rosea. Many PKS genes in T. reesei were also reported to be highly expressed during vegetative growth, suggesting diverse functional roles of polyketides48. The induction of 19 PKS genes during ageing and pigmentation of C. rosea also indicate that certain PKS genes encode enzymes involved in pigment biosynthesis, as shown for the pks4 gene in T. reesei12. Induced expression of PKS genes during pigmentation and pigment secretion were shown for several different fungi including B. cinerea49, F. graminearum7, F. fujikuroi39,40, M. purpureus50 and T. reesei41,51. For instance, C. rosea is predicted to harbour orthologs of the sorbicillinoid family-type yellow pigment sorbicillin (pks2 and pks12) gene cluster52,53, conserved and functionally active in several distantly related fungi such as Acremonium chrysogenum, P. chrysogenum, T. reesei, Chaetomium globosum and Colletotrichum graminicola30,51. In addition, C. rosea PKS1 shares similar protein domain organization (SAT-KS-AT-PT-ACP) and was shown to be phylogenetically close to the AptA PKS of A. nidulans25, known to be involved in biosynthesis of the anthraquinone-type yellow pigment asperthicin54. All three genes (pks1, pks2 and pks12) were highly induced during yellow pigment secretion in the current study suggesting biosynthesis of sorbicillin-like and asperthicin-like compounds in C. rosea.
Based on their induction during growth in PDB and SNB media, during antagonistic interactions, but not during pigmentation, we selected pks22 and pks29 for functional characterization under the hypothesis that they encode PKS proteins that biosynthesize polyketide compounds with biotic activities. Deletion of pks22 allowed us to identify several previously unknown secondary metabolites that were named Clonorosein A-D. By determining the structures of Clonorosein A and B using NMR and MS we showed that these compounds are methyl substituted octenoic or octanoic acid derivatives. The backbone of Clonorosein is likely to be assembled by PKS22, which is subsequently methylated on carbon atoms number 2, 4 and 6, possibly by the MT domain present in PKS22. Similar polyketides, i.e. fatty acid derivatives with methyl substitutions on every second carbon, have previously been described in the genus Clonostachys34,55,56,57. Taken together, deletion of pks22 combined with the identification of Clonorosein A-D as methylated fatty acid derivatives further supports that PKS22 is a PKS that is involved in the biosynthesis of Clonorosein.
A bioactivity test further proved the antifungal activity of purified Clonorosein A and B against B. cinerea and F. graminearum that fits well with the increased pks22 gene expression during interactions with the same fungi, suggesting a role of PKS22 in fungal-fungal interactions. The lack of observable phenotypic differences between Δpks22 and WT strains with regard to in vitro antagonism and in planta biocontrol may be explained by production of additional compounds or enzymes with antifungal activity by C. rosea that effectively compensate for the absence of Clonorosein.
Deletion of pks29 resulted in strains with 50% reduced ability to produce a compound with the molecular formula C15H28O3, suggesting that PKS29 is involved in the biosynthesis of this compound. However, the fact that 50% of the compound is still produced by the Δpks22 strains suggests the involvement of an additional gene in the biosynthesis of compound 5. There is an example in the human pathogen Aspergillus fumigatus where two BGCs are involved in the biosynthesis of an endocrocin polyketide through two distinct routes58. PKS29 was in fact reported to be part of a phylogenetic group with several, closely related paralogous PKSs in C. rosea25, possibly involved in producing compound 5 or the derivate of compound 5 eluted at 74 s with the same m/z.
The significant increase in B. cinerea biomass when grown in Δpks29 culture filtrates suggests a role of PKS29 and compound 5 in in vitro antagonism. This is consistent with gene expression data where pks29 showed the highest induction among all PKS genes in PDB medium. PKS29 is further required for full biocontrol ability of C. rosea against foot rot disease on barley caused by F. graminearum, again consistent with the induction of pks29 expression in C. rosea during interaction with B. cinerea and F. graminearum.
In addition to their role in antagonism, deletion of pks22 and pks29 also resulted in several phenotypic effects related to growth and reproduction in C. rosea. This plausibly is related either with the physiological costs of secondary metabolite production which otherwise are used for growth and conidiation, or with disturbance in metabolite production machinery in Δpks22 and Δpks29 deletion strains that in turn prompted the alteration in growth and conidiation59. Furthermore, our data from phenotypic analysis where deletion strains showed different phenotypic effects on PDA and CZ, with the exception of Δpks22 that showed reduced growth rate on both media, suggested that the functions of PKS22 and PKS29 are culture medium dependent and confirm the gene expression pattern where expression of pks22 or pks29 was significantly different in PDB and CZ.
The involvement of PKSs in normal mycelial growth and development, production of sexual and asexual spores, and pigmentation have previously been shown in filamentous fungi. For example disruption of GRS1 or PKS2 inhibited mycelial growth, while disruption of AUR1 and its homologue Fsr1 affected perithecium pigmentation in F. graminearum and F. verticillioides6,7. Similarly, fluP disruption mutants in A. parasiticus had significant reduction in mycelial growth and sporulation60. In T. reesei and Sordaria macrospora, loss and over-expression of pks4 resulted in disrupted sexual development8,12. Our results did not indicate any role of PKS22 or PKS29 in pigmentation, which is consistent with the lack of induction of either gene during pigmentation.
Given the large numbers of PKS genes in mycoparasitic fungi, surprisingly little is known about their functions in governing microbial interactions resulting in biological control of plant diseases. We here show that PKS gene expression in C. rosea is regulated both by nutrient conditions and by interactions with other fungi. Our results from antagonism assay using culture filtrate test showed that pks29 is involved in antagonism against B. cinerea. Furthermore, we identified two previously unknown polyketide compounds from C. rosea with antifungal activity, Clonorosein A and B.
Methods
Fungal strains and culture condition
Cultures of C. rosea IK726 WT and deletion mutants derived from it, A. alternata TUCIM 3594, B. cinerea strain B05.10, F. graminearum strain PH1, and R. solani strain SA1were maintained on PDA (Sigma-Aldrich, St. Louis, MO) plates at 25 °C.
Sequence analysis
The protein accession numbers of PKSs of C. rosea were retrieved from Karlsson et al.25, and their aa and nucleotide sequences were retrieved from GenBank at NCBI. The PKS protein domain architectures were analysed and annotated based on their aa sequence using simple modular architecture research tool (SMART)61 and conserved domain database (CDD)62. Antibiotic and secondary metabolite analysis shell (antiSMASH) version 4 web server http://antismash.secondarymetabolites.org was used to identify putative PKS BGCs in C. rosea genome63,64. The search was performed on annotated genome sequence following procedure described by the developer63,64. For each predicted ORF in clusters, a BlastP search was done using predicted aa sequences against C. rosea protein sequences to identify PKS gene part of the predicted biosynthetic gene cluster, and also to identify their predicted aa sequences.
Analysis of C. rosea antagonism in different culture media
Five different culture media: CZ (Sigma-Aldrich, St. Louis, MO); PDB (Sigma-Aldrich, St. Louis, MO), ME (Duchefa Biochemie, Haarlem, Netherlands); SMS, and SNB with different carbon and nitrogen source and pH were used to study antagonistic behaviour of C. rosea under different nutritional conditions (Table S3). C. rosea conidia (1 × 107conidia) isolated from a 2 week old plate were used to inoculate 1000 ml flasks containing 250 ml of culture medium. Flasks were incubated at 25 °C on a rotary shaker (100 rpm), and culture filtrates were obtained 4 dpi by removing fungal mycelia using vacuum filtration. Culture filtrates were passed through 0.45 µM cellulose acetate membrane syringe filters (Sarstedt Aktiengesellschaft & Co., Nümbrecht, Germany) to remove the mycelial debris before use. Harvested mycelia were washed with distilled water, frozen immediately in liquid nitrogen and stored in −70 °C for RNA extraction to use for gene expression analysis. To measure the mycelial biomass, a five mm agar plug of A. alternata, B. cinerea, F. graminearum or R. solani was inoculated into 50 ml flasks containing 10 ml of culture filtrate and were incubated at 25 °C on a rotary shaker with constant shaking. Fungus inoculated into respective fresh culture medium was used as control. Mycelial biomass was harvested 4 dpi using vacuum filtration, dried at 65 °C and weighed to determine the mycelial dry weight. The experiment was performed in five biological replicates.
Quantitative reverse transcription polymerase chain reaction
For gene expression analysis in liquid CZ, PDB and SNB culture media, C. rosea mycelia were cultivated and harvested 4 dpi as described in the previous section. C. rosea inoculated into CZ medium was used as a control treatment. For gene expression analysis during dual culture interactions with F. graminearum and B. cinerea, C. rosea was grown and harvested as described previously11,27. Mycelium harvested at same stage from C. rosea confronted with C. rosea (Cr–Cr) was used as control treatment. For gene expression analysis during pigmentation, a 5 mm agar plug of C. rosea was inoculated on PDA medium covered with cellophane membrane for easy harvesting of fungal mycelia, and incubated at 25 °C in dark to avoid conidiation. Mycelia ware harvested 10 dpi as at this stage production of yellow pigment was apparent on bottom of agar plates. Four days old C. rosea culture plates, incubated at the same conditions were used as control treatment as no pigmentation was observed at this stage.
Harvested mycelia were flash frozen in liquid nitrogen, freeze-dried (VirTis Sp scientific, Warminster, PA) at −95 °C, and then homogenized into a fine powder using Precellys 24 lysis and homogenization (Bertin Technologies, France). Immediately after homogenization, total RNA was extracted using Qiagen RNasy plant mini kit (Qiagen, Hilden, Germany). For cDNA synthesis, 1000 ng of total RNA, after treating with DNase I (Fermentas, St-Leon-Rot, Germany), was reverse transcribed (RT) in a total volume of 20 µl using iScript™ cDNA Synthesis Kit (Bio-Rad, Hercules, CA). The transcript levels were quantified by quantitative polymerase chain reaction (RT-qPCR) using gene specific primer pairs (Table S4A) in a 20 µl reaction mix as described previously65. Primer amplification efficiency of each primer pair was determined by amplifying serial dilutions of C. rosea IK726 genomic DNA. Melt curve analysis was performed after the qPCR reactions to confirm that the signal was the result from a single product amplification. Cycle threshold (Ct) values were log transformed, mean centered and autoscaled as described previously66. Relative expression levels for the target gene in relation to actin and β-tubulin11,27,65 were calculated using the 2−ΔΔCt method67. The gene expression experiments were performed in five biological replicates, and each replicate had two technical replicates.
Construction of deletion vector, transformation and mutant validation
Genomic DNA was isolated following a CTAB-based protocol68. Dream Taq DNA polymerase (Thermo Fisher Scientific) was used for PCR amplification of ~1 kbp 5′ -flank and 3′ -flank regions of the pks22 and pks29 orfs from genomic DNA using primer PKS22ups F/PKS22ups R, PKS22ds F/PKS22ds R; and PKS29ups F/PKS29ups R, PKS29ds F/PKS29ds R, respectively (Table S4B). Gateway entry clones of the purified 5′-flank and 3′-flank PCR fragments were generated as described by the manufacturer (Invitrogen, Carlsbad, CA). The entry clone of hygromycin casette (hygB) constructed during our previous studies was used69,70. The deletion vector was constructed by performing the gateway LR recombination reaction as described previously11,27 and following manufacturer’s instructions (Invitrogen, Carlsbad, CA).
ATMT was performed based on a previous protocol for C. rosea71. Transformed strains were selected on plates containing hygromycin (200 μg/ml) and cefotaxime (300 mM). Putative transformants were tested for mitotic stability, and were purified by two rounds of single spore isolation27,69. Validation of homologous integration of the deletion cassette in putative transformants was performed using a PCR screening approach as described before11,27,33,69,70 using primers specific to the hygB cassette (Hyg F/Hyg R) in combination with primers specific to sequences flanking the deletion cassette (PKS22ko F/PKS22ko R or PKS29ko F/PKS29ko R) (Table S4B, Fig. S3A). Semi-quantitative RT-PCR analysis was conducted on WT and deletion strains using RevertAid premium reverse transcriptase (Fermentas, St.Leon-Rot, Germany) and primer pairs specific to pks22 or pks29 (Table S4B).
Phenotypic analysis
All phenotypic analyses were performed using PDA and CZ media unless otherwise specified. 3 mm agar plug from the growing edge of C. rosea strains was inoculated onto PDA or CZ plates, incubated at 25 °C, and mycelial growth rate and colony morphology was recorded daily. For conidiation analysis, conidia were harvested from a 12 days old plate in 10 ml distilled water, and filtered through Miracloth to remove the mycelial debris. Conidial concentration was determined under the microscope using a bright line haemocytometer (Sigma-Aldrich, St. Louis, MO). Antagonistic behaviour of C. rosea WT and deletion strains was determined on PDA and CZ using an in vitro dual culture assay on solid medium, and a culture filtrate test in liquid culture medium. The in vitro plate confrontation assay was performed against A. altarnata, B. cinerea, F. graminearum or R. solani following procedures described previously27. Mycelial growth of C. rosea and the prey fungi was recorded daily. The culture filtrate test against B. cinerea or F. graminearum was performed as described previously11,27.
The biocontrol activity of C. rosea WT and deletion strains was assessed in an in vivo bioassay for F. graminearum foot rot disease on barley using a sand seedling test as described previously11,27. In brief, barley seeds were surface-sterilized with 2% NaOCl and air-dried on the benchtop of a laminar air-flow. The seeds were then coated with the conidial suspensions (1 × 108 conidia/ml in water) of C. rosea WT or deletion strains for 30 minutes in a rotary shaker (125 rpm). Seeds incubated with sterile distilled water were used as a control. The seeds were then sown in pre-wetted sand in the 5 × 5 × 5 cm plastic pots (3 seeds/pot). For pathogen inoculation, a 5 mm agar plug of fresh F. graminearum mycelium was placed close to the seeds in the plastic pot. A PDA plug without F. graminearum was used as a control. The pots were then incubated in a growth chamber with a photoperiod of 12 h light (150 µmol m2 s−1 light intensity)/12 h dark, 70% ± 5% relative humidity, and 15 ± 1 °C11,27. Seedlings were harvested 3 weeks post-inoculation, and disease symptoms were scored on a 0 to 4 scale as described before11,27. The experiment was performed in five biological replicates with 15 plants per replicate.
Secondary metabolite analysis
For secondary metabolite analysis, C. rosea WT and deletion strains were cultivated in 1000 ml flask containing 200 ml PDB medium for 15 days. Culture filtrates were collected by removing mycelial debris by centrifugation (13000 rpm for 5 min). Culture filtrates (injection volume 0.5 µL) were analyzed by UHPLC-MS on a reversed phase column (2.1 × 50 mm, 1.5 μm, Accucore Vanquish, Thermo Scientific, Waltham, MA, USA) using a gradient of acetonitrile (MeCN) in water, both with 0.2% formic acid (10–95% MeCN in 3 min, 95% MeCN for 1.2 min, at 0.9 mL min−1). The MS was operated in positive mode with scanning of m/z 50–1500, and the mass spectra were calibrated against sodium formate clusters. MS-MS was performed on the same instrument with 2 amu isolation width and 24 eV fragmentation energy.
For isolation and identification of secondary metabolites, cell-free culture supernatants (total volume 1000 mL) were fractionated on 4 × 10-g C-18 SPE columns. Following sample loading, each column was washed with 50 mL 25% MeCN in water and then eluted with 50 mL 95% MeCN in water. The combined 95% MeCN extract was dried under reduced pressure and re-dissolved in 4 mL 60% MeCN in water, and fractionated (4 × 1 mL injected) by gradient preparative reversed phase HPLC (21.2 × 100 mm, 5 μm, Hypersil Gold, Thermo Scientific, Waltham, MA, USA) using a gradient of MeCN in water (10–95% MeCN in 10 min, followed by 10 min at 95% MeCN, at 10 mL min−1). Fractions were analyzed by UHPLC-MS as above, and fractions containing the different target compounds, were pooled and dried under reduced pressure. The dried fractions were then subjected to isocratic reversed phase HPLC (fraction region A: 35% MeCN in water, 0.2% formic acid, 10 mL min−1; fraction region B: 47.5% MeCN in water, 0.2% formic acid, 10 mL min−1; column as above), and fractions containing the selected compounds, as indicated by UHPLC-MS (as above), were collected and dried in a vacuum centrifuge, and their structures were studied by NMR and MS.
1H and 13C NMR data were acquired in CDCl3 on a Bruker Avance III 600 MHz NMR spectrometer (Bruker Biospin GmBH, Rheinstetten, Germany) equipped with a 5-mm cryo-probe (1H, 13C, 15N, 31P). Standard pulse sequences supplied by Bruker were used for acquisition of 1D 1H, COSY, TOCSY, DEPT-HSQC, HMBC, and ROESY NMR data. Chemical shifts were determined relative to internal chloroform (δC 77.23; δH 7.27). UHPLC-MS and UHPLC-MS-MS were performed on an Agilent 1290 Infinity II UHPLC (Agilent, Palo Alto, CA, USA) connected to a Bruker maXis Impact ESI Q-TOF MS (Bruker Daltonic GmbH., Bremen, Germany). Preparative HPLC was run on a Gilson 306/306 pump system (Gilson Inc., Middleton, WI, USA) with a Gilson 119 UV/VIS detector monitoring at 210 nm. Fractions were collected with a Gilson 204 fraction collector and collection was done in polypropylene 2.2 ml square well plates (VWR, Radnor, PA, USA). MeCN of HPLC gradient grade (Sigma-Aldrich, St. Louis, MO, USA) and deionized filtered water (Millipore, Billerica, MA, USA) were used for preparation of mobile phases. Polarimetry was performed on a Perkin Elmer 341 polarimeter (λ 589 nm, path length 10.0 cm, 20 °C) with compounds dissolved in methanol.
Biological activity assay
Pure compounds dissolved in MeOH were transferred into 96-well microtiter plates, and solvents were evaporated in a fume hood at room temperature. The bioactivity of the compounds was tested at eight concentrations, 1, 2, 4, 8, 16, 32, 64, and 128 µg/ml. A suspension of 5 × 103 B. cinerea or F. graminearum spores in 100 µl of half PDB was added in each well, and plates were incubated at 25 °C temperature in dark. To evaluate the effects of traces of residual methanol that may present in well, five concentration of methanol corresponding to 1% 2%, 4%, 8%, and 16% was tested for its effect on conidial germination and germ tube growth of B. cinerea or F. graminearum. A spore suspension in half PDB only was used as control treatment. After 8 hour of incubation, photographs of the germinating conidia were recorded in a Leica DM5500M Microscope at 10 X or 20 X magnification using a Leica DFC360FX digital camera (Wetzlar, Germany). Frequency of conidial germination was determined by counting the number of germinating and non-germinating conidia, while length of the germ tube was measured using ImageJ software72. Conidia were counted as germinated if they had germ tubes at least as long as the length of the conidia. Abnormal growth and morphological differences such as lysis of the tips were also noted. Two hundred to three hundred conidia were counted and length of their germ tube was measured for each replicate. The experiment was performed in three biological replicates.
Statistical analysis
All the statistical analyses in this study were performed using Minitab 16 statistical software or Statistica version 10 (Stat-Soft, Tulsa, OK). Significant differences among the treatments were tested using one-way analysis of variance (ANOVA) while pairwise comparisons were made using Fisher’s exact test or Tukey-Kramer test with 95% level of confidence.
References
Staunton, J. & Weissman, K. J. Polyketide biosynthesis: a millennium review. Nat Prod Rep 18, 380–416 (2001).
Fujii, I. Functional analysis of fungal polyketide biosynthesis genes. J Antibiot 63, 207–218 (2010).
Keller, N. P., Turner, G. & Bennett, J. W. Fungal secondary metabolism - from biochemistry to genomics. Nat Rev Microbiol 3, 937–947 (2005).
Chooi, Y. & Tang, Y. Navigating the fungal polyketide chemical space: from genes to molecules. J Org Chem 77, 9933–9953 (2012).
Keller, N. P. Translating biosynthetic gene clusters into fungal armor and weaponry. Nat Chem Biol 11, 671–677 (2015).
Studt, L., Wiemann, P., Kleigrewe, K., Humpf, H.-U. & Tudzynski, B. Biosynthesis of Fusarubins accounts for pigmentation of Fusarium fujikuroi perithecia. Appl Environ Microbiol 78, 4468–4480 (2012).
Gaffoor, I. et al. Functional analysis of the polyketide synthase genes in the filamentous fungus Gibberella zeae (anamorph Fusarium graminearum). Eukaryot Cell 4, 1926–1933 (2005).
Schindler, D. & Nowrousian, M. The polyketide synthase genepks4 is essential for sexual development and regulates fruiting body morphology in Sordaria macrospora. Fung Genet Biol 68, 48–59 (2014).
Utermark, J. & Karlovsky, P. Role of zearalenone lactonase in protection of Gliocladium roseum from fungitoxic effects of the mycotoxin zearalenone. Appl Environ Microbiol 73, 637–642 (2007).
Kosawang, C. et al. Zearalenone detoxification by zearalenone hydrolase is important for the antagonistic ability of Clonostachys rosea against mycotoxigenic Fusarium graminearum. Fungal. Biol 118, 364–373 (2014).
Dubey, M. K., Jensen, D. F. & Karlsson, M. An ATP-binding cassette pleiotropic drug transporter protein is required for xenobiotic tolerance and antagonism in the fungal biocontrol agent Clonostachys rosea. Mol Plant-Microbe Interact 27, 725–732 (2014).
Atanasova, L., Knox, B. P., Kubicek, C. P., Druzhinina, I. S. & Baker, S. E. The polyketide synthase genepks4 of Trichoderma reesei provides pigmentation and stress resistance. . Eukaryot Cell 12, 1499–1508 (2013).
Schroers, H. J., Samuels, G. J., Seifert, K. A. & Gams, W. Classification of the mycoparasite Gliocladium roseum in Clonostachys as C. rosea, its relationship to Bionectria ochroleuca, and notes on other Gliocladium-like fungi. Mycologia 91, 365–385 (1999).
Knudsen, I. M. B., Hockenhull, J. & Jensen, D. F. Biocontrol of seedling diseases of barley and wheat caused by Fusarium culmorum and Bipolaris sorokiniana: effects of selected fungal antagonists on growth and yield components. Plant Pathol 44, 467–477 (1995).
Jensen, B., Knudsen, I. M. B., Madsen, M. & Jensen, D. F. Biopriming of infected carrot seed with an antagonist, Clonostachys rosea, selected for control of seed borne Alternaria spp. Phytopathology 94, 551–560 (2004).
Jensen, B., Lübeck, P. S. & Jørgensen, H. J. L. Clonostachys rosea reduces spot blotch in barley by inhibiting prepenetration growth and sporulation of Bipolaris sorokiniana without inducing resistance. Pest Manag Sci 72, 2231–2239 (2016).
Møller, K., Jensen, B., Andersen, H. P., Stryhn, H. & Hockenhull, J. Biocontrol of Pythium tracheiphilum in chinese cabbage by Clonostachys rosea under field conditions. Biocontrol Sci Techn 13, 171–182 (2003).
Lahlali, R. & Peng, G. Suppression of club root by Clonostachys rosea via antibiosis and induced host resistance. Plant Pathol 63, 447–455 (2014).
Iqbal, M. et al. Evaluation of Clonostachys rosea for control of plant-parasitic nematodes in soil and in roots of carrot and wheat. Phytopathology 108, 52–59 (2018).
Jensen, D. F., Karlsson, M. & Lindahl, B. Fungal-fungal interactions: from natural ecosystems to managed plant production with emphasis on biological control of plant diseases. In: Dighton, J. & White, J. (eds) The fungal community: its organization and role in the ecosystem. CRC Press, Taylor & Francis Group, Boca Raton, 549–562 (2017).
Rodriguez, M. A., Cabrera, G., Gozzo, F. C., Eberlin, M. N. & Godeas, A. Clonostachys rosea BAFC3874 as a Sclerotinia sclerotiorum antagonist: mechanisms involved and potential as a biocontrol agent. J Appl Microbiol 110, 1177–1186 (2011).
Dong, J. Y., He, H. P., Shen, Y. M. & Zhang, K. Q. Nematicidal epipolysulfanyldioxopiperazines from Gliocladium roseum. J Nat Prod 68, 1510–1513 (2005).
Thines, E., Eilbert, F., Anke, H. & Sterner, O. Glisoprenins C, D and E, new inhibitors of appressorium formation in Magnaporthe grisea, from cultures of Gliocladium roseum. J Antibiot 51, 117–122 (1998).
Okuda, T. et al. Production of TMC- 151, TMC- 154 and TMC- 171, a new class of antibiotics, is specific to ‘Gliocladium roseum’ group. Mycoscience 41, 239–253 (2000).
Karlsson, M. et al. Insights on the evolution of mycoparasitism from the genome of Clonostachys rosea. Genome Biol Evol 7, 465–480 (2015).
Nygren, K. et al. The mycoparasitic fungus Clonostachys rosea responds with both common and specific gene expression during interspecific interactions with fungal prey. Evol Appl, https://doi.org/10.1111/eva.12609 (2018).
Dubey, M., Jensen, D. F. & Karlsson, M. The ABC transporter ABCG29 is involved in H2O2 tolerance and biocontrol traits in the fungus Clonostachys rosea. Mol Genet Genomics 291, 677–686 (2016).
Kosawang, C., Karlsson, M., Jensen, D. F., Dilokpimol, A. & Collinge, D. B. Transcriptomic profiling to identify genes involved in Fusarium mycotoxin deoxynivalenol and zearalenone tolerance in the mycoparasitic fungus Clonostachys rosea. BMC Genomics 15, 55 (2014).
He, Y. & Cox, R. J. The molecular steps of citrinin biosynthesis in fungi. Chem Sci 7, 2119–2127 (2016).
Salo, O. et al. Identification of a polyketide synthase involved in sorbicillin biosynthesis by Penicillium chrysogenum. Appl Environ Microbiol 82, 3971–3978 (2016).
Wight, W. D., Kim, K. H., Lawrence, C. B. & Walton, J. D. Biosynthesis and role in virulence of the histone deacetylase inhibitor depudecin from Alternaria brassicicola. Mol Plant-Microbe Interact 22, 1258–1267 (2009).
Chen, L. et al. Genomics-driven discovery of the pneumocandin biosynthetic gene cluster in the fungus Glarea lozoyensis. BMC Genomics 14, 339 (2013).
Dubey, M. K., Jensen, D. F. & Karlsson, M. Hydrophobins are required for conidial hydrophobicity and plant root colonization in the fungal biocontrol agent Clonostachys rosea. BMC Microbiol 14, 18 (2014).
Kohno, J. et al. TMC-171A,B,C and TMC-154, novel polyketide antibiotics produced by Gliocladium sp. TC 1304 and TC 1282. J Antibiot 52, 1114–1123 (1999).
Kohno, J. et al. Isolation and structure determination of TMC-151s: novel polyketide antibiotics from Gliocladium catenulatum Gilman & Abbott tc 1280. Tetrahedron 55, 7771–7786 (1999).
Karlsson, M., Atanasova, L., Jensen, D. F. & Zeilinger, S. Necrotrophic mycoparasites and their genomes. In: Heitman, J., Howlett, B., Crous, P., Stukenbrock, E., James, T. & Gow, N. (eds) The Fungal Kingdom. ASM Press, Washington DC, 1005–1026 (2018).
Zeilinger, S., Gruber, S., Bansal, R. & Mukherjee, P. K. Secondary metabolism in Trichoderma – chemistry meets genomics. Fung Biol Rev 30, 74–90 (2016).
Bode, H. B., Bethe, B., Höfs, R. & Zeeck, A. Big effects from small changes: possible ways to explore nature’s chemical diversity. ChemBiochem 3, 619–627 (2002).
Linnemannstöns, P. et al. The polyketide synthase genepks4 from Gibberella fujikuroi encodes a key enzyme in the biosynthesis of the red pigment bikaverin. Fungal Genet Biol 37, 134–148 (2002).
Wiemann, P. et al. Biosynthesis of the red pigment bikaverin in Fusarium fujikuroi: genes, their function and regulation. Mol Microbiol 72, 931–946 (2009).
Atanasova, L. et al. Comparative transcriptomics reveals different strategies of Trichoderma mycoparasitism. BMC Genomics 14, 121 (2013).
Yao, L. et al. Isolation and expression of two polyketide synthase genes from Trichoderma harzianum 88 during mycoparasitism. Braz J Microbiol 47, 468–479 (2016).
Lutz, M. P., Feichtinger, G., Defago, G. & Duffy, B. Mycotoxigenic Fusarium and deoxynivalenol production repress chitinase gene expression in the biocontrol agent Trichoderma atroviride P1. Appl Environ Microbiol 69, 3077–3084 (2003).
Reynolds, H. T. et al. Differential retention of gene functions in a secondary metabolite cluster. Mol Biol Evol 34, 2002–2015 (2017).
Baidyaroy, D., Brosch, G., Graessle, S., Trojer, P. & Walton, J. D. Characterization of inhibitor-resistant histone deacetylase activity in plant-pathogenic fungi. Eukaryot Cell 1, 538–547 (2002).
Brakhage, A. A. Regulation of fungal secondary metabolism. Nat Rev Microbiol. 11, 21–32 (2013).
Bannister, A. J. & Kouzarides, T. Regulation of chromatin by histone modifications. Cell Res 21, 381–395 (2011).
Metz, B. et al. Expression of biomass degrading enzymes is a major event during conidia development in Trichoderma reesei. Eukaryot Cell 10, 1527–1535 (2011).
Schumacher, J. DHN melanin biosynthesis in the plant pathogenic fungus Botrytis cinerea is based on two developmentally regulated key enzyme (PKS) encoding genes. Mol Microbiol 99, 729–748 (2015).
Shimizu, T. et al. Polyketide synthase gene responsible for citrinin biosynthesis in Monascus purpureus. Appl Environ Microbiol 71, 3453–3457 (2005).
Derntl, C., Rassinger, A., Srebotnik, E., Mach, R. L. & Mach-Aigner, A. R. Identification of the main regulator responsible for the synthesis of the typical yellow pigment by Trichoderma reesei. Appl Environ Microbiol 20, 6247–6257 (2016).
Harned, A. M. & Volp, K. A. The sorbicillinoid family of natural products: isolation, biosynthesis, and synthetic studies. Nat Prod Rep 28, 1790–1810 (2011).
Guzmán-Chávez, F. et al. Mechanism and regulation of sorbicillin biosynthesis by Penicillium chrysogenum. Microb Biotechnol 10, 958–968 (2017).
Szewczyk, E. et al. Identification and characterization of the asperthecin gene cluster of Aspergillus nidulans. Appl Environ Microbiol 74, 7607–7612 (2008).
Omura, S. et al. Roselipins, novel fungal metabolites having a highly methylated fatty acid modified with a mannose and an arabinitol. J Antibiot 52, 586–589 (1999).
Tabata, N. et al. Structure elucidation of roselipins, inhibitors of diacylglycerol acyltransferase produced by Gliocladium roseum KF-1040. J Antibiot 52, 815–826 (1999).
Tomoda, H. et al. Inhibitors of diacylglycerol acyltransferase, produced by Gliocladium roseum KF-1040. J Antibiot 52, 689–694 (1999).
Throckmorton, K., Lim, F. Y., Kontoyiannis, D. P., Zheng, W. & Keller, N. P. Redundant synthesis of a conidial polyketide by two distinct secondary metabolite clusters in Aspergillus fumigatus. Environ Microbiol 18, 246–259 (2016).
Calvo, A. M., Wilson, R. A., Bok, J. W. & Keller, N. P. Relationship between secondary metabolism and fungal development. Microbiol Mol Biol Rev 66, 447–459 (2002).
Zhou, R., Rasooly, R. & Linz, J. E. Isolation and analysis of flup, a gene associated with hyphal growth and sporulation in Aspergillus parasiticus. Mol Gen Genet 264, 514–520 (2000).
Letunic, I., Doerks, T. & Bork, P. SMART 6: recent updates and new developments. Nucleic Acids Res 37, D229–D232 (2009).
Marchler-Bauer, A. et al. CDD: a Conserved Domain Database for the functional annotation of proteins. Nucleic Acids Res 39, D225–229 (2011).
Weber, T. et al. antiSMASH 3.0-a comprehensive resource for the genome mining of biosynthetic gene clusters. Nucleic Acids Res 43, W237–43 (2015).
Blin, K., Medema, M. H., Kottmann, R., Lee, S. Y. & Weber, T. The antiSMASH database, a comprehensive database of microbial secondary metabolite biosynthetic gene clusters. Nucleic Acids Res 45, D555–D559 (2017).
Kamou, N. N. et al. Investigating the compatibility of the biocontrol agent Clonostachys rosea IK726 with prodigiosin-producing Serratia rubidaea S55 and phenazine-producing Pseudomonas chlororaphis ToZa7. Arch Microbiol 198, 369–377 (2016).
Willems, E., Leyns, L. & Vandesompele, J. Standardization of real-time PCR gene expression data from independent biological replicates. Anal Bioch 379, 127–129 (2008).
Livak, K. J. & Schmittgen, T. D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 25, 402–408 (2001).
Nygren, C. M. R. et al. Growth on nitrate and occurrence of nitrate reductase-encoding genes in a phylogenetically diverse range of ectomycorrhizal fungi. New Phytol 180, 875–889 (2008).
Dubey, M. K., Ubhayasekera, W., Sandgren, M., Jensen, D. F. & Karlsson, M. Disruption of the Eng18B ENGase gene in the fungal biocontrol agent Trichoderma atroviride affects growth, conidiation and antagonistic ability. PLoS ONE 7, e36152 (2012).
Dubey, M. K. et al. The glyoxylate cycle is involved in pleotropic phenotypes, antagonism and induction of plant defence responses in the fungal biocontrol agent Trichoderma atroviride. Fungal Genet Biol 58-59, 33–41 (2013).
Utermark, J. & Karlovsky, P. Genetic transformation of filamentous fungi by Agrobacterium tumefaciens, http://www.nature.com/protocolexchange/protocols/427 (2008).
Schneider, C. A., Rasband, W. S. & Eliceiri, K. W. NIH Image to ImageJ: 25 years of image analysis. Nat Methods 9, 671–675 (2012).
Acknowledgements
The authors thank the Swedish Research Council for Environment, Agricultural Sciences and Spatial Planning (FORMAS) (grant numbers 220-2014-289 and 942-2015-1128), the Plant Protection Platform at the Swedish University of Agricultural Sciences and the Centre for Biological Control (CBC) for financial support for the study. We thank Lea Atanasova, Institute of Food Technology, University of Natural Resources and Life Sciences, Vienna for providing fungal strain Alternaria alternata TUCIM 3594.
Author information
Authors and Affiliations
Contributions
M.D., M.K. and D.F.J. conceived and designed the experiments. U.F., A.B. and M.D. performed the experiments and analysed the data. D.F.J. and A.B. contributed reagents/materials/analysis tools. M.D., A.B. and M.K. wrote the manuscript. All authors read and approved the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Fatema, U., Broberg, A., Jensen, D.F. et al. Functional analysis of polyketide synthase genes in the biocontrol fungus Clonostachys rosea. Sci Rep 8, 15009 (2018). https://doi.org/10.1038/s41598-018-33391-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-33391-1
Keywords
This article is cited by
-
RNA silencing is a key regulatory mechanism in the biocontrol fungus Clonostachys rosea-wheat interactions
BMC Biology (2024)
-
Characterization and control of Rhizoctonia solani affecting lucky bamboo (Dracaena sanderiana hort. ex. Mast.) using some bioagents
Scientific Reports (2023)
-
Understanding the mechanisms underlying biological control of Fusarium diseases in cereals
European Journal of Plant Pathology (2023)
-
De novo genome assembly and functional annotation for Fusarium langsethiae
BMC Genomics (2022)
-
Endophytic fungi from the common walnut and their in vitro antagonistic activity against Ophiognomonia leptostyla
Biologia (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
