
Abstract
The prion protein (PrPC), a mainly neuronal protein, is known to modulate glucose homeostasis in mouse models. We explored the underlying mechanism in mouse models and the human pancreatic β-cell line 1.1B4. We report expression of PrPC on mouse pancreatic β-cells, where it promoted uptake of iron through divalent-metal-transporters. Accordingly, pancreatic iron stores in PrP knockout mice (PrP−/−) were significantly lower than wild type (PrP+/+) controls. Silencing of PrPC in 1.1B4 cells resulted in significant depletion of intracellular (IC) iron, and remarkably, upregulation of glucose transporter GLUT2 and insulin. Iron overloading, on the other hand, resulted in downregulation of GLUT2 and insulin in a PrPC-dependent manner. Similar observations were noted in the brain, liver, and neuroretina of iron overloaded PrP+/+ but not PrP−/− mice, indicating PrPC-mediated modulation of insulin and glucose homeostasis through iron. Peripheral challenge with glucose and insulin revealed blunting of the response in iron-overloaded PrP+/+ relative to PrP−/− mice, suggesting that PrPC-mediated modulation of IC iron influences both secretion and sensitivity of peripheral organs to insulin. These observations have implications for Alzheimer’s disease and diabetic retinopathy, known complications of type-2-diabetes associated with brain and ocular iron-dyshomeostasis.
Similar content being viewed by others


Introduction
Type-2-diabetes is a metabolic disorder characterized by hyperglycemia resulting from decreased secretion of insulin due to pancreatic β-cell dysfunction and resistance of peripheral organs to available insulin. The underlying pathobiology is complex, and includes a combination of host genetics and environmental factors1. Among the latter, a positive correlation between systemic iron and type-2-diabetes has been recognized for some time, but the underlying mechanism is not clear2,3,4,5,6,7,8,9. This correlation takes on an increased significance since type-2-diabetes is a known risk for Alzheimer’s disease (AD)10,11, a common dementia of the elderly associated with impaired neuronal glucose metabolism and brain iron dyshomeostasis12. Likewise, diabetic retinopathy (DR), another complication of type-2-diabetes, is fueled by iron dyshomeostasis13. The possibility that iron serves as the pathogenic link between type-2-diabetes, AD, and DR is intriguing, and offers untapped opportunities for a better understanding of disease pathogenesis and unconventional therapeutic options through iron chelation.
Interestingly, prion protein (PrPC), a mainly neuronal protein14,15,16, has been reported to influence glucose homeostasis in mouse models17,18,19 and facilitate iron uptake by functioning as a ferrireductase (FR) partner for divalent metal transporters20. Though apparently disconnected, it is likely that PrPC modulates blood glucose by altering the expression of glucose transporter 2 (GLUT2) on pancreatic β-cells through iron, a bidirectional glucose transporter that regulates the release of insulin21,22,23. A similar function of PrPC on neuronal cells might induce neurotoxicity by the combined effect of iron-mediated oxidative stress and glucose deprivation in disorders associated with brain and ocular iron dyshomeostasis such as AD, sporadic Creutzfeldt-Jakob-disease (sCJD), and DR24. Experimental proof of this hypothesis, however, is lacking.
Here, we explored the correlation between PrPC-mediated change in IC iron and expression of glucose transporters in pancreatic β-cells, hepatocytes, neuronal cells, and the retina in mouse and cell models expressing variable levels of PrPC in the absence or presence of excess iron. We report that PrPC influences glucose homeostasis by modulating the expression of glucose transports through iron. Implications for AD and DR, common complications of long-standing type-2-diabetes associated with brain and ocular iron dyshomeostasis are discussed.
Results
The following mouse lines were used in this study; F2 generation of wild-type C57BL/6 (labeled as C6 PrP+/+) crossed with PrP-knock out on 129/Ola background developed by Manson et al.25 (labeled as C6 PrP−/−), and FVB/NJ PrP−/− developed by Fischer et al. (labeled as PrP−/−)26 that were used to develop FVB/NJ Tg40 mice that express 2x human PrP (labeled as Tg40 PrP).
PrPC is expressed mainly on β-cells of mouse pancreas
To evaluate the expression and post-translational processing of PrPC in mouse pancreas, immunohistochemistry and Western blotting was performed with two PrP-specific antibodies; 3F4 that reacts with human PrP, not mouse PrP, and 8H4 that reacts with both human and mouse PrP. Both antibodies react with unglycosylated and post-translationally glycosylated full-length (FL) PrP forms. However, their reactivity with post-translationally processed α-cleaved (C1) and β-cleaved (C2) C-terminal fragments of PrP differs27. 3F4 reacts with C2, not C1 where its epitope is lost, whereas 8H4 reacts with both C1 and C2. Thus, differential antibody reactivity combined with migration on SDS gels based on molecular mass provides a convenient method for distinguishing between α- (C1) and β-cleaved (C2) forms of PrP (Fig. 1a).

PrP is expressed on insulin positive β-cells in mouse pancreas, primary cells from mouse pancreas, and human insulin producing β-cell line 1.1B4: (a) Schematic representation of full length (FL), α-cleaved (C1), and β-cleaved (C2) forms of PrP and antibody reactivity. (b) Probing of pancreatic lysates for PrP shows the expected glycoforms in C6 PrP+/+ samples and no reactivity in C6 PrP−/− samples (lanes 1 & 2). β-actin provides a loading control. (c) Probing of lysates from 1.1B4 cells for PrP shows the expected glycoforms that are down-regulated in cells transfected with PrP-specific siRNA. α-tubulin provides a loading control. The full-length blots are provided in Supplementary File (Raw data). (d) Immunoreaction of 1.1B4 cells for PrP shows plasma membrane and endosomal reaction (panels 1 & 2). Scale bar: 20 μm. (e) Immunoreaction of pancreatic sections from Tg40 PrP mice with 3F4 shows a positive reaction for PrP that co-localizes with insulin staining on β-cells (panels 1 & 2). No reaction for PrP is detected in PrP−/− samples that show a robust reaction for insulin (panels 3 & 4). (f) Primary cultures from Tg40 PrP pancreas show a positive reaction for PrP in insulin positive β-cells (panels 1 & 2). No reaction for PrP is detected in cells from PrP−/− pancreas though insulin positive β-cells are prominent (panels 3 & 4). Scale bar: 20 μm.
Probing of Western blots of pancreatic lysates from C6 PrP+/+ and C6 PrP−/− mice with 8H4 revealed unglycosylated and glycosylated FL, and C2 and C1 forms similar to neuronal PrPC 27 in C6 PrP+/+, and no reactivity in C6 PrP−/− samples (Fig. 1b, lanes 1 & 2). Probing of 1.1B4 cell lysates with 3F4 revealed the unglycosylated and glycosylated FL PrPC as above, and almost complete loss of reactivity by silencing PrP (Fig. 1c, lanes 1–3). Immunoreaction of permeabilized 1.1B4 cells with 3F4 followed by Alexa Fluor-conjugated secondary antibody revealed prominent reaction on the plasma membrane and endocytic vesicles as in neuronal cells (Fig. 1d, panels 1 & 2)28.
Immunoreaction of fixed sections of pancreas from Tg40 PrP and matching PrP−/− mice with 3F4 and insulin-specific antibody showed a strong reaction for PrPC on insulin positive β-cells in Tg40 PrP samples, and a positive reaction for insulin, but not 3F4 in PrP−/− samples (Fig. 1e, panels 1–4). A similar evaluation of cultured primary cells from freshly harvested pancreas showed a positive reaction for PrPC in insulin positive cells from Tg40 PrP samples, a reaction for insulin in PrP−/− samples that did not react with 3F4 as expected (Fig. 1f, panels 1–4).
Most of the PrPC in pancreatic β-cells is cleaved at the β-site
Under physiological conditions, PrPC expressed on neuronal cells recycles from the plasma membrane and undergoes α-cleavage in an endocytic compartment, resulting in the generation of C127. Cleavage at the β-site is typical of disease-associated PrP-scrapie (PrPSc)14,29,30,31, and is triggered by exposure to reactive oxygen species32.
To evaluate whether post-translational processing of PrPC on pancreatic β-cells differs from neurons27,33, pancreatic lysates from C6 mice and human brain (as a representative tissue for neurons) were deglycosylated and probed with 8H4. Surprisingly, pancreatic lysates from C6 PrP+/+ samples revealed barely detectable FL PrPC, most of which was detected as C2 and C1 fragments. Human brain sample, on the other hand, showed equivalent amounts of FL and C1, and C6 PrP−/− samples did not show any reactivity as expected (Fig. 2a, lanes 1–3).

Iron uptake is coupled with β-cleavage of PrPC: (a) Probing of deglycosylated pancreatic lysates from C6 PrP+/+ mice with 8H4 shows β-cleavage of majority of PrP. No reactivity is detected in C6 PrP−/− samples as expected. Most of the PrP from human brain homogenate is cleaved at the α-site (lanes 3). (All samples were fractionated and processed on the same gel. Complete gel is included in supplementary file-raw data). *Represents a non-specific band. (b) Probing of deglycosylated pancreatic lysates from Tg40 PrP mice with 3F4 shows β-cleavage of majority of PrP (lanes 1–6). Systemic iron overload increases FL and the β-cleaved form of PrP (lanes 2, 4 & 6 vs. 1, 3 & 5). Most of the PrP in human brain homogenate is FL (lanes 8 & 9). Pancreatic lysates from PrP−/− mice show no reactivity for 3F4 as expected. (c) The ratio of C2 vs. full-length PrP is significantly higher in pancreatic lysates relative to the brain. Values are mean + SEM of the indicated n. ***p < 0.001. (d) Exposure of PrP-GFP expressing 1.1B4 cells to ferric ammonium citrate (FAC) results in the loss of GFP tag from the plasma membrane within 20 minutes (panels 1–4). (e) Exposure of 1.1B4 cells expressing GFP-tagged DMT-1, ZIP8, or ZIP14 to FAC, on the other hand, shows no change in localization or intensity of GFP fluorescence up to 20 minutes (panels 1–6). Scale bar 20 μm. (f) Probing of deglycosylated lysates from 1.1B4 cells expressing vector, DMT-1, ZIP8, and ZIP14 with 3F4 reveals significant increase in the ratio of C2/FL in cells expressing DMT-1 and ZIP14 (lane 5 vs. 6 & 8; g). (The expression of GFP-tagged transfected proteins was equivalent (at 15–17%) as determined by manual counting of 20 random fields of each at 5×)- (see Supplementary Fig. S3). (g) The ratio of C2 vs. FL after normalization with transfection efficiency and β-actin shows a significant increase in cells expressing DMT-1 and ZIP14. Values are mean + SEM of the indicated n. *p < 0.05. The full-length blots are provided in Supplementary File (Raw data).
Similar results were obtained from deglycosylated pancreatic lysates from Tg40 PrP mice probed with 3F4. Majority of PrPC in the pancreas exhibited the mobility of C2. Systemic iron overload induced by injecting 36 µg/22 g mouse weight ferric ammonium citrate (FAC) upregulated FL PrPC to some extent, but the major difference was in the intensity of C2. (Fig. 2b, lanes 1–6; Fig. 2c). Pancreatic lysates from PrP−/− mice showed no reactivity with 3F4, while brain lysates revealed mainly FL PrPC and minimal reactivity for C2 (Fig. 2b, lanes 7–9; Fig. 2c). The small increase in C2 in iron over-loaded pancreas (Fig. 2c) indicated iron-mediated cleavage of PrP in vivo, a phenomenon that was investigated further in the 1.1 B4 cells in vitro. Similar results were obtained from probing the membrane with 8H4 (see Supplementary Fig. S2).
Accordingly, 1.1B4 cells expressing green-fluorescent protein (GFP) tagged PrPC (PrPC-GFP) were exposed to FAC, and live cells were imaged every 5 min for up to 20 min. Since the GFP tag is inserted between N-terminal residues 37 and 38 of PrP20,34, the premise was that the tag would be lost following α- or β-cleavage of PrPC. Representative images from time zero (T-0) and 20 min (T-20) time point are shown (Fig. 2d, panels 1–4). In control cells exposed to vehicle, the signal from PrPC-GFP was prominent at the plasma membrane, peri-nuclear Golgi region, and endocytic vesicles at T-0 and T-20 (Fig. 2d, panels 1 & 2). In FAC exposed cells, the distribution of PrPC-GFP at T-0 was similar to controls (Fig. 2d, panels 1 & 3). However, at T-20 most of the GFP signal was lost from the plasma membrane, though peri-nuclear and endosomal signal remained (Fig. 2d, panel 4). (The intensity of green fluorescence in the inset was increased 5-fold in panels 3 and 4). Thus, the GFP carrying N-terminus of PrPC is lost within 20 min of exposure to FAC. Based on Western blot results in Fig. 2b and published reports32,35, it is likely that FAC induces β-cleavage of PrPC.
To evaluate whether β-cleavage of PrPC is coupled with iron transport through divalent metal transporter-1 (DMT-1) or members of the Zrt Irt-like protein (ZIP) family, in particular ZIP1420,36,37, 1.1B4 cells transfected with GFP-tagged DMT-1, ZIP8, and ZIP14 were exposed to FAC and imaged at T-0 and T-20. Unlike PrPC-GFP (Fig. 2d), there was no difference in the intensity or localization of DMT-1-GFP, ZIP8-GFP, or ZIP14-GFP following exposure to exogenous FAC (Fig. 2e, panels 1–6). However, overexpression of these transporters in 1.1B4 cells resulted in increased β-cleavage of PrPC in cells transfected with DMT-1 and ZIP14 (Fig. 2f, lanes 6 & 8; Fig. 2g). Since ZIP14 is known to mediate uptake of non-transferrin bound iron (NTBI) by pancreatic β-cells38 (see Supplementary Fig. S1), these results suggest that PrPC functions as a FR partner for ZIP14, and is cleaved at the β-site during this process.
PrPC mediates uptake of iron by pancreatic β-cells in vitro and in vivo
To confirm the facilitative role of PrPC in iron uptake by the pancreas39,40,41, 1.1B4 cells were transfected with siRNA to silence PrPC, and exposed to FAC. Following an incubation of 16 h, control and experimental cell lysates were processed for Western blotting and probed for ferritin. Silencing of PrPC resulted in significant downregulation of ferritin relative to non-transfected and scrambled siRNA transfected controls (Fig. 3a, lanes 1–3; Fig. 3b). Exposure to FAC caused significant upregulation of ferritin in controls as expected, but had minimal effect in the absence of PrPC (Fig. 3a, lanes 4–6); Fig. 3b).

PrP mediates iron uptake by pancreatic β-cells: (a) Non-transfected and 1.1B4 cells transfected with scrambled or PrP-specific siRNA were cultured in the absence (−FAC) or presence of ferric ammonium citrate (+FAC) for 16 h, and lysates were processed for Western blotting. Probing with 3F4 shows PrP glycoforms, and down-regulation by PrP-specific siRNA. Probing for ferritin shows significant down-regulation in cells where PrP had been silenced. Exposure to exogenous iron upregulates ferritin in untreated controls and cells transfected with scrambled siRNA, but has minimal effect on cells where PrP is silenced. (b) Quantification of ferritin expression by densitometry after normalization with β-actin. Values are mean ± SEM of the indicated n. *Represents change in ferritin relative to untreated, non-transfected control. ##Represents change in ferritin relative to FAC exposed non-transfected control. *p < 0.05, ##p < 0.01. (c) Western blotting shows upregulation of PrP in iron-overloaded C6 PrP+/+, no signal in C6 PrP−/− samples, and the expected glycoforms in human brain sample (lane 9). Ferritin is significantly higher in iron-overloaded relative to untreated C6 PrP+/+ samples and matched C6 PrP−/− samples. There is no change in ferritin iron overloaded C6 PrP−/− samples relative to untreated controls. Probing for TfR shows significant reduction in iron-overloaded relative to untreated C6 PrP+/+ samples and matched C6 PrP−/− samples. There is minimal change in TfR expression in iron-overloaded C6 PrP−/− samples relative to untreated controls. (d) Density of protein bands after normalization with β-actin. Values are mean ± SEM of the indicated n. *Represents change in expression relative to untreated C6 PrP+/+ samples. *p < 0.05. Full-length blots are provided in Supplementary File (Raw data). (e) Immunoreaction of fixed pancreatic sections from the above mice for ferritin shows a positive reaction in mainly β-cell rich endocrine islets (panels 1–4). Iron over-loading increases ferritin reactivity in C6 PrP+/+ sections, but shows minimal change in C6 PrP−/− samples (panels 3 & 4). Reactivity for TfR is also localized to the endocrine islets (panels 5–8). Iron-overloading down-regulates TfR expression in C6 PrP+/+ samples (panel 6 vs. 5), but has minimal effect on C6 PrP−/− samples (panels 7 & 8). Scale bar 20 μm.
The above observations were substantiated in C6 PrP+/+ and C6 PrP−/− mice injected with iron to create systemic iron overload (Fig. 3c–e). Evaluation of pancreatic lysates by Western blotting revealed upregulation of PrPC in iron-overloaded C6 PrP+/+ mice as in Fig. 2b above (Fig. 3c, lanes 1, 2, 5, 6; Fig. 3d). Probing for ferritin revealed significantly less ferritin in C6 PrP−/− relative to C6 PrP+/+ controls. Overloading with iron caused significant upregulation of ferritin in C6 PrP+/+, but not in C6 PrP−/− samples (Fig. 3c, lanes 1–8; Fig. 3d). Expression of transferrin receptor (TfR) was reduced in iron overloaded C6 PrP+/+ mice, but showed minimal change in similarly treated C6 PrP−/− mice (Fig. 3c, lanes 1–8; Fig. 3d). Similar results were observed in the Tg40 PrP and PrP−/− mice (see Supplementary Fig. S4).
Immunohistochemistry of fixed sections of pancreas from the above mice revealed reactivity for ferritin in the islets of both C6 PrP+/+ and C6 PrP−/− samples (Fig. 3e, panels 1 & 3), and a significant increase in iron overloaded C6 PrP+/+ relative to C6 PrP−/− samples (Fig. 3e, panels 2 & 4). Reaction for (TfR) showed significant downregulation in iron overloaded C6 PrP+/+, but minimal change in C6 PrP−/− samples (Fig. 3e, panels 5–8).
Together, the above results leave little doubt that PrPC mediates iron uptake in pancreatic β-cells. Since increased systemic iron is associated with the risk of type-2-diabetes, further studies were directed at whether PrP-mediated change in pancreatic β-cell iron influences insulin production and/or secretion and blood glucose levels.
PrPC-mediated increase in intracellular iron downregulates glucose transporters in the pancreas, liver, and retina
Pancreatic β-cells sense blood glucose levels through GLUT2, a bidirectional glucose transporter, and release insulin to maintain glucose concentrations within a defined range22,23. To evaluate whether PrPC-mediated increase in β-cell iron alters the expression of GLUT2 (Fig. 4) and insulin (Fig. 6), pancreas from iron over-loaded Tg40 PrP and corresponding PrP−/− mice were subjected to Western blotting and immunohistochemistry (Fig. 4a–c).

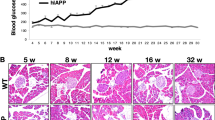
PrP-mediated increase in pancreatic β-cell iron downregulates GLUT2: (a) Pancreatic lysates from control and iron overloaded mice were subjected to Western blotting and probed for GLUT2. Samples from control PrP−/− samples show significant upregulation of GLUT2 relative to Tg40 PrP samples (lanes 3 & 7 vs. 1 & 5; B). Iron overloading downregulates GLUT2 in Tg40 PrP samples (*star), but has minimal effect on similarly treated PrP−/− samples (arrowhead) (lanes 2 & 6 vs. 4 & 8; B). (b) Densitometric analysis of GLUT2 expression after normalization with β-actin. Values are mean ± SEM of the indicated n. *p < 0.05, **p < 0.01, ***p < 0.001, ns, not significant. The full-length blots are provided in Supplementary File (Raw data). (c) Immunoreaction of pancreatic sections from the above mice mirrors the results in panel a. Immunoreaction for GLUT2 is higher in PrP−/− relative to Tg40 PrP samples (panels 1 & 3). Iron overloading reduces reactivity for GLUT2 in Tg40 PrP, but not in PrP−/− samples (panels 1 vs. 2 & 3 vs. 4). Reaction for PrP is limited to pancreatic islets as for GLUT2, and iron overloading upregulates PrP (panels 5 & 6). PrP−/− samples show no reactivity for PrP as expected (panels 7 & 8). H&E sections show no obvious toxicity due to iron overloading (panels 9–12). Scale bar 20 μm.
Probing of pancreatic lysates for GLUT2 revealed significantly higher expression in PrP−/− relative to Tg40 PrP samples. Overloading with iron downregulated GLUT2 in Tg40 PrP, but had no influence on PrP−/− mice (Fig. 4a, lanes 1–8, Fig. 4b).
Immunohistochemistry of pancreas mirrored the Western blot results. Iron overloading decreased GLUT2 reactivity in Tg40 PrP, but had minimal effect on PrP−/− samples (Fig. 4c, panel 1–4). Expression of PrPC was upregulated by excess iron as in Fig. 2 above, and PrP−/− samples showed no reactivity as expected (Fig. 4c, panels 5–8). Staining with H & E showed no damage or change in the architecture of pancreatic tissue due to iron overloading (Fig. 4c, panels 9–12).
To evaluate whether PrPC-mediated increase in IC iron alters glucose transporters on neuronal cells, in particular glucose transporter 3 (GLUT3)42, brain lysates from Tg40 PrP and PrP−/− mice were subjected to Western blotting and probed for GLUT3. As noted in pancreatic β-cells, expression of GLUT3 was upregulated in PrP−/− relative to Tg40 PrP samples (Fig. 5a, lanes 1–4; Fig. 5b).

PrP-mediated increase in IC iron downregulates glucose transporters in the brain, neuroretina, and the liver: (a) Probing of Western blots of brain lysates from Tg40 PrP and PrP−/− mice for GLUT3 shows a significant increase in PrP−/− relative to Tg40 PrP samples (lanes 2 & 4 vs. 1 & 3). (b) Density of protein bands after normalization with β-actin. Values are mean ± SEM of the indicated n. *p < 0.05. (c) Immunoreaction of M17 cells for GLUT3 shows increased reactivity in vector-transfected relative to PrPC over-expressing cells (panels 1 & 3). Exposure to iron downregulates GLUT3 significantly more in PrPC relative to vector controls (panels 2 & 4). Scale bar 25 μm. (d) Western blotting of lysates from control and iron-exposed PrPC and vector expressing cells mirrors the results in panel c. (e) Density of protein bands after normalization with β-actin. Values are mean ± SEM of the indicated n. *p < 0.05, ***p < 0.001, ns, not significant. (f) A similar evaluation of retinal lysates shows upregulation of GLUT1 in PrP−/− relative to Tg40 PrP samples (lanes 1 & 3). Iron overloading downregulates GLUT1 in Tg40 PrP samples relative to untreated controls, but has minimal effect on similarly treated PrP−/− samples (lanes 2 & 4). (g) Density of protein bands after normalization with β-actin. Values are mean ± SEM of the indicated n. *p < 0.05, **p < 0.01, ns, not significant. (h) Probing of Western blots of liver lysates for GLUT2 mirrors the results in pancreatic, brain, and neuroretinal lysates (lanes 1–4). (i) Density of protein bands after normalization with β-actin. Values are mean ± SEM of the indicated n. *p < 0.05, **p < 0.01, ns, not significant. The full-length blots are provided in Supplementary File (Raw data).
Further confirmation of this phenomenon was obtained in M17 cells, a neuroblastoma cell line transfected to over-expressing PrPC and the respective vector control. Immunoreaction of fixed, permeabilized cells for GLUT3 revealed significantly less reactivity in PrPC-expressing cells relative to vector controls (Fig. 5c, panels 1 & 2). Exposure to exogenous iron decreased GLUT3 reactivity in both cell lines, but significantly more in PrPC-expressing cells relative to vector controls (Fig. 5c, panels 3 & 4). These results were confirmed by Western blotting of similarly treated cell lysates. Probing for GLUT3 revealed significantly lower expression in PrPC-expressing cells relative to vector controls (Fig. 5d, lanes 2 & 3; Fig. 5e). Exposure to exogenous iron downregulated GLUT3 in both cell lines, but significantly more in PrPC- expressing cells relative to vector controls (Fig. 5d, lanes 1 & 4; Fig. 5e).
Evaluation of samples from the liver and neuroretina of Tg40 PrP and PrP−/− mice mimicked the results obtained in the pancreas, the brain, and neuronal cells. Western blotting showed downregulation of glucose transporter 1 (GLUT1) in the neuroretina and GLUT2 in the liver of Tg40 PrP relative to PrP−/− samples (Fig. 5f and h, lanes 1 & 3). Iron overloading resulted in the downregulation of GLUT1 and GLUT2 in Tg40 PrP mice, but not in PrP−/− mice (Fig. 5f and h, lanes 2 & 4; Fig. 5g and i).
All the above investigation was also carried out in the C6 mice in both the strains; C6 PrP+/+ and C6 PrP−/−, with similar results in all 4 tissues (see Supplementary Fig. S5).
PrP-mediated increase in β-cell iron downregulates insulin
To evaluate whether expression of GLUT2 influences insulin levels in β-cells, pancreatic lysates from control and iron overloaded Tg40 PrP mice were subjected to Western blotting and probed for insulin with two different antibodies43,44. Essentially the same observations were noted as for GLUT2 (Fig. 4). Reactivity for insulin was significantly higher in PrP−/− relative to Tg40 PrP samples. Iron overloading decreased insulin in Tg40 PrP, but had minimal effect on PrP−/− samples (Fig. 6a, lanes 1–8, upper and lower panels; Fig. 6b). This experiment was also conducted in C6 mice in both the strains; C6 PrP+/+ and C6 PrP−/−, with similar results (see Supplementary Fig. S6).

PrP-mediated increase in β-cell iron decreases insulin: (a) Probing of Western blots of pancreatic lysates from control and iron overloaded Tg40 PrP and PrP−/− mice with antibodies specific for insulin dimer and pentamer (upper and lower panels) shows decreased expression in Tg40 PrP relative to PrP−/− samples (lanes 1 & 5 vs. 3 & 7; b). Iron overloading decreases insulin levels in Tg40 PrP (lanes 1vs. 2 & 5 vs. 6; b), but has minimal effect on PrP−/− samples (lanes 3 vs. 4 & 7 vs. 8; b). (b) Density of protein bands after normalization with β-actin. Values are mean ± SEM of the indicated n. *p < 0.05, ***p < 0.001, ns, not significant. The full-length blots are provided in Supplementary File (Raw data). (c) Immunohistochemistry of pancreas shows relatively higher reactivity for insulin in PrP−/− relative to Tg40 PrP sections (panels 1 & 3). Iron overloading decreases insulin reactivity in Tg40 PrP but not in PrP−/− samples (panels 1 vs. 2 & 3 vs. 4). Reaction for glucagon is higher in iron overloaded Tg40 PrP relative to control samples (panels 5 & 6). The difference is minimal in PrP−/− samples (panels 7 & 8). (d) Immunostaining of 1.1B4 cells for insulin shows decreased reactivity after 2 and 4 hours of exposure to FAC (panels 1–3). (e) Silencing of PrP with siRNA abrogates iron-mediated decrease in insulin reactivity (panels 1–4). Scale bar 20 μm.
Immunoreaction of fixed pancreas for insulin showed reactivity on β-cells as expected. The intensity of insulin reactivity was higher in PrP−/− relative to Tg40 PrP samples, and excess iron reduced the reaction in Tg40 PrP but had minimal effect on PrP−/− samples, consistent with Western blot results in panel A (Fig. 6c, panels 1–4). A similar analysis for glucagon showed increased reactivity in iron overloaded Tg40 PrP samples, indicating proliferation of glucagon producing α-cells by this treatment45. PrP−/− samples, on the other hand, showed minimal reactivity in untreated and iron overloaded samples (Fig. 6c, panels 5–8).
To confirm a causal relationship between β-cell iron levels and insulin, 1.1B4 cells were exposed to FAC for 2 and 4 hours, and immunostained for insulin. Reactivity for insulin decreased after 2 h, and was negligible after 4 h. Nuclear staining revealed normal nuclear morphology with no obvious signs of toxicity (Fig. 6d, panels 1–3). Downregulation of PrPC by siRNA, however, reversed the negative effect of iron on insulin reactivity (Fig. 6e, panels 1–4).
Together, the above observations demonstrate that increase in β-cell iron results in downregulation of GLUT2 and insulin, and PrPC plays a significant role in this process by facilitating iron uptake.
PrPC-mediated dysregulation of blood glucose is exacerbated by iron
To evaluate whether decreased reactivity for insulin in PrPC-expressing β-cells and exaggeration of this phenotype by excess iron is due to decreased synthesis or rapid release from cells, control and iron overloaded C6 PrP+/+ and C6 PrP−/− mice were injected with glucose or insulin according to guidelines for conducting glucose tolerance test (GTT) and insulin tolerance test (ITT) in laboratory mice46. Blood glucose was measured and recorded at the indicated times (Fig. 7). As expected, both mouse lines showed a spike in blood glucose after 15 min (T-15), and a gradual decline to basal levels (T-0) after 180 min (Fig. 7a). It is notable that blood glucose was significantly higher in C6 PrP+/+ samples at T-60 relative to C6 PrP−/− samples (Fig. 7a). This difference was more widespread and exaggerated following over-loading, and the blood glucose of C6 PrP+/+ mice was significantly higher than C6 PrP−/− at all the time-points tested (Fig. 7b). The results from ITT were more dramatic. Following an injection of insulin, the blood in C6 PrP+/+ mice remained significantly higher than C6 PrP−/− samples at all the time points tested regardless of iron overloading (Fig. 7c & d).

PrP-mediated increase in IC iron blunts the systemic response to peripheral challenge with glucose and insulin: (a) GTT in C6 PrP+/+ and C6 PrP−/− mice shows an initial spike in blood glucose in both mouse lines after 15 min, followed by a gradual decline to control levels by 180 min. Blood glucose is significantly higher in C6 PrP+/+ relative to C6 PrP−/− mice at the 60 min time point. (b) Iron overloading decreases glucose tolerance in C6 PrP+/+ relative to C6 PrP−/− mice at all-time points tested. (c) ITT shows a hypoglycemic response in C6 PrP−/− mice relative to that C6 PrP+/+ controls. (d) Iron overloading increases insulin resistance in C6 PrP+/+ mice, but has minimal effect on C6 PrP−/− mice. The data represent mean ± SEM of values from two different strains of mice. *p < 0.05, **p < 0.01, ***p < 0.001.
Relatively poor glucose tolerance in C6 PrP+/+ mice especially after iron overloading suggests reduced levels of circulating insulin in response to glucose, consistent with decreased levels of GLUT2 and insulin in pancreatic β-cells observed above. Impaired response of C6 PrP+/+ mice to injected insulin regardless of iron overload suggests peripheral resistance to insulin that increases after iron overload.
Together, the above observations indicate that C6 PrP+/+ mice display a phenotype of type-2-diabetes, i.e. impaired synthesis of insulin and peripheral resistance to available insulin47, exacerbated further by iron overload. C6 PrP−/− mice, on the other hand, are relatively resistant to iron-mediated fluctuations in blood glucose. These experiments were also conducted on Tg40 PrP and PrP−/− mice with similar trend in GTT and ITT results.
Discussion
Our data demonstrate a direct correlation between PrPC and IC iron, and a converse relationship between IC iron and glucose uptake in pancreatic β-cells, neuronal cells, hepatocytes, and the retina. In pancreatic β-cells, PrPC-mediated increase in IC iron and downregulation of GLUT2 reduced the synthesis and release of insulin. In peripheral organs, iron overloading increased peripheral resistance to insulin in a PrPC-dependent manner, suggesting a dual role of PrPC-mediated increase in systemic iron on glucose homeostasis. Experimental conditions show that stimulated iron uptake by the pancreas resulted in β-cleavage of PrPC, indicating regulation of iron uptake through this process. Together, these observations explain the mechanism underlying PrPC-mediated modulation of blood glucose17, and confirm the positive correlation between iron and type-2-diabetes2 (Fig. 8). Since GLUT3 is downregulated in AD and prion disease affected brains48,49, conditions associated with brain iron dyshomeostasis50, it is likely that PrPC induces toxicity by the combined effect of oxidative stress and glucose deprivation51.

Graphical representation of PrPC-mediated modulation of glucose homeostasis through iron. (1 & 2) PrPC facilitates cellular uptake of Tf-Fe3+ and non-Tf-bound iron (Fe3+) by functioning as a ferrireductase partner for DMT-1 and ZIP1420. (3) The increase in intracellular (IC) iron down-regulates HIF1α53, resulting in the downregulation of GLUT2 in pancreatic β-cells and hepatocytes (black), GLUT 3 in neuronal cells (red), and GLUT1 at the blood-retinal barrier (blue). (4) Reduced uptake of glucose downregulates insulin, resulting in hyperglycemia. (5) Down-regulation or deletion of PrPC decreases IC iron, resulting in the upregulation of HIF1α53 and glucose transporters. (6) Increased uptake of glucose stimulates insulin secretion with resultant hypoglycemia.
It was surprising that PrPC had a significant effect on IC iron in pancreatic β-cells despite relatively low expression17. PrPC was localized to insulin-producing β-cells in mouse pancreas, and showed the expected glycoforms as in neuronal cells. However, unlike neuronal cells, most of the PrPC on pancreatic β-cells and 1.1B4 cells was cleaved at the β-site, a processing event that was triggered by exogenous iron and over-expression of divalent metal transporters DMT-1 and ZIP14. The concomitant increase in cellular ferritin suggested that β-cleavage of PrPC is coupled with iron uptake32, and proteolytic cleavage of PrPC could serve to regulate iron uptake35. Further exploration is necessary to understand this phenomenon. The expression and functional activity of ZIP14 and DMT-1 on human and mouse pancreatic β-cells has been described recently38,52, and supports the above assumption.
A positive correlation between systemic iron and type-2-diabetes is well-established, and several studies have addressed the underlying mechanism2,4. The increase in intracellular (IC) iron down-regulates hypoxia inducible factors 1 (HIF1α)53, resulting in the downregulation of GLUTs. The inverse scenario where hyperglycemia results in iron dysregulation has also been studied54. Our data strengthen these observations, and provide a novel mechanism of glucose modulation through PrPC, a mainly neuronal protein, thereby linking both peripheral and neuronal glucose homeostasis to IC iron. Given the relatively low expression of PrPC on pancreatic β-cells in comparison to neurons, reduced expression of GLUT2 and insulin in the pancreas of Tg40 PrP relative to PrP−/− mice was intriguing, and suggested that even minor changes in β-cell IC iron are sufficient to alter glucose homeostasis. Systemic iron overload amplified this effect and increased peripheral resistance to insulin47, simulating the phenotype of type-2-diabetes. PrP−/− mice, on the other hand, remained unaffected, demonstrating a direct correlation between iron, GLUT2, and insulin. The dose of iron used in our study was within the recommended range for the treatment of anemia, making it unlikely that the observed effects are due to iron-induced β-cell toxicity4. Knock-out of ZIP14 in mouse pancreas has been reported to induce hyperinsulinemia and hypoglycemia due to iron deficiency as in PrP−/− mice, lending support to our observations55. Similar observations were noted in the liver of Tg40 PrP and PrP−/− mice, indicating that the inverse correlation between IC iron and glucose transport is not limited to the pancreas, and relatively low expression of PrPC is sufficient to induce this change. Our results differ from a previous report indicating impaired or delayed response of PrP−/− mice to hyperglycemia17, a discrepancy that is difficult to explain from our data. However, diverse observations regarding PrPC and glucose homeostasis have been reported in the literature56,57,58,59,60,61,62 requiring further exploration on this subject.
Reduced expression of GLUT3 in brain homogenates and GLUT1 in the neuroretina of Tg40 PrP relative to PrP−/− suggests similar regulation of IC iron and glucose transporters by PrPC as in pancreatic β-cells. These observations have significant implications for neuronal cells that express high levels of PrPC and require glucose for their high metabolic demands. Since several neurodegenerative diseases including AD and Creutzfeldt–Jakob disease (sCJD) are associated with neuronal iron dyshomeostasis12, it is likely that in pathological conditions such as these, PrPC accentuates neuronal injury by the combined effect of iron-mediated oxidative stress and glucose deprivation. It is notable that GLUT3 is downregulated in scrapie infected animal brains49 and human cases of CJD63. A similar downregulation of GLUT3 has been observed in AD brains48, a known long-term complication of type-2-diabetes10. Whether PrPC is the principal regulator of GLUT3 expression in neurons is not clear from our data, and remains an open question. However, considering our observations on pancreatic β-cells and the similarities between GLUT2 and GLUT321, it is tempting to speculate that PrPC plays a similar role in neurons, and is likely to influence neuronal health under normal and pathological conditions by modulating iron and glucose uptake24. Likewise, PrPC is expressed widely in the neuroretina, including retinal pigment epithelial cells in the outer blood retinal barrier where it mediates uptake of iron by the neuroretina. PrPC is therefore likely to modulate both iron and glucose homeostasis in the retina, that, like the brain, has a high metabolic rate and depends on glucose for normal function.
In conclusion, our observations reveal a novel function of PrPC in regulating blood glucose through iron, and reaffirm the correlation between systemic iron and type-2-diabetes. The relatively high expression of PrPC in the brain and the neuroretina underscores its significance as a key protein that regulates iron and glucose homeostasis, vital processes critical for the functioning of these metabolically active tissues. The correlation between PrPC, iron, and glucose homeostasis is likely to provide new, untapped therapeutic opportunities for type-2-diabetes, AD, and other neurodegenerative conditions associated with iron dyshomeostasis.
Materials and Methods
Ethics statement
All animals were housed in the Association for Assessment and Accreditation of Laboratory Animal Care International (AAALAC)-approved Animal Resource Center (ARC) at Case Western Reserve University (CWRU) School of Medicine (SOM) under a 12h day-night cycle, and provided ad libitum access to food and water. All experimental procedures were reviewed and approved by the CWRU IACUC committee in accordance with provisions of the Animal Welfare Act and Guide for the Care and Use and of Laboratory Animals as well as the U.S. Government Principles for the Utilization and Care of Vertebrate Animals Used in Testing, Research, and Training (animal protocol # 2015–0027). The ARC at CWRU is directed by Dr. Durfee, DVM, Diplomate ACLAM, and daily animal care is provided by two full-time veterinarians. The CWRU PHS Assurance number A-3145–01 is valid until 04/30/19.
Mouse strains
Four mouse strains were used to improve the validity of this study; PrP-knock out (PrP−/−) mice deposited by25 to Jackson Laboratories (cat # 129-Prnptm2Edin/J Stock No: 012938) and crossed with C57BL/6 wild-type mice for 10 generations. F2 generation of wild type (C6 PrP+/+) and corresponding PrP−/− (C6 PrP−/−) were used for these studies. FVB/NJ Tg40 (Tg40 PrP) that express 2 × human PrP were generated from FVB/Prnp00 mice26 kindly provided by the Prusiner laboratory and used as corresponding controls for Tg40 PrP mice64. All mouse lines were fed regular chow (Prolab Isopro RMH 3000 from www.labdiet.com) and maintained under similar conditions. All experiments were conducted on 6–8 week old male mice since females show less pronounced phenotype of glucose intolerance17, and carried out at the same time of the day.
Chemicals
Alexa Fluor 546-tagged secondary antibodies (A11071, A11018) were from Southern Biotech, USA and Molecular Probes, USA respectively. PNGase F (P0704S) was from New England Biolabs (NEB), USA, Lipofectamine 3000 was from Invitrogen, USA. Ferric ammonium citrate (FAC) (F5879) and desferrioxamine (D9533) were from Sigma Aldrich, USA, D-glucose (dextrose-15023021) was from ThermoFisher Scientific, USA, and Humulin-R was from Eli Lilly, USA. siRNA against PrP (sc36318) and scrambled siRNA (sc37007) were from Santa Cruz Biotechnology Inc, USA.
Scrambled siRNA sequence: Sense: UUCUCCGAACGUGUCACGUtt
Antisense: ACGUGACACGUUCGGAGAAtt
PrP siRNA (h) is a pool of 3 different siRNA duplexes: sc-36318A: Sense: GUGACUAUGAGGACCGUUAtt
Antisense: UAACGGUCCUCAUAGUCACtt sc-36318B: Sense: GAGACCGACGUUAAGAUGAtt
Antisense: UCAUCUUAACGUCGGUCUCtt sc-36318C: Sense: GUUGAGCAGAUGUGUAUCAtt
Antisense: UGAUACACAUCUGCUCAACtt
Note: all sequences are in 5′ → 3′ orientation.
Plasmid encoding DMT-1-GFP (-IRE) was from Jerry Kaplan (University of Utah). PrPC and PrPC-GFP constructs were prepared by ligating cDNA from previously reported constructs in modified PiggyBac vectors (System Biosciences, Mountain view, CA)34. Plasmids encoding ZIP14 and ZIP8 tagged with GFP were obtained from Mitchell D. Knutson, PhD (University of Florida)65. Transfection was carried out using Lipofectamine3000 transfection kit (Invitrogen, USA).
Cell lines, transfection, and RNAi knockdown
Insulin producing human pancreatic β-cell line 1.1B4 was obtained from Sigma Aldrich (Cat. No: 10012801), USA and cultured as described66. This is a PANC-1 hybrid human pancreatic beta cell line that secretes insulin. Cells were cultured in RPMI-1640 supplemented with 2 mM Glutamine and 10% heat inactivated FBS. Cells were passaged every fourth day. For silencing PrPC, 1.1B4 cells were transfected with siRNA against PrPC or scrambled control using Lipofectamine RNAimax (Invitrogen). After 72 h, cells were exposed to 16.7 mM of glucose for 1 h and processed for Western blotting. Human neuroblastoma cells (M17) were purchased from ATCC. Cells expressing vector or PrPC were generated described in previous reports67,68, and cultured in DMEM supplemented with 10% FBS. All cell lines were maintained at 37 °C in a humidified atmosphere containing 5% CO2.
Antibodies
PrPC-specific antibodies 3F4 and 8H4 were from Signet laboratories (Dedham, MA) and Sigma Aldrich respectively. Other antibodies were obtained from the following sources: ferritin specific for heavy and light chain (F5012) from Sigma Aldrich, USA, GLUT-1 (NB110-39113) from Novus Biologicals, GLUT-2 (ab54460), and GLUT-3 (ab41525) from Abcam, USA, TfR (13-6800) from Invitrogen, USA, insulin from Santa Cruz Biotechnology Inc. (sc-9168) and Novus Biologicals (NBP2-34260), USA (recognize insulin and a 51-amino acid polypeptide composed of A and B chains connected through the C-peptide), glucagon (sc-13091) from Santa Cruz Biotechnology Inc, USA, and β-actin (MAB1501) from Millipore, USA. HRP-conjugated secondary antibodies (anti-mouse, NA931V, anti-rabbit, NA934V) were from GE Healthcare, UK.
Iron treatment
1.1B4 and M17 cells cultured in complete medium were exposed to vehicle or 30 μM of ferric ammonium citrate (FAC) for 16 h at 37 °C before processing for immunostaining and Western blot as described69. To create systemic iron overload, age and sex-matched C6 PrP+/+, C6 PrP−/−, Tg40 PrP and PrP−/− mice were injected intraperitoneally with 36 µg/22 g mouse weight of FAC and euthanized after 24 h for further analysis37. The dose of iron used (1.6 mg/kg body weight) is well below the recommended ~10 mg/kg/day used for the treatment of anemia in an average adult weighing 75 kg70.
Western blotting and Immunostaining
Western blotting and immunostaining were carried out as described69. In short, cells and tissues were lysed in RIPA lysis buffer (50 mM Tris-HCl pH 7.4, 100 mM NaCl, 1% NP-40, 0.5% deoxycholate), boiled in reducing gel-loading buffer for 5 min at 100 °C, and fractionated by SDS-PAGE. Fractionated proteins were transferred to PVDF membranes and probed for specific proteins. Quantification of protein bands was performed by densitometry using UN-SCAN-IT gels (version6.1) software (Silk Scientific) and analyzed graphically using GraphPad Prism (Version 5.0) software (GraphPad Software Inc.).
Glucose Tolerance Test (GTT) and Insulin Tolerance Test (ITT)
GTT and ITT were performed in age and sex-matched mice at the same time of day as described46. For GTT, the mice were fasted overnight with ad libidum access to water, and 1 g glucose/kg body weight was injected intraperitoneally. Blood glucose was monitored at 0, 15, 30, 60, 120, and 180 min post-injection with a glucometer (EasyMax-Diabetic Promotions, USA). For ITT, the mice had ad libidum access to food and water because PrP−/− mice went into hypoglycemic shock after insulin injection. Accordingly, non-fasted animals were injected with 0.75 U insulin/kg body weight intraperitoneally, and blood glucose was monitored as above at 0, 15, 30, 45, 60 and 120 min post-injection.
Statistical analysis
Data were analyzed using GraphPad Prism5 (GraphPad Software, Inc., La Jolla, CA) and presented as Mean ± SEM. Level of significance was calculated by Two-way ANOVA between the control and experimental groups.
References
Skyler, J. S. et al. Differentiation of diabetes by pathophysiology, natural history, and prognosis. Diabetes 66, 241–255 (2017).
Simcox, J. A. & McClain, D. A. Iron and diabetes risk. Cell metabolism 17, 329–341 (2013).
Swaminathan, S., Fonseca, V. A., Alam, M. G. & Shah, S. V. The Role of Iron in Diabetes and Its Complications. Diabetes Care 30, 1926–1933, https://doi.org/10.2337/dc06-2625 (2007).
Rajpathak, S. N. et al. The role of iron in type 2 diabetes in humans. Biochimica et Biophysica Acta (BBA)-General Subjects 1790, 671–681 (2009).
Fernandez-Real, J. M., Lopez-Bermejo, A. & Ricart, W. Iron stores, blood donation, and insulin sensitivity and secretion. Clin Chem 51, 1201–1205, https://doi.org/10.1373/clinchem.2004.046847 (2005).
Cario, H., Holl, R. W., Debatin, K. M. & Kohne, E. Insulin sensitivity and beta-cell secretion in thalassaemia major with secondary haemochromatosis: assessment by oral glucose tolerance test. Eur J Pediatr 162, 139–146, https://doi.org/10.1007/s00431-002-1121-7 (2003).
McClain, D. A. et al. High prevalence of abnormal glucose homeostasis secondary to decreased insulin secretion in individuals with hereditary haemochromatosis. Diabetologia 49, 1661–1669, https://doi.org/10.1007/s00125-006-0200-0 (2006).
Acton, R. T. et al. Relationships of serum ferritin, transferrin saturation, and HFE mutations and self-reported diabetes in the Hemochromatosis and Iron Overload Screening (HEIRS) study. Diabetes Care 29, 2084–2089, https://doi.org/10.2337/dc05-1592 (2006).
Lee, D. H., Folsom, A. R. & Jacobs, D. R. Jr. Dietary iron intake and Type 2 diabetes incidence in postmenopausal women: the Iowa Women’s Health Study. Diabetologia 47, 185–194, https://doi.org/10.1007/s00125-003-1307-1 (2004).
Cheng, D. et al. Type 2 diabetes and late-onset Alzheimer’s disease. Dementia and geriatric cognitive disorders 31, 424–430 (2011).
Correia, S. C. et al. Insulin signaling, glucose metabolism and mitochondria: Major players in Alzheimer’s disease and diabetes interrelation. Brain Research 1441, 64–78, https://doi.org/10.1016/j.brainres.2011.12.063 (2012).
Belaidi, A. A. & Bush, A. I. Iron neurochemistry in Alzheimer’s disease and Parkinson’s disease: targets for therapeutics. Journal of neurochemistry 139, 179–197 (2016).
Ciudin, A., Hernández, C. & Simó, R. Iron overload in diabetic retinopathy: a cause or a consequence of impaired mechanisms? Experimental diabetes research 2010 (2010).
Prusiner, S. B. P. Proceedings of the National Academy of Sciences 95, 13363–13383 (1998).
Wulf, M.-A., Senatore, A. & Aguzzi, A. The biological function of the cellular prion protein: an update. BMC biology 15, 34 (2017).
Gains, M. J., Roth, K. A. & LeBlanc, A. C. Prion protein protects against ethanol-induced Bax-mediated cell death in vivo. Neuroreport 17, 903–906, https://doi.org/10.1097/01.wnr.0000220144.14166.e2 (2006).
Strom, A., Wang, G.-S. & Scott, F. W. Impaired glucose tolerance in mice lacking cellular prion protein. Pancreas 40, 229–232 (2011).
Strom, A., Wang, G. S., Reimer, R., Finegood, D. T. & Scott, F. W. Pronounced cytosolic aggregation of cellular prion protein in pancreatic beta-cells in response to hyperglycemia. Lab Invest 87, 139–149, https://doi.org/10.1038/labinvest.3700500 (2007).
de Brito, G. et al. Loss of prion protein is associated with the development of insulin resistance and obesity. Biochem J 474, 2981–2991, https://doi.org/10.1042/BCJ20170137 (2017).
Tripathi, A. K. et al. Prion protein functions as a ferrireductase partner for ZIP14 and DMT1. Free Radical Biology and Medicine 84, 322–330 (2015).
Mueckler, M. & Thorens, B. The SLC2 (GLUT) family of membrane transporters. Molecular aspects of medicine 34, 121–138 (2013).
Thorens, B. GLUT2, glucose sensing and glucose homeostasis. Diabetologia 58, 221–232 (2015).
Sansbury, F. et al. SLC2A2 mutations can cause neonatal diabetes, suggesting GLUT2 may have a role in human insulin secretion. Diabetologia 55, 2381–2385 (2012).
Singh, N. et al. Iron in neurodegenerative disorders of protein misfolding: a case of prion disorders and Parkinson’s disease. Antioxidants & redox signaling 21, 471–484 (2014).
Manson, J. C. et al. 129/Ola mice carrying a null mutation in PrP that abolishes mRNA production are developmentally normal. Molecular neurobiology 8, 121–127, https://doi.org/10.1007/bf02780662 (1994).
Fischer, M. et al. Prion protein (PrP) with amino-proximal deletions restoring susceptibility of PrP knockout mice to scrapie. The EMBO journal 15, 1255–1264 (1996).
Chen, S. G. et al. Truncated forms of the human prion protein in normal brain and in prion diseases. The Journal of biological chemistry 270, 19173–19180 (1995).
Singh, A. et al. Prion Protein Modulates Cellular Iron Uptake: A Novel Function with Implications for Prion Disease Pathogenesis. PLOS ONE 4, e4468, https://doi.org/10.1371/journal.pone.0004468 (2009).
Tank, E. M., Harris, D. A., Desai, A. A. & True, H. L. Prion protein repeat expansion results in increased aggregation and reveals phenotypic variability. Molecular and cellular biology 27, 5445–5455 (2007).
Ma, J. & Wang, F. Prion disease and the ‘protein-only hypothesis’. Essays in biochemistry 56, 181–191 (2014).
Masujin, K. et al. Detection of atypical H-type bovine spongiform encephalopathy and discrimination of bovine prion strains by real-time quaking-induced conversion. Journal of clinical microbiology 54, 676–686 (2016).
Watt, N. T. et al. Reactive oxygen species-mediated β-cleavage of the prion protein in the cellular response to oxidative stress. Journal of Biological Chemistry 280, 35914–35921 (2005).
Mangé, A. et al. Alpha‐and beta‐cleavages of the amino‐terminus of the cellular prion protein. Biology of the Cell 96, 125–132 (2004).
Gu, Y. et al. Mutant prion protein-mediated aggregation of normal prion protein in the endoplasmic reticulum: implications for prion propagation and neurotoxicity. J Neurochem 84, 10–22 (2003).
Asthana, A. et al. Prion protein facilitates retinal iron uptake and is cleaved at the β-site: Implications for retinal iron homeostasis in prion disorders. Scientific Reports 7, 9600, https://doi.org/10.1038/s41598-017-08821-1 (2017).
Arredondo, M., Mendiburo, M. J., Flores, S., Singleton, S. T. & Garrick, M. D. Mouse divalent metal transporter 1 is a copper transporter in HEK293 cells. BioMetals 27, 115–123, https://doi.org/10.1007/s10534-013-9691-6 (2014).
Haldar, S. et al. Prion protein promotes kidney iron uptake via its ferrireductase activity. The Journal of biological chemistry 290, 5512–5522, https://doi.org/10.1074/jbc.M114.607507 (2015).
Coffey, R. & Knutson, M. D. The plasma membrane metal-ion transporter ZIP14 contributes to nontransferrin-bound iron uptake by human β-cells. American Journal of Physiology - Cell Physiology 312, C169–C175, https://doi.org/10.1152/ajpcell.00116.2016 (2017).
Singh, A. et al. Prion protein (PrP) knock-out mice show altered iron metabolism: a functional role for PrP in iron uptake and transport. PLoS One 4, e6115 (2009).
Pushie, M. J. et al. Prion protein expression level alters regional copper, iron and zinc content in the mouse brain. Metallomics 3, 206–214 (2011).
Singh, A. et al. Prion protein regulates iron transport by functioning as a ferrireductase. Journal of Alzheimer’s Disease 35, 541–552 (2013).
Simpson, I. A. et al. The facilitative glucose transporter GLUT3: 20 years of distinction. American Journal of Physiology-Endocrinology and Metabolism 295, E242–E253 (2008).
Chakraborty, D., Samadder, A., Dutta, S. & Khuda-Bukhsh, A. R. Antihyperglycemic potentials of a threatened plant, Helonias dioica: antioxidative stress responses and the signaling cascade. Experimental Biology and Medicine 237, 64–76, https://doi.org/10.1258/ebm.2011.011161 (2012).
Dosanjh, J. K. Protein damage during purification: understanding the effects of size exclusion chromatography on the structure of biosynthetic human insulin (BHI), UCL (University College London), (2011).
Takeda, Y. et al. Reduction of both beta cell death and alpha cell proliferation by dipeptidyl peptidase-4 inhibition in a streptozotocin-induced model of diabetes in mice. Diabetologia 55, 404–412, https://doi.org/10.1007/s00125-011-2365-4 (2012).
Ayala, J. E. et al. Standard operating procedures for describing and performing metabolic tests of glucose homeostasis in mice. Disease Models & Mechanisms 3, 525–534, https://doi.org/10.1242/dmm.006239 (2010).
Chatterjee, S., Khunti, K. & Davies, M. J. Type 2 diabetes. The Lancet 389, 2239–2251, https://doi.org/10.1016/S0140-6736(17)30058-2 (2017).
Simpson, I. A., Chundu, K. R., Davies‐Hill, T., Honer, W. G. & Davies, P. Decreased concentrations of GLUT1 and GLUT3 glucose transporters in the brains of patients with Alzheimer’s disease. Annals of neurology 35, 546–551 (1994).
Yan, Y. E. et al. Significant reduction of the GLUT3 level, but not GLUT1 level, was observed in the brain tissues of several scrapie experimental animals and scrapie-infected cell lines. Molecular neurobiology 49, 991–1004, https://doi.org/10.1007/s12035-013-8574-8 (2014).
Blasco, G. et al. Brain iron overload, insulin resistance, and cognitive performance in obese subjects: a preliminary MRI case-control study. Diabetes Care 37, 3076–3083 (2014).
Singh, N. The role of iron in prion disease and other neurodegenerative diseases. PLoS pathogens 10, e1004335 (2014).
Hansen, J. B. et al. Divalent metal transporter 1 regulates iron-mediated ROS and pancreatic β cell fate in response to cytokines. Cell metabolism 16, 449–461 (2012).
Cheng, K. et al. Hypoxia-inducible factor-1alpha regulates beta cell function in mouse and human islets. J Clin Invest 120, 2171–2183, https://doi.org/10.1172/JCI35846 (2010).
Rowe, P. A., Kavanagh, K., Zhang, L., Harwood, H. J. & Wagner, J. D. Short-term hyperglycemia increases arterial superoxide production and iron dysregulation in atherosclerotic monkeys. Metabolism 60, 1070–1080 (2011).
Aydemir, T. B. et al. Zinc transporter ZIP14 functions in hepatic zinc, iron and glucose homeostasis during the innate immune response (endotoxemia). PLoS One 7, e48679 (2012).
Li, Q. Q. et al. Cellular prion protein promotes glucose uptake through the Fyn-HIF-2alpha-Glut1 pathway to support colorectal cancer cell survival. Cancer science 102, 400–406, https://doi.org/10.1111/j.1349-7006.2010.01811.x (2011).
Kuwahara, C. et al. Enhanced expression of cellular prion protein gene by insulin or nerve growth factor in immortalized mouse neuronal precursor cell lines. Biochem Biophys Res Commun 268, 763–766, https://doi.org/10.1006/bbrc.2000.2152 (2000).
Bitel, C. L., Feng, Y., Souayah, N. & Frederikse, P. H. Increased expression and local accumulation of the prion protein, Alzheimer Aβ peptides, superoxide dismutase 1, and nitric oxide synthases 1 & 2 in muscle in a rabbit model of diabetes. BMC physiology 10, 18 (2010).
Pham, N., Dhar, A., Khalaj, S., Desai, K. & Taghibiglou, C. Down regulation of brain cellular prion protein in an animal model of insulin resistance: possible implication in increased prevalence of stroke in pre-diabetics/diabetics. Biochemical and biophysical research communications 448, 151–156 (2014).
Zhu, C., Schwarz, P., Abakumova, I. & Aguzzi, A. Unaltered prion pathogenesis in a mouse model of high-fat diet-induced insulin resistance. PloS one 10, e0144983 (2015).
Nielsen, D., Gyllberg, H., Ostlund, P., Bergman, T. & Bedecs, K. Increased levels of insulin and insulin-like growth factor-1 hybrid receptors and decreased glycosylation of the insulin receptor alpha-and beta-subunits in scrapie-infected neuroblastoma N2a cells. Biochemical Journal 380, 571 (2004).
Östlund, P., Lindegren, H., Pettersson, C. & Bedecs, K. Altered insulin receptor processing and function in scrapie-infected neuroblastoma cell lines. Molecular Brain Research 97, 161–170 (2001).
Kim, E. J. et al. Glucose metabolism in sporadic Creutzfeldt–Jakob disease: a statistical parametric mapping analysis of 18F‐FDG PET. European journal of neurology 19, 488–493 (2012).
Kong, Q. et al. Chronic Wasting Disease of Elk: Transmissibility to Humans Examined by Transgenic Mouse Models. The Journal of Neuroscience 25, 7944–7949, https://doi.org/10.1523/jneurosci.2467-05.2005 (2005).
Coffey, R. & Knutson, M. D. The plasma membrane metal-ion transporter ZIP14 contributes to nontransferrin-bound iron uptake by human β-cells. American Journal of Physiology-Cell Physiology 312, C169–C175 (2017).
Vasu, S., McClenaghan, N. H., McCluskey, J. T. & Flatt, P. R. Mechanisms of toxicity by proinflammatory cytokines in a novel human pancreatic beta cell line, 1.1B4. Biochimica et Biophysica Acta (BBA) - General Subjects 1840, 136–145, https://doi.org/10.1016/j.bbagen.2013.08.022 (2014).
Gu, Y., Fujioka, H., Mishra, R. S., Li, R. & Singh, N. Prion peptide 106-126 modulates the aggregation of cellular prion protein and induces the synthesis of potentially neurotoxic transmembrane PrP. The Journal of biological chemistry 277, 2275–2286, https://doi.org/10.1074/jbc.M104345200 (2002).
Jin, T. et al. The chaperone protein BiP binds to a mutant prion protein and mediates its degradation by the proteasome. The Journal of biological chemistry 275, 38699–38704, https://doi.org/10.1074/jbc.M005543200 (2000).
Baksi, S., Tripathi, A. K. & Singh, N. Alpha-synuclein modulates retinal iron homeostasis by facilitating the uptake of transferrin-bound iron: Implications for visual manifestations of Parkinson’s disease. Free Radical Biology and Medicine 97, 292–306, https://doi.org/10.1016/j.freeradbiomed.2016.06.025 (2016).
Koch, T. A., Myers, J. & Goodnough, L. T. Intravenous iron therapy in patients with iron deficiency anemia: dosing considerations. Anemia 2015 (2015).
Acknowledgements
This study was supported by grant NS092145 from the National Institutes of Health, NINDS, to NS. Neena Singh is the guarantor of this work and, as such, had full access to the data in the study and takes full responsibility for the integrity and accuracy of the data. We thank Stuti Dalal for assistance in experimental work.
Author information
Authors and Affiliations
Contributions
N.S. Conceived and designed the study, supervised experimental plan and data collection, wrote and edited the manuscript; A.A. designed the study, planned and executed experiments, analyzed the data, wrote and edited the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ashok, A., Singh, N. Prion protein modulates glucose homeostasis by altering intracellular iron. Sci Rep 8, 6556 (2018). https://doi.org/10.1038/s41598-018-24786-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-24786-1
This article is cited by
-
Cellular prion protein offers neuroprotection in astrocytes submitted to amyloid β oligomer toxicity
Molecular and Cellular Biochemistry (2023)
-
Mixed pathologies in pancreatic β cells from subjects with neurodegenerative diseases and their interaction with prion protein
Acta Neuropathologica Communications (2021)
-
Cellular prion protein activates Caspase 3 for apoptotic defense mechanism in astrocytes
Molecular and Cellular Biochemistry (2021)
-
Altered cellular localisation and expression, together with unconventional protein trafficking, of prion protein, PrPC, in type 1 diabetes
Diabetologia (2021)
-
Cellular prion protein dysfunction in a prototypical inherited metabolic myopathy
Cellular and Molecular Life Sciences (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
