
Abstract
The outbreak of multidrug-resistant pathogenic bacteria made the discovery of novel control strategies necessary. Phages have regained attention for their specific lytic activity against pathogenic bacterium. A newly isolated phage infecting the clinical Escherichia coli isolates, including several multidrug-resistant strains, was isolated, and this phage showed high control effects against the tested pathogenic E. coli strains. Host range analysis revealed that although the phage exhibited broad lytic spectrum against the tested E. coli strains, it could not lyse strains from the other species. Comparative genomic analysis showed that phages had undergone at least three genome recombination events during the evolutionary process at the position of the three phage tail genes, which was reported to be associated with the host range determination of the phage. The recombinant tail proteins contained functional domains that were highly similar with genes of the Salmonella phage and genes of Pseudomonas and Neisseria. The findings of this study not only provide resources for developing phage therapy against E. coli, but also showed the highly variable genome structure of the phage.
Similar content being viewed by others
Introduction
Escherichia coli is a type of Gram-negative bacterium of the Enterobacteriaceae family, the majority of which reside in the human gastrointestinal tract as a part of the normal flora. However, some E. coli have evolved into virulent straains, and are responsible for a variety of diseases, including intestinal diarrhea, urinary tract infections (UTI), septicemia, pneumoniae, and meningitis1,2. Like the other widely distributed multidrug-resistant pathogenic bacterial species, the multidrug resistance of E. coli has high prevalence.
The development of multidrug-resistant E. coli gained extra concern with respect to human health, and infections caused by E. coli are usually harder to treat, resulting in increased severity and duration of infection. Production of extended-spectrum β-lactamases (ESBL), found in some strains of Enterobacteriaceae, are one of the resistant mechanisms, which act as a worldwide threat. These species are chief culprits that cause treatment failure, including infections of the urogenital tract, abdomen, and bloodstream3. In addition to the production of ESBL, these strains are prevalently related to other mechanisms of resistance, making them multidrug resistant (MDR)4. The development of antimicrobial resistance can occur via two processes: mutation in certain genes and acquisition of resistant genes by horizontal transfer5. Therefore, it can contribute to the spread of antimicrobial resistance between bacterial species as well as to other members of the same species. Globally, antibiotic resistance analysis reveals that this serious threat is no longer a worry for the future, which occurring now in every region of the world, urging the exploration of new agents to resolve this situation.
Phages have gained growing attention as an alternative candidate to antibiotics. Phages possess unique advantages, and nature provides an almost inexhaustible supply, and no two identical phages have ever been found. However, in consideration of the potential for immunogenicity, rapid toxin release by lytic action, development of bacterial resistance and other else during the use of phages for therapeutic purposes, phage therapy was still actively pursued6,7. For example, through shedding the receptor on the cell surface that the virus used to enter, a bacterium becomes resistant to one phage. It can be overcome by adding more phages to the viral cocktails that patients receive8. In previous study, the phage cocktails consisting of 10 and 16 T4 type phage isolates could lyse one half to two thirds of the E. coli strains, representing five main pathotypes isolated from diarrheal patients9. Galtier M. et al. utilized a single dose of a cocktail, which consisted of three phages, led to a sharp decrease of E. coli levels throughout the gut10. These studies suggest that the E. coli phage has high potential to be used in controlling pathogenic E. coli, especially multidrug resistant E. coli.
To exploit more efficient phages that target clinically relevant E. coli, a newly isolated phage named vB_EcoS_HSE2 was isolated from hospital sewage, its biological and genomic characters were investigated and its potential usage in controlling pathogenic E. coli was evaluated. The finding of this study increases our understanding on E. coli phage diversity and provides resource for controlling pathogenic E. coli infections.
Results
Isolation and virion morphology observation of phage vB_EcoS_HSE2
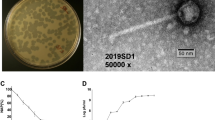
By using hospital sewage samples, one phage infecting E. coli strain 40371 was isolated, and the phage was named vB_EcoS_HSE2. Phage vB_EcoS_HSE2 formed transparent, small, round plaques of approximately 0.5 mm in diameter on the lawn of E. coli strain 40371 (Fig. 1A). TEM observation of the phage virion revealed that the phage had an icosahedral capsid with a diameter of 56 nm and a non-contractile tail that was approximately 178 nm in length and 12 nm in diameter (Fig. 1B). Based on the morphology, the phage was classified as a Siphoviridae family phage.

Morphology observation and characterization of phage vB_EcoS_HSE2. (A) Plaque morphologies of phage vB_EcoS_HSE2. (B) Virion morphology of phage vB_EcoS_HSE2. The phage virion was stained with potassium phosphotungstate and observed using transmission electron microscopy. (C) One step growth curve of the phage. (D) Thermal tolerance of the phage. (E) Tolerance of the phage to different pH treatment.
One-step growth curve
The one-step growth curve of phage vB_EcoS_HSE2 was analyzed by infecting the exponential growth strains with the phage at an MOI of 1.0. The latent period of the phage, which was the period between the absorption of the phage to the host bacterium and the beginning of the lysis of the host bacterium, was approximately 30 min, and the final concentration of the phage reached up to 1011 PFU/ml (Fig. 1C). Based on the final concentration of the phage and the concentration of the bacterial cell that was infected by the phage, the calculated burst size of phage vB_EcoS_HSE2 was about 86 PFU per bacterial cell.
Highly specific lytic spectrum
In this study, the host range of the phage vB_EcoS_HSE2 was tested against 18 clinical pathogenic strains, including 11 E. coli strains, and strains of Pseudomonas aeruginosa, Salmonella enterica, Neisseria gonorrhoeae, Klebsiella pneumoniae, Bacillus cereus, and Staphylococcus aureus. Host range testing revealed that vB_EcoS_HSE2 was highly specific. The phage could only infect six of the tested E. coli strains, but not the other tested E. coli strains and strains from other species (Table 1). Among the six E. coli strains sensitive to phage vB_EcoS_HSE2, strain 40199 was isolated from infected wound secretion of the patient, and the other strains were isolated from the urine samples of symptomatic patients. The antibiotic resistance analysis of the phage sensitive strains revealed that all the strains were resistant to more than ten of the tested antibiotics (Table 1).
High thermal and pH stability
The thermal and pH tolerance of the phage was analyzed. The phage was highly thermal tolerant, and no significant decrease in phage viability was observed by treating the phage with temperatures below 50 °C for 30 min, while by treating the phage at 60 °C for 30 min, the viability of the phage decreased sharply, and only 26.7% of the phage was alive (Fig. 1D). The phage lost its infective ability after been treated at 70 °C for 30 min. The phage also showed high resistance against pH treatment (Fig. 1E). The phage was stable when treating the phage with pH between 3 and 9, while treating the phage with pH 11, resulting in the phages losing their infective ability.
Generation of phage resistant mutant strain to phage vB_EcoS_HSE2
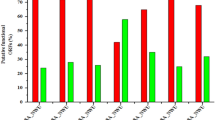
Due to the co-evolution of phage and host bacterium, phage-resistant bacteria were generated at high frequency, which would influence the efficiency of phage therapy11. In this study, the generation of phage resistant bacteria was analyzed. By co-incubating the phage with the strain, the phage significantly inhibited the growth of the bacteria during the first 7 hours (Fig. 2A). However, after that, the turbidity of the cultures increased fast, which might be due to the generation of cell debris or the growth of phage-resistant strains. To figure out the reason for the increased turbidity, the numbers of the viable cells in each culture were determined. The result showed that the final strain concentrations in the cultures treated with phage at an MOI of 0.001, 0.1, 1, and 10, were approximately 1.43%, 0.98%, 0.011%, and 0.0056%, respectively, compared to cultures that have not been treated by phage, suggesting that the phage effectively control the growth of pathogenic E. coli (Fig. 2B). The micro-examination of the cultures also showed that the phage treated cultures contain fewer bacterial cells, but more cell debris (data not shown). The treatment of the bacterium using phage also reduced the number of phage resistant strains in the culture. For example, the phage-resistant strains in the culture treated by the phage of MOI 1 was 30.9% compared to that in the culture without treatment by phage.

Control effect of phage vB_EcoS_HSE2 to the pathogenic E. coli strain 40371. (A) Growth curve of strain 40371 co-cultivated with phage vB_Ecos_HSE2. The strains were treated with the phage of different MOI and strain without treating by phage was used as control. (B) Counts of the total strains and phage resistant strains. The total strain concentrations and concentrations of phage resistant strains were tested after treating the strain with different concentrations of phage for 6 hours. The ratios of phage resistant strain were analyzed by calculating the ratio of phage resistant strains to the total strains.
General features of vB_EcoS_HSE2 genome
The genome of phage vB_EcoS_HSE2 was a linear genome with a genome size of 41,371 bp. The G + C content of the genome was 50.87%, which was almost the same as the genome of the E. coli strains (GenBank accession number NC_002695.1). Fifty-eight open reading frames (ORFs), including 24 functionally annotated ORFs, were predicted in the phage genome. No tRNAs and repeat sequences were found in the genome. The ORFs were mainly annotated as DNA metabolism associated proteins, cell lysis related proteins, and structural proteins (Fig. 3). Based on the locations of the functional genes, the genome of phage vB_EcoS_HSE2 showed modular genome structure and genes with associated functions were mainly located in the same gene cluster.

Genome structure of phage vB_EcoS_HSE2 and comparative genomic analysis with phage P AB-2017, K1-dep(4) and LPSE1. The outermost ring presented the CDSs of the linear vB_EcoS_HSE2 genome (blue) and the adjacent three rings showed BLASTN homology between vB_EcoS_HSE2 and P AB-2017 (pale pink), K1-dep(4) (pale green), and LPSE1(pale blue). The middle ring indicated the GC content (black), and the innermost ring represented the GC skew of vB_EcoS_HSE2 genome. The predicted functions of the CDSs were indicated.
Numerous nucleotide metabolism associated genes
At least eleven genes encoded by phage vB_EcoS_HSE2 were predicted to play roles in the process of phage nucleotide metabolism, including a helicase, a helicase-primase, a DNA polymerase, a nuclease superfamily protein, a transcriptional repressor, a transcriptional regulator, a replication protein, a VRR-NUC domain protein, a helix-destabilizing protein, and two alleles of HNH endonuclease (Fig. 3). The highly present of the DNA metabolism associated genes in vB_EcoS_HSE2 genome might reduce the dependence of the phage nucleotide metabolism on the host bacterium.
Three component host lysis system
Three genes that might be involved in the lysis of the bacterial cell were found in phage vB_EcoS_HSE2 genome. Gene gp24 and gp25, encoding holin-like protein and endolysin, respectively, might form a holin-dependent phage host lysis system to lyse the host cell at the end of the phage life cycle to release progeny phage virions12. According to previous reports, the spanin complex conducts the final step in host lysis by disrupting the outer membrane after holin and endolysin have permeabilized the inner membrane and degraded the host peptidoglycans, respectively13. One spanin protein encoding gene (gp36) was found located downstream of the phage host lysis system and might facilitate the release of the phage virion from the out membrane.
Mosaic structure of phage vB_EcoS_HSE2 structural proteins
Totally, twelve ORFs in phage vB_EcoS_HSE2 genome were found to encode phage structural proteins and these genes were scattered in the genome (Fig. 3). These eleven ORFs mainly encode proteins that function as terminases (Gp30 and Gp31), tail proteins (Gp01, Gp02, Gp34, Gp43, Gp45, Gp53), head proteins (Gp33 and Gp39), and two putative structural proteins (Gp32 and Gp35). In additional to the protein Gp01, Gp02, and Gp53, the other annotated structural proteins showed similarities that were higher than 95% with structural proteins of the other E. coli phages.
BLASTP analysis of Gp01 reveals that the protein showed highest similarity with the tail fiber proteins of Salmonella phages (Fig. 4A), including the Salmonella phage SS3e, which shows extremely broad host spectrum against strains of various Salmonella serovars, E. coli, Shigella sonnei, Enterobacter cloacae, and Serratia marcescens14 and the gene might be obtained from the Salmonella phage genome by horizontal gene transfer during their co-infection of the Salmonella strain or E. coli strain. BLASTP analysis of GP02 showed that the protein had a mosaic structure. The N-terminal 267 amino acids (from residues 1–267) of the protein showed high similarities with the tail proteins of Escherichia phage K1ind3 (42% similarity) and Escherichia phage G AB2017 (68% similarity), while the 492 amino acids at the C-terminus (from residues 243–734) only showed similarities with proteins of Pseudomonas sp. strain (41% similarity) and Neisseria sp. strain (37% similarity), but not the protein from the E. coli phage (Fig. 4C). Functional prediction of the C-terminus of protein Gp02 revealed that the residues between 243–327 were similar to the endopolygalacturonase (PDB entry 2iq7) in structure15 and contained the polysaccharide ligand binding residues (Fig. 4D). The residues between 519–624 were similar with the cellulosome protein dockerin carbohydrate binding module (PDB entry 2wz8) in structure16. Several phages infect the host bacteria by binding to the polysaccharides on the bacterial cell surface and some phages also contain virion-associated enzymes to overcome the carbohydrate barriers during infection17,18. The containment of polysaccharide binding domain in the phage structural might facilitate the infection of the phage.

Phylogenetic and functional analysis of the phage structural proteins. (A) and (B) Phylogenetic analysis of protein Gp01 and Gp53. The proteins that showed similarity with protein Gp01 and Gp53 were collected from GenBank and used for phylogenetic tree construction using Mega6.0 with neighbor-joining method with the bootstrap replicate of 1000. (C) Schematic diagram of the similarity and domain composition of Gp02. The N-terminus and C-terminus were similar with E. coli phages and Pseudomonas sp. proteins, respectively. The protein Gp02 contained three functional predicted domains, including the domain similar to the tailspike protein, endopolygalacturonase (indicated as PDB entry 2IQ7), and cellulosome protein dockerin carbohydrate binding module (indicated as PDB entry 2WZ8). (D) Protein structure modeling of residues 243–327 from protein Gp02. The structure of the protein endopolygalacturonase (PDB entry 2IQ7) was used as template for structure modeling and the polysaccharide was indicated.
Except for gp02, the gene gp53, which encodes the tape measure protein, was the longest gene in vB_EcoS_HSE2 genome and also had a mosaic nature. The tape measure protein dictates the tail length and facilitates DNA transit to the bacterial cell cytoplasm during infection19,20,21. BLASTN analysis of gp53 reveals that the 3′ termini 610-bp of the gene showed the highest similarity (83%) with the tape measure protein gene of Escherichia phages, while the 5′ terminal 1577-bp terminus of the gene showed the highest similarity (78%) with the tape measure protein gene of Salmonella phages. BLASTP analysis of Gp53 revealed that the protein showed the highest similarity (82% similarity) with the Salmonella phage SETP7, while the highest similarity of thisprotein to protein encoded by the E. coli phage is 55% (E. coli phage P AB-2017). Phylogenetic analysis of Gp53 showed that the protein was close in evolution with the tape measure proteins from the Salmonella phages. The results indicated that the gene gp53 might also have encountered horizontal gene transfer and gene recombination during the evolutionary process of the phage.
Phylogenetic and comparative genomic analysis of phage vB_EcoS_HSE2
BLASTN analysis of the phage vB_EcoS_HSE2 genome showed that the genome of phage vB_EcoS_HSE2 was highly similar (similarity approximately 50%) with the genomes of the E. coli phage K1H, K1ind2, K1G, K1ind3, L AB-2017, P AB-2017, and G AB-2017. The phage also showed similarity with Salmonella phages, such as phage BPS11Q3, Jersey, and SS3e (Fig. 5). Phylogenetic analysis of the phage vB_EcoS_HSE2 revealed that though the phage was close in relationship with the E. coli phages, it was clustered into the single branch (Fig. 6A). The core genes of the nine phages closely in evolution were analyzed and the result showed that 30 proteins were found to be conserved in all the six phages (Fig. 6B). The functions of these proteins were mainly related to the phage structure proteins and DNA replication, as well as host lysis, which were essential for the phage life cycle (Table S2).

Comparative genomic analysis of phages that were similar with phage vB_EcoS_HSE2. The phages with genome similarity higher than 20% were collected from GenBank and used for dot plot analysis. The dot plot analysis was performed by using Gepard.

Phylogenetic and comparative genomic analysis of the phage vB_EcoS_HSE2. (A) Phylogenetic analysis of phages that showed similarity with the phage vB_EcoS_HSE2. The phage genomes that showed similarity higher than 20% with the genome of phage vB_EcoS_HSE2 were collected and used for phylogenetic tree construction. (B) Core-genome analysis of phages from the same evolutionary branch with phage vB_EcoS_HSE2. The numbers of core genes for the 9 phages were indicated in the central circle, and the number of specific genes were also shown. (C) Dot plot analysis of phage vB_EcoS_HSE2 with phage K1ind1 and Jersery. The genes that were absent in phage K1ind1 genome are shown.
Based on the phylogenetic analysis of phage vB_EcoS-HSE2, the genomes of E. coli phage K1ind1 and Salmonella phage Jersey, accompany the genome of phage vB_EcoS_HSE2, were used for comparative genomic analysis (Fig. 5D). The result of the comparative genomic analysis showed that the genome of phage vB_EcoS_HSE2 showed a highly co-linear relationship with the genomes of phage K1ind1 and Jersey, except for the deletion of gene gp02, gp07-gp09, gp39, and gp51-gp53. Meanwhile, the genes gp07-gp09 and genes gp51-gp53, which were absent in phage K1ind1 genome, show homologues with the genes of the Salmonella phage Jersey.
Discussion
The outbreak of multidrug resistance E. coli has greatly limited antibiotic therapy and made it urgent to develop novel control strategies. The phage regained attention for its’ specific lytic activity and special control mechanisms against pathogenic bacterium. Numerous phages infecting E. coli have been isolated and their potential in controlling pathogenic multidrug resistant E. coli strains have been evaluated22,23,24. However, the generation of phage resistance was even faster than the generation of antibiotic resistance and, as described in this study, the phage-resistant bacteria even existed before the use of phage preparation. Construction of phage cocktail containing phages of different genetic backgrounds could efficiently reduce the generation of phage resistance and increase the efficiency of phage therapy. Thus, it is valuable to isolate phages with novel genetic backgrounds. In this study, an E. coli phage vB_EcoS_HSE2 that could efficiently control pathogenic E. coli by reducing both the cell concentrations of phage sensitive bacterium and the phage resistant bacterium was isolated and characterized. The phage exhibited highly specific host range and could only infect partial of the tested E. coli strains, but not the other E. coli strains or strains from the other species. Besides, no toxin gene, antibiotic resistance gene, phage lysogeny factor and other pathogen-related genes were found in the phage genome, suggesting that the phage was biosafe for being used in control pathogenic E. coli.
Comparative genomic analysis revealed that, though the phage showed the highest similarity with E. coli phage, several of its structural proteins, especially the tail proteins (Gp01 and Gp53), were similar with that of Salmonella phages. According to previous reports, the tail fiber protein and tail spike protein of Salmonella phage shows high diversity due to the gene recombination at the C-termini25,26,27. The tail fiber and tail spike are appendages in the phage tail that facilitates the initial binding of the phage to the bacterial host and have roles in host specificity determination28. As shown in our study, highly variable tail fiber protein typically presented a fairly conserved N-terminus, with low identities in the C terminus. The conservations of the N-terminal residues were consistent with the fact that this region attaches to the baseplate, while the remainder of the protein projects from the surface of the phage tail distal and appears to be involved in the initial binding to the host. The gene gp02, which was located immediately after gp01, was found to be highly similar to the genes from strains of Pseudomonas sp. and Neisseria sp., which are both human pathogens29,30. Gp02 was the phage tail spike protein and functional analysis of Gp02 revealed that the C-termini of the protein contained a polysaccharide binding domain that might work as a phage receptor binding protein. Further functional analysis of these two proteins and the interaction between the phage vB_EcoS_HSE2 and the host bacterium might figure out the roles of these two proteins in phage host range determination.
The genes gp01 and gp02 were located at the very beginning of the 5′-termini of the phage genome, and the genes downstream of these two genes were transcribed in different directions as these two genes, suggesting that these two genes might be obtained by genome recombination. The horizontal gene transfer is one of the most frequently approach for microbial genome mutation and the phage was thought to be the most important vector to delivery horizontal gene transfer31,32. In consideration of the high similarity of gp01 with genes of Salmonella phages and the high similarity of gp02 with genes Pseudomonas sp. and Neisseria sp., it is rational to speculate that the phage had undergone at least two genome recombination events during its evolutionary process. The 3′-termini of gene gp02 might be obtained from Pseudomonas sp. or Neisseria sp. during infection of the ancestral phage to one of these two strains. Subsequently, or antecedently, the recombinant phage co-infected with the other Salmonella phages obtained the gene gp01. Except for the gene gp01 and gp02, the gene gp53, which was located near the 3′-termini of the phage genome, also showed low similarity with the E. coli phage, but highly similar 3′-termini with the genes of Salmonella phages. The results indicating that the 3′-termini of the phage gene gp53 might also be acquired from Salmonella phages by genome recombination during co-infection with Salmonella phages. In summary, during the evolutionary process, the phage vB_EcoS_HSE2 underwent at least three genome recombination events by at least two infection processes to the non-E. coli strains. The phage was isolated from hospital sewage, which contained several pathogenic bacteria, including strains of E. coli, Pseudomonas sp., Neisseria sp., and Salmonella sp., which provided benefits for the recombination of the phage.
In conclusion, this study isolated a phage that might be used for pathogenic E. coli control. Genomic analysis of the phage revealed that the phage had undergone at least three rounds of genome recombination events during the evolutionary process. The finding of this study not only provides resources for developing phage therapy against E. coli, but also showed the highly variable genome structure of the phage.
Materials and Methods
Strain and antimicrobial susceptibility test
The E. coli strains used in this study were isolated from urine or wound secretion of symptomatic patients in Liyuan Hospital (Wuhan, China) and stored by our laboratory. Antimicrobial susceptibility tests of these strains were conducted using the Kirby-Bauer disk diffusion method according the Clinical and Laboratory Standards Institute (CLSI) guidelines 201733. E. coli strains were cultured in Luria-Bertani (LB) broth medium at 37 °C. E. coli strain 40371 was used as an indicator strain for phage isolation.
Phage isolation and propagation
Hospital sewage samples were collected from Liyuan hospital and were centrifuged at 12,000 × g for 10 min to remove the solid impurities. The supernatants were filtered through a 0.22-μm pore-size membrane filter to remove bacterial debris. For each sample, 200 μL of supernatant was added into 4 ml exponential growth strain 40371 and incubated for additional 8 h. After incubation, the supernatant of the culture was collected by centrifugation at 8,000 × g at 4 °C for 30 min and filtered through a 0.22-μm pore-size membrane filter. Then, 100 μl of the filtered supernatant was mixed with 200 μl of exponential growth 40371. After binding for 10 min, the mixture was added into 4 ml of melt semisolid LB medium and then overlaid onto an LB agar plate. Phage propagation, purification, and the efficiency-of-plating test were carried out by double-agar overlay assay as previously described34.
Electron microscopy observation of phage virions
To prepare phage for transmission electron microscopy (TEM) observation, the purified phage suspension was deposited on the cuprum grid with carbon-coated Formvar film and stained with 2% potassium phosphotungstate (pH 7.2). After air drying, the sample was observed with a TEM (H-7000FA; Hitachi, Tokyo, Japan) at an acceleration voltage of 100 kV.
Host range determination
The lytic activity of vB_EcoS_HSE2 was tested against 13 species as determined by standard spot tests35. The test strains were grown overnight in LB broth and 200 μL of the strain was mixed with 4 ml melt semisolid medium to pour the double-layer medium. Then, 1 μL purified phage suspensions (approximately 108 Pfu/ml) were spotted onto the upper layer medium and left to incubate overnight. Bacterial sensitivity to phage coincided with the formation of the spot where the phage suspension was deposited. Each strain was tested three times at 37 °C.
Phage stability assay
The purified phage suspensions were stored at −80, 4, and 30 °C, respectively, and the phage titer was determined by double-agar overlay assay immediately and every week after storage36. For the thermal stability assay, equal volumes of phage (approximately 108 Pfu/ml) were added into the 1.5 ml tube and incubated at 28 °C, 37 °C, 50 °C, 60 °C, and 70 °C for 30 min. After treatment, the tube was cooled slowly and placed in an ice water bath, and then these samples were assayed to determine the surviving phages. For the pH stability assay, after treating the phage at a pH of 3.0, 5.0, 7.0, 9.0, and 11.0 for 30 min, the phage suspensions were used for phage titer determination immediately after the finish of the treatment. The results were expressed as a percentage of the initial viral counts. Each assay was performed as three repetitions and the values represented are the means.
One-step growth curve analysis
To analyze the one step growth curve of the phage, phage was added into the exponential growth strain at an MOI of 1.0 at 37 °C for 5 min for phage adsorption. The non-adsorbed phage was removed by centrifugation at 10,000 × g for 1 min, and the pellet was resuspended in 50 ml of LB broth. The phage titer in the culture was determined every 10 min. The burst size of the phage was determined as previously described37.
Assay of the appearance ratio of phage resistant bacteria
To determine the phage-resistant ratio of the strain, the exponential growth strains were cultivated with the phage with a multiplicity of infection (MOI) of 0.01, 0.1, 1, and 10, and added into LB broth at a ratio of 1%. Cultures that had not been added phage were used as control. The strains were incubated at 37 °C with moderate shaking for 6 hours and used for phage-resistant strain counting. To count the number of phage resistant bacterial cells, the phage was mixed with the upper semisolid medium with a final concentration of 108 PFU/ml, which could inhibit the growth of the sensitive strain, and poured onto the plate. The strain cultures were gradient diluted and spread onto the upper medium. After overnight cultivation at 37 °C, the concentration of the phage-resistant strain were recorded and calculated. The gradient diluted cultures were also spread onto an LB agar plate to determine the total bacteria concentrations.
Phage genome extraction and sequencing
The purified phage suspension was used for phage genome purification. Phage genome extraction was performed as described before by phenol-chloroform extraction with protease K-sodium dodecyl sulfate (SDS) treatment38. The phage genome was sequenced using an Illumina HiSeq. 2500 sequencer. The reads obtained by sequencing were assembled into contigs using SPAdes-3.5.0 software39. The coding sequences (CDSs) of the phage genomes were predicted using the FGENE SV0 software (Softberry, http://www.softberry.com/berry.phtml?topic=virus0&group=programs&subgroup=gfindv) and by visual inspection. The putative function of each gene was predicted by performing a search in the National Center for Biotechnology Information nonredundant (NR) database and the CDD databases using the basic local alignment search tool (BLAST)40. The motif and functional domain composition of the predicted protein were analyzed by searching the Pfam database and by using HHpred41,42. Phage genome annotation was visualized by using CGview43. The genes encoding the putative tRNAs were analyzed using tRNAScan44. Tandem repeat and insert sequences in the phage genome were analyzed by using Tandem Repeat Finder and Repeat Masker45,46. Comparative genome analysis of the phages was carried out using Gepard 1.347,48. Core gene analysis of phages was performed by CoreGenes 3.0 and genes with scores above 75 were regarded as the core genes49. The phylogenetic trees in the present study were constructed using Mega 6.050.
Nucleotide sequence accession number
The nucleotide sequence of the phage vB_EcoS_HSE2 genome was deposited in GenBank under the accession number MG252615.
References
Bolocan, A. S., Callanan, J., Forde, A., Ross, P. & Hill, C. Phage therapy targeting Escherichia coli-a story with no end? FEMS Microbiology Letters 363, https://doi.org/10.1093/femsle/fnw256 (2016).
Brussow, H. Targeting the gut to protect the bladder: Oral Phage therapy approaches against urinary Escherichia coli infections? Environmental Microbiology 18, 2084–2088, https://doi.org/10.1111/1462-2920.13310 (2016).
Anonymous. Antimicrobial resistance: global report on surveillance. World Health Organisation, Geneva, Switzerland (2014).
Mathers, A. J., Peirano, G. & Pitout, J. D. The role of epidemic resistance plasmids and international high-risk clones in the spread of multidrug-resistant Enterobacteriaceae. Clinical Microbiology Reviews 28, 565–591, https://doi.org/10.1128/CMR.00116-14 (2015).
Bowen, M. Antimicrobial stewardship: Time for change. Equine Veterinary Journal 45, 127–129, https://doi.org/10.1111/evj.12041 (2013).
Yosef, I., Manor, M., Kiro, R. & Qimron, U. Temperate and lytic bacteriophages programmed to sensitize and kill antibiotic-resistant bacteria. Proceedings of the National Academy of Sciences of the United States of America 112, 7267–7272, https://doi.org/10.1073/pnas.1500107112 (2015).
Minsuk Kong, S. R. Bacteriophage PBC1 and Its Endolysin as an Antimicrobial Agent against Bacillus cereus. Applied and Environmental Microbiology 81, 2274–2283 (2015).
Reardon, S. Phage therapy gets revitalized. Nature 510, 15–16, https://doi.org/10.1038/510015a (2014).
Denou, E. et al. T4 phages against Escherichia coli diarrhea: potential and problems. Virology 388, 21–30, https://doi.org/10.1016/j.virol.2009.03.009 (2009).
Galtier, M. et al. Bacteriophages to reduce gut carriage of antibiotic resistant uropathogens with low impact on microbiota composition. Environmental Microbiology 18, 2237–2245, https://doi.org/10.1111/1462-2920.13284 (2016).
Scanlan, P. D. & Buckling, A. Co-evolution with lytic phage selects for the mucoid phenotype of Pseudomonas fluorescens SBW25. The ISME journal 6, 1148–1158, https://doi.org/10.1038/ismej.2011.174 (2012).
Catalao, M. J., Gil, F., Moniz-Pereira, J., Sao-Jose, C. & Pimentel, M. Diversity in bacterial lysis systems: bacteriophages show the way. FEMS Microbiology Review 37, 554–571 (2013).
Summer, E. J. et al. Rz/Rz1 lysis gene equivalents in phages of Gram-negative hosts. Journal of Molecular Biology 373, 1098–1112, https://doi.org/10.1016/j.jmb.2007.08.045 (2007).
Kim, S. H. et al. Complete genome sequence of Salmonella bacteriophage SS3e. Journal of Virology 86, 10253–10254, https://doi.org/10.1128/JVI.01550-12 (2012).
Bonivento, D. et al. Crystal structure of the endopolygalacturonase from the phytopathogenic fungus Colletotrichum lupini and its interaction with polygalacturonase-inhibiting proteins. Proteins 70, 294–299, https://doi.org/10.1002/prot.21610 (2008).
Correia, M. A. et al. Signature active site architectures illuminate the molecular basis for ligand specificity in family 35 carbohydrate binding module. Biochemistry 49, 6193–6205, https://doi.org/10.1021/bi1006139 (2010).
Latka, A., Maciejewska, B., Majkowska-Skrobek, G., Briers, Y. & Drulis-Kawa, Z. Bacteriophage-encoded virion-associated enzymes to overcome the carbohydrate barriers during the infection process. Applied Microbiology and Biotechnology 101, 3103–3119, https://doi.org/10.1007/s00253-017-8224-6 (2017).
Prokhorov, N. S. et al. Function of bacteriophage G7C esterase tailspike in host cell adsorption. Molecular Microbiology, https://doi.org/10.1111/mmi.13710 (2017).
Zivanovic, Y. et al. Insights into bacteriophage T5 structure from analysis of its morphogenesis genes and protein components. Journal of Virology 88, 1162–1174, https://doi.org/10.1128/JVI.02262-13 (2014).
Mahony, J. et al. Functional and structural dissection of the tape measure protein of lactococcal phage TP901-1. Scientific Reports-UK 6, 36667, https://doi.org/10.1038/srep36667 (2016).
Cumby, N., Reimer, K., Mengin-Lecreulx, D., Davidson, A. R. & Maxwell, K. L. The phage tail tape measure protein, an inner membrane protein and a periplasmic chaperone play connected roles in the genome injection process of E. coli phage HK97. Molecular Microbiology 96, 437–447, https://doi.org/10.1111/mmi.12918 (2015).
Gundogdu, A., Bolkvadze, D. & Kilic, H. In vitro Effectiveness of Commercial Bacteriophage Cocktails on Diverse Extended-Spectrum Beta-Lactamase Producing Escherichia coli Strains. Frontiers in Microbiology 7, https://doi.org/10.3389/Fmicb.2016.01761 (2016).
Amarillas, L. et al. Isolation and Characterization of phiLLS, a Novel Phage with Potential Biocontrol Agent against Multidrug-Resistant Escherichia coli. Frontiers in Microbiology 8, https://doi.org/10.3389/Fmicb.2017.01355 (2017).
Pallavali, R. R., Degati, V. L., Lomada, D., Reddy, M. C. & Durbaka, V. R. P. Isolation and in vitro evaluation of bacteriophages against MDR-bacterial isolates from septic wound infections. Plos One 12, https://doi.org/10.1371/journal.pone.0179245 (2017).
Moreno Switt, A. I. et al. Genomic characterization provides new insight into Salmonella phage diversity. BMC Genomics 14, 481, https://doi.org/10.1186/1471-2164-14-481 (2013).
Li, E. et al. Isolation and characterization of a bacteriophage phiEap-2 infecting multidrug resistant Enterobacter aerogenes. Scientific Reports -UK 6, 28338, https://doi.org/10.1038/srep28338 (2016).
Hooton, S. P., Timms, A. R., Rowsell, J., Wilson, R. & Connerton, I. F. Salmonella Typhimurium-specific bacteriophage PhiSH19 and the origins of species specificity in the Vi01-like phage family. Virology Journal 8, 498, https://doi.org/10.1186/1743-422X-8-498 (2011).
Vinga, I. et al. Role of bacteriophage SPP1 tail spike protein gp21 on host cell receptor binding and trigger of phage DNA ejection. Molecular Microbiology 83, 289–303, https://doi.org/10.1111/j.1365-2958.2011.07931.x (2012).
Juan, C., Pena, C. & Oliver, A. Host and Pathogen Biomarkers for Severe Pseudomonas aeruginosa Infections. The Journal of Infectious Diseases 215, S44–S51, https://doi.org/10.1093/infdis/jiw299 (2017).
Soriani, M. Unraveling Neisseria meningitidis pathogenesis: from functional genomics to experimental models. F1000Research 6, 1228, https://doi.org/10.12688/f1000research.11279.1 (2017).
Touchon, M., Moura de Sousa, J. A. & Rocha, E. P. Embracing the enemy: the diversification of microbial gene repertoires by phage-mediated horizontal gene transfer. Current Opinion in Microbiology 38, 66–73, https://doi.org/10.1016/j.mib.2017.04.010 (2017).
Wotzka, S. Y., Nguyen, B. D. & Hardt, W. D. Salmonella Typhimurium Diarrhea Reveals Basic Principles of Enteropathogen Infection and Disease-Promoted DNA Exchange. Cell Host & Microbe 21, 443–454, https://doi.org/10.1016/j.chom.2017.03.009 (2017).
CLSI. Performance Standards for Antimicrobial Susceptibility Testing, 27th ed. (Clinical and Laboratory Standards Institute, 2017).
Yuan, Y. et al. Effects of actin-like proteins encoded by two Bacillus pumilus phages on unstable lysogeny, revealed by genomic analysis. Applied and Environmental Microbiology 81, 339–350, https://doi.org/10.1128/AEM.02889-14 (2015).
Kutter, E. Phage host range and efficiency of plating. Methods in Molecular Biology 501, 141–149, https://doi.org/10.1007/978-1-60327-164-6_14 (2009).
Chen, M., Xu, J. T., Yao, H. C., Lu, C. P. & Zhang, W. Isolation, genome sequencing and functional analysis of two T7-like coliphages of avian pathogenic Escherichia coli. Gene 582, 47–58 (2016).
Catalao, M. J., Gil, F., Moniz-Pereira, J. & Pimentel, M. The mycobacteriophage Ms6 encodes a chaperone-like protein involved in the endolysin delivery to the peptidoglycan. Molecular Microbiology 77, 672–686, https://doi.org/10.1111/j.1365-2958.2010.07239.x (2010).
Yuan, Y. H., Gao, M. Y., Wu, D. D., Liu, P. M. & Wu, Y. Genome Characteristics of a Novel Phage from Bacillus thuringiensis Showing High Similarity with Phage from Bacillus cereus. Plos One 7 (2012).
Bankevich, A. et al. SPAdes: A New Genome Assembly Algorithm and Its Applications to Single-Cell Sequencing. Journal of Computional Biology 19, 455–477 (2012).
Marchler-Bauer, A. et al. CDD: conserved domains and protein three-dimensional structure. Nucleic Acids Research 41, D348–352, https://doi.org/10.1093/nar/gks1243 (2013).
Finn, R. D. et al. The Pfam protein families database: towards a more sustainable future. Nucleic Acids Research 44, D279–285, https://doi.org/10.1093/nar/gkv1344 (2016).
Soding, J. Protein homology detection by HMM-HMM comparison. Bioinformatics 21, 951–960, https://doi.org/10.1093/bioinformatics/bti125 (2005).
Stothard, P. & Wishart, D. S. Circular genome visualization and exploration using CGView. Bioinformatics 21, 537–539, https://doi.org/10.1093/bioinformatics/bti054 (2005).
Schattner, P., Brooks, A. N. & Lowe, T. M. The tRNAscan-SE, snoscan and snoGPS web servers for the detection of tRNAs and snoRNAs. Nucleic Acids Research 33, W686–689, https://doi.org/10.1093/nar/gki366 (2005).
Benson, G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Research 27, 573–580 (1999).
Tarailo-Graovac, M. & Chen, N. Using RepeatMasker to identify repetitive elements in genomic sequences. Current Protocols in Bioinformatics Chapter 4, Unit 4 10, https://doi.org/10.1002/0471250953.bi0410s25 (2009).
Krumsiek, J., Arnold, R. & Rattei, T. Gepard: a rapid and sensitive tool for creating dotplots on genome scale. Bioinformatics 23, 1026–1028, https://doi.org/10.1093/bioinformatics/btm039 (2007).
Zafar, N., Mazumder, R. & Seto, D. CoreGenes: a computational tool for identifying and cataloging “core” genes in a set of small genomes. BMC Bioinformatics 3, 12 (2002).
Mahadevan, P., King, J. F. & Seto, D. CGUG: in silico proteome and genome parsing tool for the determination of “core” and unique genes in the analysis of genomes up to ca. 1.9 Mb. BMC Research Notes 2, 168, https://doi.org/10.1186/1756-0500-2-168 (2009).
Tamura, K. et al. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Molecular Biology and Evolution 28, 2731–2739, https://doi.org/10.1093/molbev/msr121 (2011).
Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 31500155).
Author information
Authors and Affiliations
Contributions
Y.Y. designed the study. Q.P. and Y.Y. performed the experiments, analyzed the data, and wrote the manuscript. All authors gave final approval of the version to be published.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Peng, Q., Yuan, Y. Characterization of a newly isolated phage infecting pathogenic Escherichia coli and analysis of its mosaic structural genes. Sci Rep 8, 8086 (2018). https://doi.org/10.1038/s41598-018-26004-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-26004-4
This article is cited by
-
Phage tailspike modularity and horizontal gene transfer reveals specificity towards E. coli O-antigen serogroups
Virology Journal (2023)
-
Characterization of bacteriophage vB_KleM_KB2 possessing high control ability to pathogenic Klebsiella pneumoniae
Scientific Reports (2023)
-
Characterization and synergy studies of Caudoviricete Escherichia phage FS2B infecting multi-drug resistant uropathogenic Escherichia coli isolates
International Microbiology (2023)
-
Isolation and identification of the broad-spectrum high-efficiency phage vB_SalP_LDW16 and its therapeutic application in chickens
BMC Veterinary Research (2022)
-
Isolation and characterization of a novel Escherichia coli O157:H7-specific phage as a biocontrol agent
Journal of Environmental Health Science and Engineering (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
