
Abstract
Trypanosoma cruzi is the agent of Chagas disease, transmitted by hematophagous triatomine vectors. Establishing transmission cycles is key to understand the epidemiology of the disease, but integrative assessments of ecological interactions shaping parasite transmission are still limited. Current approaches also lack sensitivity to assess the full extent of this ecological diversity. Here we developed a metabarcoding approach based on next-generation sequencing to identify triatomine gut microbiome, vertebrate feeding hosts, and parasite diversity and their potential interactions. We detected a dynamic microbiome in Triatoma dimidiata, including 23 bacterial orders, which differed according to blood sources. Fourteen vertebrate species served as blood sources, corresponding to domestic, synantropic and sylvatic species, although four (human, dog, cow and mice) accounted for over 50% of blood sources. Importantly, bugs fed on multiple hosts, with up to 11 hosts identified per bug, indicating very frequent host-switching. A high clonal diversity of T. cruzi was detected, with up to 20 haplotypes per bug. This analysis provided much greater sensitivity to detect multiple blood meals and multiclonal infections with T. cruzi, which should be taken into account to develop transmission networks, and characterize the risk for human infection, eventually leading to a better control of disease transmission.
Similar content being viewed by others

Introduction
Trypanosoma cruzi is the agent of Chagas disease, a major vector-borne parasitic disease in the Americas. T. cruzi parasite transmission from triatomine vectors to vertebrate hosts occurs via the insect’s feces through a complex succession of events, making transmission from vector to vertebrate host rather unlikely1. The parasite is nonetheless able to infect a very large variety of mammalian hosts covering many orders, including Marsupialia, Rodentia, Lagomorpha, Chiroptera, Carnivora and Primata. The identification of the host species involved in transmission cycles is key to understand the epidemiology of the disease and the risk for human transmission. The identification of triatomine host feeding patterns and their variations2,3,4,5,6,7 is starting to provide useful information to assess the role of different hosts species as parasite reservoirs or for contributing to increasing the risk for human infection. Separate or overlapping transmission cycles among species communities may also be evidenced7,8,9,10. Nonetheless, host feeding sources and feeding behavior of triatomines remain poorly understood as current techniques based on direct sequencing of PCR products can only identify the dominant sequence/host in each sample. The addition of a cloning step prior to sequencing allowed detecting up to 3–5 host species in some bugs4,5,6, suggesting that multiple blood meals may be more frequent than previously acknowledged. Such multiple blood meals are key to evaluate the potential for parasite transmission between hosts species.
Similarly, while parasite genetic diversity has been known for a long time and has led to the division of T. cruzi into seven discrete typing units (DTUs TcI to TcVI and Tcbat)11,12, the geographic distribution of the different DTUs as well as their association with transmission cycles restricted to specific habitats and hosts is beginning to be challenged13,14,15. Indeed, while it was believed that TcI DTU was largely predominant in Mexico and Central America (over 95% of the strains)12,16, recent studies have documented the presence of non-TcI parasite strains in triatomines from different regions in Mexico and Central America at high frequencies2,14,17. Similarly in the southern US, initial work reported only TcI and TcIV DTUs18, but recent studies also indicate the presence of additional non-TcI DTUs at important frequencies19,20. Together, these observations indicate clearly that T. cruzi genotype distribution in many countries and regions needs to be re-examined16. Also, potential parasite genetic differentiation and adaptation to specific hosts should be examined to establish the epidemiologic relevance of T. cruzi genotypes in the transmission of Chagas disease. As for the identification of blood feeding source mentioned above, current PCR genotyping methods are also limited to detecting the dominant genotype in biological samples, and the multiclonality of infections is mostly overlooked. Indeed, the genetic analysis of multiple parasite clones isolated from single hosts revealed concurrent infections with up to 10 parasite genotypes per opossum individual21. More recently, the analysis of sequence variation in GP63 through deep sequencing also revealed a large number of gene variants present in Chagasic patients22. The multiclonality of infection in humans is also evidenced in longitudinal studies of patients, a large proportion of which present changes in the dominant DTU identified before and after drug treatment, indicating that infection with multiple DTUs and heterogeneous drug response may be occurring23. Multiclonal infections may thus be the norm rather than the exception, and the interactions among parasite genotypes within vectors and hosts are still mostly unknown.
Parasite development and the vectorial capacity of vectors may also be affected by the bacteria present in the insect gut24,25. For example, the development of Trypanosoma brucei, the agent of African trypanosomiasis, in its tsetse fly vector, is directly influenced by a microbiome-regulated gut immune barrier26. Gut microbiome similarly modulates dengue virus infection in Aedes aegypti mosquitos27,28, and microbiome manipulation may be used to control virus transmission29. Triatomines harbor a diverse gut microbiome that is beginning to be identified30,31, but its influence on T. cruzi transmission remains unclear. In Triatoma infestans, infection by T. cruzi induces the overexpression of antimicrobial peptides modulating the microbiome, and this inhibition of the bacterial microflora is important for parasite establishment in the vector32. Nonetheless, our understanding of triatomine microbiome is still too limited to further assess its role in vectorial capacity and T. cruzi transmission, or to take full advantage of its potential manipulation in novel parasite control strategies such as para-transgenesis as suggested before33,34.
Finally, while some of the aspects mentioned above have begun to be evaluated in an isolated manner, there has been no or little effort to provide an integrative assessment of the ecological interactions that combine to shape T. cruzi transmission. Such interactions may however be critical for parasite transmission dynamics and Chagas disease epidemiology, and may lead to novel disease control approaches. Our objective was thus to develop an integrative and highly sensitive molecular approach using metabarcoding based on next-generation sequencing for the identification of triatomine genotype, gut microbiome, vertebrate feeding hosts, and parasite genetic diversity and their potential interactions.
Materials and Methods
Triatoma dimidiata collections and DNA extraction
A total of 14 bugs identified as Triatoma dimidiata were used in this study. They were collected during entomological surveillance following a pilot vector control intervention during years 2013–2015, in the villages of Bokoba, Sudzal and Teya35, located in the Yucatan peninsula, Mexico, about 15–20 km apart36, as well as in the sylvatic habitat surrounding these villages (up to 8 km from the villages)(Supplementary Table S1). The study was approved by both the World Health Organization and the Autonomous University of Yucatan institutional bioethics committees. DNA vas purified from the distal part of the abdomen of individual bugs using Qiagen DNAeasy Blood & Tissue kit following the instructions from the manufacturer, eluted in 20 µl of water, and stored at −20 °C until used.
DNA markers amplification
We used triatomine Internal Transcribed Spacer (ITS)-2 nuclear marker for T. dimidiata genotyping and sibling species identification, as before37,38,39. Briefly, a 320 bp sequence was amplified (35 cycles) using the previously reported primers ITS2_200F (5′-TCGYATCTAGGCATTGTCTG-3′) and ITS2_200R (5′-CTCGCAGCTACTAAGGGAATCC-3′) and PCR conditions40,41.
To identify the microbiome composition, a 140 bp fragment of the bacterial 16 S rRNA gene was PCR amplified (35 cycles) using primers E786F (5′-GATTAGATACCCTGGTAG-3′) and U926R (5′-CCGTCAATTCCTTTRAGTTT-3′) as described before42.
For the identification of vertebrate blood meals, a 215 bp fragment of the 12 S rRNA gene was amplified (35 cycles) with the primers L1085 (5′-CCCAAACTGGGATTAGATACCC-3′ and H1259 (5′-GTTTGCTGAAGATGGCGGTA-3′) as described before5,41,43.
The presence of T. cruzi parasites in bugs was detected by PCR amplification (35 cycles) of parasite satellite DNA with primers TcZ44,45. Parasite genotyping was performed by multiplex PCR (40 cycles) using a mixture of three primers able to amplify T. cruzi DTUs TcI and TcBat (350 bp), TcII, TcV and TcVI (300 bp), and TcIV (380–400 bp), based on the mini-exon gene46. DNA samples purified from reference strains from the relevant T. cruzi DTUs were used as controls.
All PCR reactions were run using Thermo Scientific™ DreamTaq™ DNA polymerase. Positive and negative PCR controls were included in each batch of PCR reactions. A DNA extraction control using only water was also PCR amplified with each for the DNA marker used to detect potential contamination during DNA extraction. All PCR products were separated in 2% agarose gels, stained with ethidium bromide and visualized under UV light, to ensure the presence of bands of the expected size.
Next generation sequencing
PCR products for triatomine ITS2 gene, bacterial 16 S gene, vertebrate 12 S rRNA gene, and T. cruzi parasite markers were pooled for each bug, and cleaned using Promega Wizard® SV Gel and PCR Clean-Up System. Following end-repair and indexing, libraries were prepared and sequenced on a MiSeq (Illumina) or Ion Torrent (Life Technologies) platforms. From 100,000 to 900,000 reads were obtained from each bug after quality and size filtering. Sequence data were deposited in the SRA database under accession number SRR6337087 to SRR6337099. Reads were aligned to reference sequences for each of the target markers in Geneious 9.1 using the Geneious algorithm, corresponding to a depth ranging from 130 to 340,000 reads per marker (Supplementary Table S1). Sequences were trimmed of primer sequences for further analysis.
Sequence and data analysis
Sequences for bacterial 16 S gene and vertebrate 12 S rRNA gene were screened for chimeras using UChime47, and chimeric sequences were discarded form further analysis. Sequences for each marker were then aligned using Muscle as implemented in Geneious to assess sequence diversity. Sequence variants were identified using SNP/variant tool, to distinguish between sequencing errors/artefacts and significant sequence variants. For triatomine ITS-2 sequence analysis, variant haplotypes were aligned with reference ITS-2 sequences representing the proposed sibling species of the T. dimidiata complex37,38 for a precise taxonomic identification of the triatomines. Templeton, Crandall and Sing (TCS) haplotype networks48 of ITS-2 sequences were constructed in PopArt 1.7. New ITS-2 haplotype sequences were deposited in Genbank database under accession numbers: MF767417 to MF767433.
For microbiome composition, bacterial 16 S rRNA sequences were analyzed using a Bayesian classifier from the Ribosomal Database Project49,50 with update 5, release 11 of the sequence database. Taxonomic identification of bacteria was made at a threshold of >96% sequence identity, and the frequency of sequences for each taxonomic unit was considered as a proxy of its abundance in triatomine gut. Rarefaction curves were elaborated, at the individual and group level, to estimate species richness of our sampling. For blood meal sources, 12 S rRNA sequences were analyzed by MEGABLAST against the entire “nr” GenBank database (version of March 2017). Sequence match with >96% identity was used for species or genus identification, with an E value of at least 6.09E-77, and sequences representing less than 0.5% of a blood meal were discarded from the analysis. Rarefaction curves were also constructed. Sequences identified from the same species were further aligned to detect sequence haplotype variants as an indicator of feeding on different hosts of the same species. A feeding network and parasite transmission pathways was constructed using Cytoscape 3.5, to visualize the frequency of the respective feeding sources as well as possible pathways for parasite transmission among species when multiple blood meals were detected, since feeding sources can be used as evidence of vector-host contact. Nodes of the network represent the various species detected as feeding sources, and edges link hosts that were found in the same individual gut content. Network statistics were calculated as implemented in Cytoscape, and included the average number of neighbors, network density, network centralization, average clustering coefficient, and neighborhood connectivity distribution.
Parasite sequences from the mini-exon gene were aligned with sequences from reference strains covering all DTUs (USAOPOSSUM (TcI), Tu18 (TcII), AF1Cl7 (TcII), M6531 (TcIII), M6241 (TcIII), MT4167 (TcIV), CanIII (TcIV), SC43 (TcV), MN (TcV), CL Brener (TcVI) and VSC (TcVI)), and TCS haplotype networks were constructed using PopArt, taking into account the observed frequency of the different haplotypes within bugs, to visualize parasite diversity infecting T. dimidiata. Due to large differences between TcI and non-TcI sequences, these were also analyzed separately. T. cruzi mini-exon sequences were deposited in Genbank database under accession numbers: MF770768 to MF770822. To assess if parasites haplotypes were consistently associated within bugs, or rather circulated independently from one another, we determined which haplotypes are shared between bugs, and which are unique. This allowed identifying sets of haplotypes that can be transmitted independently from each others among vectors and hosts. Assuming that no parasite selection occurs in triatomines and a good sensitivity of our method allowing to detect rare haplotypes, this is likely to reflect independent infection events of the bugs, allowing to estimate the possible number of infection events of bugs (alternatively, it may also indicate infection on a single mammal with a multi-clonal infection).
To further describe microbiome, blood meal sources and parasite genotype sequence diversity, Shannon diversity index (H = -Σ(ni/N).ln(ni/N)) as well as Margalef Richness index ((S-1)/ln N) were calculated, with ni representing the number of individuals of species/taxa i, N the total number of individuals, and S the total number of species/taxa. Indices were compared using Student’s t test, and linear regression was used to assess the potential associations among biodiversity indicators. Frequency data were compared by χ2 or Fisher’s Exact tests.
Results
Triatoma dimidiata ITS-2
Over 260,000 partial ITS-2 sequences (209 bp long) were analyzed, corresponding to an average of 18,627 sequences per bug (N = 14). Four bugs presented a single ITS-2 sequence, and eight presented two sequence haplotypes at a frequency of about 50% each, suggesting that these corresponded to heterozygote bugs. Interestingly, one bug presented four haplotypes, at frequencies of 46, 39, 9 and 6%, respectively. Two of these four sequences were novel and not found in other bugs, thus were not a contamination from other samples, but we could not exclude that they may represent chimeric sequences. Overall, these data confirmed the extensive concerted evolution of this multi-copy gene, which mostly behaves as a single copy sequence. It also confirmed the filtering out of errors/artefacts throughout our amplification, sequencing and analysis process.
Comparison of ITS-2 haplotypes with reference haplotypes confirmed that all analyzed bugs belonged to the previously described Group 3 of this species complex37,38, and the haplotype of most bugs corresponded to known ITS-2 haplotypes (Fig. 1). Eleven bugs (Bok006, Bok011, Bok012, Sud059, Sud061, Sud094, Tey011, Tey012, Tey015, Tey016, Tey139) presented haplotypes identical to H28, H29 and H30. Four new haplotypes were identified differing in 1 nucleotide each: Sud036, also observed in three other bugs (Sud059, Sud094, and Tey139), and three additional haplotypes were all observed in bug Sud054. There was no association between the ITS-2 haplotype and the village of collection of the bugs (Fisher’s Exact Test, P = 0.8368).

ITS-2 haplotypes of T. dimidiata. Partial ITS-2 sequences were aligned with reference haplotypes H19, H20, H21, H23, H24, H28, H29, H30, H31, H32, covering Groups 2 and 3 from Genbank, and a TCS network was constructed in PopArt. Circles represent the respective haplotypes, their size proportional to the number of sequences. Ticks on branches indicate the number of mutations separating haplotypes. ITS-2 Group 2 (yellow) and Group 3 (green) are clearly separated by at least 11 mutations as described before. Haplotypes labeled in bold font correspond to new haplotypes.
Microbiome composition
About 500,000 partial 16 S rRNA sequences (110 bp long) were analyzed, corresponding to an average of 37,654 sequences per bug (N = 13). A total of 23 bacterial orders were identified with high confidence, and rarefaction curve indicated that most of the richness had been identified through our sampling (Supplementary Figure S1), although some additional low abundance taxa may remain to be detected. The overall Shannon diversity index (H) was 1.41 and Margalef richness index was 1.90. The most abundant orders were Bacillales, followed by Actinomycetales, Enterobacteriales and Burkholderiales, which accounted for over 70% of T. dimidiata microbiome (Fig. 2A), and are commonly found in other insect gut microbiome including triatomines31,51,52. The main bacteria genus identified were Rhodococcus, Corynebacterium, Actinomycetospora and Arthrobacter (from the Actnomycetales order) and Bacillus, Staphylococcus and Anoxybacillus (from the Bacillales order). Importantly, Wolbachia was not detected in any of the samples, indicating that it might be absent from this insect species.

Gut microbiome composition of Triatoma dimidiata. The average composition of the microbiome from 14 individuals is shown, to the level of bacterial order (A). There are significant differences between male and female microbiomes, with females presenting a greater diversity of orders. (B) Microbiome composition is also significantly different depending on the dominant blood meal present in triatomine gut, which was identified by analysis of 12 S rARN vertebrate sequences (see below).
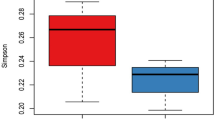
Interestingly, the composition of bacterial orders was significantly different between males and females (χ2 = 141.3, d.f. = 18, P < 0.0001), with females presenting a larger diversity of orders in their microbiome (Fig. 3A). Margalef Richness index was indeed higher in females compared to males (1.49 ± 0.16 vs 0.97 ± 0.21, respectively, P = 0.041), and Shannon Index tended to be higher in females (1.70 ± 0.14 vs 1.40 ± 0.29, P = 0.19). Oceanospirillales, Pseudomonadales, Xanthomonadales, Flavobacteriales, Caulobacterales, Burkholderiales and Actinomycetales were more abundant in females, while Bacillales, Bacteroidales, Enterobacteriales, Neisseriales and Rhodobacterales were more abundant in males. More strikingly, microbiome composition differed strongly based on the dominant blood meal source of the bugs (Fig. 2B), with Burkholderiales predominating in bugs fed on dogs, Bacillales in those fed on humans, and Enterobacteriales for those fed on porcupine, respectively (χ2 = 519.5, d.f. = 36, P < 0.0001).

Blood feeding sources of Triatoma dimidiata. The sources of blood were identified to the genus or species level by analysis of 12 s rRNA sequences obtained by NGS, and corresponded to 14 genus/species (A). Females tended to feed on a wider diversity of species, but this did not reach significance. (B) Feeding on multiple species or individuals was detected in most individual bugs (bugs 1–14), with an average of 4.9 ± 0.7 hosts/bug. (C) Multiple dog 12 S haplotypes detected in a single bug (Bok012), which had feed on at least 4 dogs. Nucleotides highlighted in color indicate sequence variants. These data indicate very frequent host-switching when feeding, within and among species.
Vertebrate host feeding
About 218,000 partial vertebrate 12 S rRNA sequences (about 170 bp long) were analyzed for the identification of blood sources of triatomines, corresponding to an average of 15,573 sequences per bug (N = 14). MEGABLAST analysis revealed a diverse range of at least 14 host species, including the expected domestic and synantropic species such as dog (Canis lupus familiaris), cow (Bos taurus), cat (Felis spp.), mouse (Mus musculus), rat (Rattus spp.), pig (Sus scrofa), turkey (Meleagris gallopavo) and chicken (Gallus gallus), in addition to humans (Homo sapiens) (Supplementary Table 2 for BLAST scores). Sylvatic species were also detected, corresponding to porcupine (Coendou spp.), squirrel (Sciurus spp.), fruit bat (Artibeus spp.), kinkajou (Potos flavus), and dove (Zenaida spp.) (Supplementary Table 2 for BLAST scores), with a small proportion of sequences with ambiguous or unidentified origin (Fig. 3A). Only four species (human, dog, cow and mice) accounted for over 50% of blood sources, and may thus play a key role in parasite transmission cycles. There were small differences in blood sources of male and female triatomines, with a tendency of females to have a somewhat wider range of hosts (Fig. 3A), but this did not reach statistical significance (χ2 = 12.5, d.f. = 14, P = 0.56). Shannon diversity index for the blood sources of female and male bugs was also similar (0.44 ± 0.18 vs 0.39 ± 0.16, respectively, P = 0.43), as well as the Margalef richness index (0.43 ± 0.18 vs 0.34 ± 0.10, respectively, P = 0.33).
At the level of individual bugs, variations in sequence frequency allowed to easily identify a dominant (and likely last) blood meal, accounting for 50–100% of the sequences, while lower frequency sequences may correspond to older or partial blood meals. Thus, multiple host species were detected in all bugs except one, with up to 7 identified host species in a single bug (Fig. 3B). Within species, multiple haplotypes were also detected (Fig. 3C), so that the average number of hosts/bug reached 4.9 ± 0.7. Based on the persistence of blood DNA in triatomine gut for about 5–6 weeks after a blood meal53, these data indicate an average of about 50 hosts/year for each bug, up to about 115 hosts/year. These results also show frequent feeding on different hosts by individual bugs, which suggested an opportunist rather than specialized feeding behavior of T. dimidiata. The mixture of domestic and sylvatic species as blood sources among several individual bugs further confirmed the occurrence of widely overlapping parasite transmission cycles among species.
To further assess potential transmission cycles of T. cruzi parasites by T. dimidiata among vertebrate host species, a feeding and transmission network was constructed (Fig. 4). Feeding frequency on each host is indicated by the size of the node, to reflect the more common feeding sources, while connecting edges between species are based on the observed occurrence of multiple feeding sources within single bugs. Since birds cannot carry T. cruzi parasites, they only play a role as blood source for triatomines, which is indicated by dotted edge connections between hosts, the solid lines between mammals indicate potential parasite transmission pathways. The observed network shows a strong connectivity (average number of neighbors: 7.57, network density 0.58) and strong clustering (average clustering coefficient: 0.85) with a medium centralization (network centralization: 0.49) (Fig. 4), which illustrates the frequent host-switching behavior of T. dimidiata. Analysis of neighborhood connectivity distribution showed that neighborhood connectivity decreased strongly with the number of neighbors (Supplementary Figure S2, R2 = 0.96, P < 0.0001), indicating that highly connected nodes predominate in shaping the network. Indeed, the network shows the limited role of birds as feeding sources, while a mixture of domestic and sylvatic mammalian species contribute to potential parasite transmission pathways (Fig. 4). Humans may thus become infected by T. cruzi parasites originating from dogs, cows and mice, as well as from sylvatic hosts such as porcupines, squirrels and fruit bats. Particularly, dogs can play a key role as domestic host/reservoir favoring parasite transmission to humans, while, cats, rats and pigs play a secondary role in parasite transmission.

Feeding and possible parasite transmission network of T. dimidiata. Blood source nodes correspond to domestic (green symbols) and sylvatic (orange symbols) host species, as well as humans (blue), with the size proportional to the feeding frequency on each host. Diamond shaped nodes represent birds, which do not carry T. cruzi parasites, and circles represent mammals, which can be infected by T. cruzi. Edges link species which are found together in multiple blood meals within individual bugs, and the width of the lines is proportional to the frequency of the association between species. Solid dark gray lines link mammalian species, among which T. cruzi may circulate, while dotted light gray lines involve bird species, which only serve as blood sources for the bugs. Humans may thus become infected by T. cruzi parasites originating from dogs, cows and mice, as well as from sylvatic hosts such as porcupines, squirrels and fruit bats. Dogs can play a key role as domestic host/reservoir favoring parasite transmission to humans. On the other hand, cats, rats and pigs play a secondary role in parasite transmission.
Trypanosoma cruzi infection
About 50,000 partial mini-exon sequences (260–300 bp long) from T. cruzi were analyzed from infected T. dimidiata bugs for the characterization of parasite diversity in triatomines, corresponding to an average of 8,420 sequences per bug (N = 6). This allowed the detection of an important haplotype diversity within each individual triatomine, ranging from 2 to 20 parasite haplotypes, with frequencies ranging from 0.1% to 65% (Tables 1 and 2). TCS network analysis indicated that haplotype diversity covered several DTUs, including TcI, as well as TcII-TcV-TcVI DTUs, but TcIII, TcIV and TcBat were not detected (Fig. 5A). For a finer resolution, TcI and non-TcI sequences were thus analyzed separately. For TcI, a total of 20 haplotypes were detected, with up to 12 in a single bug (Bok012), and only 2 in Tey011 bug (Fig. 5B). For TcII, TcV and TcVI, 20 haplotypes were detected, all also present in a single bug (Tey016). Several of these haplotypes were identical or clearly related to TcII DTU, and others were somewhat closer to TcV and TcVI reference sequences, but could not be easily assigned to a specific DTU. One bug (Sud036) harbored haplotypes from three TcI and one TcII DTUs. Some haplotypes were shared among bugs (for example TcI-H1 and H2 shared by 5 and 4 bugs, respectively), while others were more exclusive, suggesting the independent circulation of many groups of haplotypes among bugs (Fig. 5B,C). While the mini-exon gene is a multi-copy sequence54, which may thus present some level of paralogous sequence variation within a single parasite clone/genome, the detection of independent groups of haplotypes circulating among bugs and of multiple parasite DTUs strongly support at least some level of multiclonality of T. cruzi infection in T. dimidiata. Based on the independent groups of haplotypes, the number of likely infection events could be estimated for individual bugs, and up to 4 infection events were detected in some cases.

Trypanosoma cruzi genetic structure in Triatoma dimidiata. TCS network were constructed based on sequences from the mini-exon gene. Circles represent unique T. cruzi haplotypes, with the size of the circle proportional to their frequency, and the colors correspond to the indicated bugs harboring the parasites. Tey000 are bugs from the village of Teya, while Sud036 is from the village of Sudzal, and Bok012 from the village of Bokoba. Ticks on branches between haplotypes indicate the number of nucleotide substitutions separating each haplotypes. (A) Complete network shows the diversity of parasite DTUs present, including TcI and TcII-V-VI DTUs, which are not resolved in this global analysis. (B) Detailed network of TcI haplotypes, showing that some haplotypes are found in most bugs (H1 and H2), while others are found in only a few or even a single bug. The independent occurrence of haplotypes in different bugs, and at variable frequencies, suggest the occurrence of multiple infection events. (C) Detailed network analysis of TcII, TcV and TcVI DTU sequences.
Shannon diversity index was then used to compare the diversity of T. cruzi parasites with the number of infection events, feeding sources and gut microbiome diversity in individual T. dimidiata bugs (Fig. 6). As expected, parasite diversity significantly increased with the number of infection events (R2 = 0.69, P = 0.039). Unexpectedly, we could not detect an association between parasite diversity and blood meal species diversity (P = 0.96), possibly because of our small sample size combined with the fact that only recent blood meals can be detected, while bugs remain infected with T. cruzi for their lifespan. Also, parasite diversity tended to increase with microbiome diversity, but this did not reached statistical significance (R2 = 0.25, P = 0.38).

Association between parasite diversity, feeding sources and microbiome diversity. Shannon diversity index was used to compare the overall diversity of T. cruzi parasite, feeding sources and gut microbiome diversity in T. dimidiata bugs. (A) Parasite diversity significantly increased with the number of infection events (R2 = 0.69, P = 0.039). (B) There was no association between parasite diversity and blood meal species diversity (P = 0.96). (C) Parasite diversity tended to increase with microbiome diversity, but this did not reach statistical significance (R2 = 0.25, P = 0.38).
Discussion
Molecular approaches are providing very relevant information on T. cruzi transmission and the risk for human infection in many settings. However, most of these approaches have limited sensitivity to assess the full diversity of triatomine feeding hosts and parasite genotypes, or lack integration. We developed here a metabarcoding strategy based on next-generation sequencing to successfully identify a wide range of ecological associations of T. dimidiata in the Yucatan peninsula, Mexico. Analysis of triatomine ITS-2 sequences confirmed that our study focused on ITS-2 Group 3 of T. dimidiata species complex. This group is clearly differentiated from the other ITS-2 group37,38, with a different ecological niche at the regional level55, although it appears to have a comparable epidemiological role as other sub-species from the dimidiata complex39,56,57. The lack of ITS-2 sequence variants in most bugs also confirmed the strong concerted evolution of this multi-copy marker, as well as the reliability of our methodological approach.
We also obtained extensive information on T. dimidiata gut microbiome for the first time. We identified that the most common bacterial orders present are similar to those found in the microbiome of other insect vectors such as Aedes, as well as other triatomine species. However, there were also some differences among T. dimidiata and other triatomines. For example, Clostridiales and Rhodocyclales are more frequent in Triatoma infestans, Rhodnius prolixus, and Panstrongylus megistus but marginally present in T. dimidiata51. Enterobateriales predominate in Rhodnius prolixus52, while Bacillales are more abundant in T. dimidiata. Rhizobiales, Burkholderiales, Sphingomonadales, Pseudomonadales, Caulobacterales, Lactobacilales and Xanthomonadales are also relatively abundant in T. dimidiata, but have not been reported in other triatomines51, although they have been observed in mosquitoes such as Aedes spp., Culex spp.58,59,60 or ants61. Remarkably, Wolbachia was not detected in T. dimidiata, in agreement with its absence from triatomine species such as Triatoma infestans, Triatoma vitticeps, Dipetalogaster maximus, Panstrongylus maximus30, Triatoma brasiliensis and Triatoma pseudomaculata31, and Rhodnius remains the only triatomine genus in which Wolbachia has been detected in both natural and laboratory populations62.
T. dimidiata microbiome appeared as very dynamic, with differences between males and females, which may reflect differences in nutrient needs. Blood feeding sources may also affect microbiome composition, as suggested by our observation of differing microbiomes among bugs with different dominant blood sources, and further studies are needed to clarify this point. In mosquitoes, the microbiome has been found to be similarly very dynamic, showing seasonal variations63,64, as well as changes due to the environment, diet, and aging65, although a constant core microbiome could be identified. Further studies should help clarify the variations in T. dimidiata microbiome composition according to its ecological conditions.
Blood feeding was observed on at least 14 host species, most of which had been reported before as feeding sources for T. dimidiata2,6,66,67,68, with the exception of some of the sylvatic hosts such as the kinkajou, porcupine, squirrel and fruit bat. However, the most striking observation was that all bugs except one had blood from more than one host, with up to 11 hosts detected in a single bug. This is considerably higher that what has been observed with previous molecular approaches, which at best can detect 3–5 hosts/bug4,5,6, and most studies report unmixed/single blood meals7. This highlights the high sensitivity of our deep sequencing approach. We were thus able to identify not only the dominant blood meal, likely corresponding to the most recent/abundant meal, but also the remaining older or partial blood meals present in much lower amount. The identification of blood from different hosts in a bug represents key evidence of host-switching, which appears to be very frequent for T. dimidiata. Feeding frequency of triatomines has been a long sought parameter, as it contributes to transmission dynamics69. Estimates from field studies suggested that it was around a blood meal every 1.7–7 days for T. infestans7,70. Our data suggest about one (at least partial) blood meal every week, up to one every 3 days, which is very consistent with these previous studies. We thus estimated that T. dimidiata could feed on up to 115 hosts per year, which may provide many opportunities for parasite circulation among hosts and individuals and/or species. Such a large number of feeding hosts is also considerably higher than for other vector species such as mosquitoes, which at best feed on a handful of hosts during their lifespan69, and this is likely to strongly affect parasite transmission dynamics71. In addition, the mixture of blood meals on domestic and sylvatic hosts confirmed the strong overlap of domestic and sylvatic transmission cycles, which may reflect a high mobility of both vectors and hosts between these habitats. This is in agreement with the intrusive behavior of T. dimidiata in this region72,73,74,75,76,77.
Information on multiple blood meals was used to construct potential T. cruzi transmission network as it evidences chains of vector-host contacts. Dogs emerged as a key host for parasite transmission to humans, as observed in other regions78,79,80, and also in agreement with a relatively high prevalence of infection of this host81,82. This observation strengthens the rationale for controlling T. cruzi infection in dogs as part of an integral control intervention. This may be achieved by insecticide treatment83,84,85 or a vaccine86,87. Mice also seem to play an important role in parasite transmission, although their infection rate by T. cruzi may be highly variable in the region81,88,89. On the other hand, our network indicated that rats and cats played a limited role in parasite transmission to humans, in spite of a significant prevalence of T. cruzi infection observed in previous studies81,90. Nonetheless, rodent control has been successfully tested as part of a Chagas disease control intervention in Guatemala91. Cows rather than pigs are other domestic animals that appear to contribute to the circulation of T. cruzi in our setting, although limited information is available about T. cruzi infection in these hosts92. Importantly, several sylvatic host species including porcupine, squirrel and fruit bat, reported here for the first time as important blood sources for T. dimidiata, were also strongly connected to potential parasite transmission to humans and other domestic hosts, emphasizing the direct links between sylvatic and domestic hosts of T. cruzi. Importantly, fruit bats have previously been found to be infected by T. cruzi (about 3% for Artibeus spp.) in the southern part of the Yucatan peninsula89.
Unexpectedly, no blood meals were detected from Didelphis opossums, which have been reported with a very high T. cruzi infection rate in the region (12–54%)81,89,93. This may be due to the limited number of bugs analyzed, but infection through other routes such as oral or through anal gland may also play a role for these hosts. PCR primer bias may also occur, although we successfully amplified control opossum DNA from Yucatan with our primers, and opossum has been previously reported using the same primers6. Analysis of additional populations of T. dimidiata, and of the seasonal variations in feeding profiles using our approach should provide additional information to refine feeding and transmission networks, and of the potential source of parasites infecting humans.
Analysis of T. cruzi genetic diversity revealed a very high level of parasite diversity in T. dimidiata, with up to 20 parasite haplotypes per bug, based on the analysis of the mini-exon marker. While this diversity may be due to sequence variants in paralogous copies within clones/genomes of this repeated sequence, variations in haplotype frequencies, their apparent independent circulation among bugs, and the presence of several well established parasite DTUs all support extensive multiclonality of infection. While most studies focus on identifying the main dominant genotype circulating in vectors and overlook this multiclonality14,89,94,95,96, our data emphasize the very high sensitivity of our approach, which provided detailed information on the composition of parasite populations infecting bugs, not only at the level of sequence haplotype, but also on their relative frequency. Indeed, we were able to detect even very low frequency haplotypes, representing as little as 0.1% of the parasite haplotypes present in T. dimidiata. Some of this diversity may be due to contamination among samples following incorrect indexing, which would tend to minimize differences among bugs, or may also represent chimeric sequences and/or sequencing artefacts. However, we also detected rare parasite haplotypes in a single bug (for example H18-TcI (0.2%) is unique to Tey015, or H41-NoTcI (0.1%) is unique to Tey016), which are more likely to be true sequence variants.
A high clonal diversity of T. cruzi in T. dimidiata is in agreement with the extensive host switching behavior described above, as bugs feeding on a large variety of hosts may likely get infected with a large variety of parasites. Using Shannon diversity index as an indicator of parasite diversity, we could confirm the significant correlation between parasite genetic diversity with the number of infection event of bugs. However, we could not detect any correlation between blood sources diversity and parasite diversity. As mentioned above, this may be due to small sample size, combined with the fact that blood meals only reflect recent feeding host, while T. cruzi infection lasts for the entire lifespan of the bugs. On the other hand, parasite diversity may be associated with microbiome diversity, and future studies should help establish the critical interactions between triatomine microbiome and T. cruzi development51.
TcII DTU was detected in T. dimidiata for the first time in the region, and possibly additional non-TcI DTUs are also present since our analysis revealed a large number of haplotype variant within the closely related TcV and TcVI DTUs. Although the reference sequences from TcII, TcV and TcVI DTUs are clearly separated, we observed many intermediate sequences much harder to classify. Further studies using additional markers should help refine parasite genetic structure, and the population dynamics resulting from interactions among parasite genotypes. Overall, our results are in agreement with the large diversity of T. cruzi strains and DTUs observed in T. dimidiata in Veracruz, Mexico13, and Guatemala17, as well as in vertebrate hosts from the Yucatan89, even though these studies only documented the dominant genotypes as mentioned above. Our observations raise the questions of the nature of the interactions among parasite haplotypes within vectors, which may play an important role in shaping parasite transmission and the epidemiology of T. cruzi infection97,98. In any case, it is clear that future studies should take into account the multiclonality of T. cruzi infections to further understand parasite transmission networks and the epidemiology of Chagas disease.
In conclusion, we described here a powerful new approach based on metabarcoding through next generation sequencing to provide detailed information on ecological associations of T. dimidiata bugs in an integrative manner. Importantly, our analysis provided much greater sensitivity than conventional molecular approaches to detect multiple blood meals in bugs as well as multiclonal infections with T. cruzi. Moreover, our observations confirm the presence of a very dynamic triatomine microbiome, which may be modulated by blood sources and may interact with T. cruzi parasite. This extensive diversity of feeding sources and parasite infections allowed documenting transmission network. Expanding these studies to additional T. dimidiata populations and at different times of the year will provide unprecedented data to characterize T. cruzi parasite transmission and the risk for human infection, and which ultimately will lead to a better control of disease transmission.
References
Nouvellet, P., Dumonteil, E. & Gourbiere, S. The improbable transmission of Trypanosoma cruzi to human: The missing link in the dynamics and control of Chagas disease. PLoS neglected tropical diseases 7, e2505 (2013).
Torres-Montero, J., López-Monteon, A., Dumonteil, E. & Ramos-Ligonio, A. House infestation dynamics and feeding sources of Triatoma dimidiata in central Veracruz, Mexico. The American journal of tropical medicine and hygiene 86, 677–682 (2012).
Klotz, S. A. et al. Free-roaming kissing bugs, vectors of Chagas disease, feed often on humans in the Southwest. The American journal of medicine 127, 421–426 (2014).
Gorchakov, R. et al. Trypanosoma cruzi Infection Prevalence and Bloodmeal Analysis in Triatomine Vectors of Chagas Disease From Rural Peridomestic Locations in Texas, 2013–2014. J Med Entomol 53, 911–918 (2016).
Waleckx, E., Suarez, J., Richards, B. & Dorn, P. L. Triatoma sanguisuga blood meals and potential for Chagas disease, Louisiana, USA. Emerging infectious diseases 20, 2141–2143 (2014).
Stevens, L., Monroy, M. C., Rodas, A. G. & Dorn, P. L. Hunting, swimming, and worshiping: human cultural practices illuminate the blood meal sources of cave dwelling Chagas vectors (Triatoma dimidiata) in Guatemala and Belize. PLoS neglected tropical diseases 8, e3047 (2014).
Gurtler, R. E. et al. Domestic animal hosts strongly influence human-feeding rates of the Chagas disease vector Triatoma infestans in Argentina. PLoS neglected tropical diseases 8, e2894 (2014).
Buitrago, R. et al. Blood meal sources of wild and domestic Triatoma infestans (Hemiptera: Reduviidae) in Bolivia: connectivity between cycles of transmission of Trypanosoma cruzi. Parasites & vectors 9, 214 (2016).
Almeida, C. E., Faucher, L., Lavina, M., Costa, J. & Harry, M. Molecular Individual-Based Approach on Triatoma brasiliensis: Inferences on Triatomine Foci, Trypanosoma cruzi Natural Infection Prevalence, Parasite Diversity and Feeding Sources. PLoS neglected tropical diseases 10, e0004447 (2016).
Gottdenker, N. L., Chaves, L. F., Calzada, J. E., Saldana, A. & Carroll, C. R. Host life history strategy, species diversity, and habitat influence Trypanosoma cruzi vector infection in Changing landscapes. PLoS neglected tropical diseases 6, e1884 (2012).
Zingales, B. et al. A new consensus for Trypanosoma cruzi intraspecific nomenclature: second revision meeting recommends TcI to TcVI. Memorias do Instituto Oswaldo Cruz 104, 1051–1054 (2009).
Zingales, B. et al. The revised Trypanosoma cruzi subspecific nomenclature: rationale, epidemiological relevance and research applications. Infection, genetics and evolution 12, 240–253 (2012).
Ramos-Ligonio, A., Torres-Montero, J., Lopez-Monteon, A. & Dumonteil, E. Extensive diversity of Trypanosoma cruzi discrete typing units circulating in Triatoma dimidiata from central Veracruz, Mexico. Infection, genetics and evolution 12, 1341–1343 (2012).
Ibanez-Cervantes, G. et al. Identification by Q-PCR of Trypanosoma cruzi lineage and determination of blood meal sources in triatomine gut samples in Mexico. Parasitology international 62, 36–43 (2013).
Risso, M. G. et al. Immunological Identification of Trypanosoma cruzi Lineages in Human Infection Along the Endemic Area. The American journal of tropical medicine and hygiene 84, 78–84 (2011).
Breniere, S. F., Waleckx, E. & Barnabe, C. Over Six Thousand Trypanosoma cruzi Strains Classified into Discrete Typing Units (DTUs): Attempt at an Inventory. PLoS neglected tropical diseases 10, e0004792 (2016).
Pennington, P. M., Paiz, C., Grajeda, L. M. & Cordon-Rosales, C. Short report: concurrent detection of Trypanosoma cruzi lineages I and II in domestic Triatoma dimidiata from Guatemala. The American journal of tropical medicine and hygiene 80, 239–241 (2009).
Roellig, D. M. et al. Molecular typing of Trypanosoma cruzi isolates, United States. Emerging infectious diseases 14, 1123–1125 (2008).
Herrera, C. P., Licon, M. H., Nation, C. S., Jameson, S. B. & Wesson, D. M. Genotype diversity of Trypanosoma cruzi in small rodents and Triatoma sanguisuga from a rural area in New Orleans, Louisiana. Parasites & vectors 8, 123 (2015).
Garcia, M. N. et al. Molecular identification and genotyping of Trypanosoma cruzi DNA in autochthonous Chagas disease patients from Texas, USA. Infection, genetics and evolution 49, 151–156 (2017).
Llewellyn, M. S. et al. Extraordinary Trypanosoma cruzi diversity within single mammalian reservoir hosts implies a mechanism of diversifying selection. International journal for parasitology 41, 609–614 (2011).
Llewellyn, M. S. et al. Deep sequencing of the Trypanosoma cruzi GP63 surface proteases reveals diversity and diversifying selection among chronic and congenital Chagas disease patients. PLoS neglected tropical diseases 9, e0003458 (2015).
Martinez-Perez, A. et al. Prevalence of Trypanosoma cruzi’s Discrete Typing Units in a cohort of Latin American migrants in Spain. Acta tropica 157, 145–150 (2016).
Azambuja, P., Garcia, E. S. & Ratcliffe, N. A. Gut microbiota and parasite transmission by insect vectors. Trends in parasitology 21, 568–572 (2005).
Kelly, P. H. et al. The Gut Microbiome of the Vector Lutzomyia longipalpis Is Essential for Survival of Leishmania infantum. mBio 8 (2017).
Weiss, B. L., Wang, J., Maltz, M. A., Wu, Y. & Aksoy, S. Trypanosome infection establishment in the tsetse fly gut is influenced by microbiome-regulated host immune barriers. PLoS pathogens 9, e1003318 (2013).
Hill, C. L., Sharma, A. A., Shouche, Y. & Severson, D. W. Dynamics of midgut microflora and dengue virus impact on life history traits in Aedes aegypti. Acta tropica 140, 151–157 (2014).
Ramirez, J. L. et al. Reciprocal tripartite interactions between the Aedes aegypti midgut microbiota, innate immune system and dengue virus influences vector competence. PLoS neglected tropical diseases 6, e1561 (2012).
Jupatanakul, N., Sim, S. & Dimopoulos, G. The insect microbiome modulates vector competence for arboviruses. Viruses 6, 4294–4313, https://doi.org/10.3390/v6114294 (2014).
da Mota, F. F. et al. Cultivation-independent methods reveal differences among bacterial gut microbiota in triatomine vectors of Chagas disease. PLoS neglected tropical diseases 6, e1631 (2012).
Gumiel, M. et al. Characterization of the microbiota in the guts of Triatoma brasiliensis and Triatoma pseudomaculata infected by Trypanosoma cruzi in natural conditions using culture independent methods. Parasites & vectors 8, 245 (2015).
Buarque, D. S. et al. A new antimicrobial protein from the anterior midgut of Triatoma infestans mediates Trypanosoma cruzi establishment by controlling the microbiota. Biochimie 123, 138–143 (2016).
Durvasula, R. V. et al. Prevention of insect-borne disease: an approach using transgenic symbiotic bacteria. Proceedings of the National Academy of Sciences of the United States of America 94, 3274–3278 (1997).
Taracena, M. L. et al. Genetically modifying the insect gut microbiota to control Chagas disease vectors through systemic RNAi. PLoS neglected tropical diseases 9, e0003358 (2015).
Waleckx, E. et al. An innovative ecohealth intervention for Chagas disease vector control in Yucatan, Mexico. Transactions of the Royal Society of Tropical Medicine and Hygiene 109, 143–149 (2015).
Ramirez-Sierra, M. J., Herrera-Aguilar, M., Gourbière, S. & Dumonteil, E. Patterns of house infestation dynamics by non-domiciliated Triatoma dimidiata reveal a spatial gradient of infestation in rural villages and potential insect manipulation by Trypanosoma cruzi. Tropical medicine & international health: TM & IH 15, 77–86 (2010).
Bargues, M. D. et al. Phylogeography and genetic variations of Triatoma dimidiata, the main Chagas disease vector in Central America, and its position within the genus Triatoma. PLoS neglected tropical diseases 2, e233 (2008).
Dorn, P. L. et al. Two distinct Triatoma dimidiata (Latreille, 1811) taxa are found in sympatry in Guatemala and Mexico. PLoS neglected tropical diseases 3, e393 (2009).
Herrera-Aguilar, M. et al. Identification of a large hybrid zone between sympatric sibling species of Triatoma dimidiata in the Yucatan peninsula, Mexico, and its epidemiological importance. Infection, genetics and evolution 9, 1345–1351 (2009).
Richards, B., Rua, N. M., Monroy, C., Stevens, L. & Dorn, P. L. Novel polymerase chain reaction-restriction fragment length polymorphism assay to determine internal transcribed spacer-2 group in the Chagas disease vector, Triatoma dimidiata (Latreille, 1811). Memorias do Instituto Oswaldo Cruz 108, 395–398 (2013).
Wong, Y. Y. et al. Molecular epidemiology of Trypanosoma cruzi and Triatoma dimidiata in costal Ecuador. Infection, genetics and evolution 41, 207–212 (2016).
Baker, G. C., Smith, J. J. & Cowan, D. A. Review and reanalysis of domain-specific 16s primers. J Microbiol Meth 55, 541–555 (2003).
Pellecer, M. J., Dorn, P. L., Bustamante, D. M., Rodas, A. & Monroy, M. C. Vector blood meals are an early indicator of the effectiveness of the Ecohealth approach in halting Chagas transmission in Guatemala. The American journal of tropical medicine and hygiene 88, 638–644 (2013).
Moser, D. R., Kirchhoff, L. V. & Donelson, J. E. Detection of Trypanosoma cruzi by DNA amplification using the polymerase chain reaction. Journal of clinical microbiology 27, 1477–1482 (1989).
Dumonteil, E. et al. Geographic distribution of Triatoma dimidiata and transmission dynamics of Trypanosoma cruzi in the Yucatan peninsula of Mexico. The American journal of tropical medicine and hygiene 67, 176–183 (2002).
Souto, R. P., Fernandes, O., Macedo, A. M., Campbell, D. A. & Zingales, B. DNA markers define two major phylogenetic lineages of Trypanosoma cruzi. Molecular and biochemical parasitology 83, 141–152 (1996).
Edgar, R. C., Haas, B. J., Clemente, J. C., Quince, C. & Knight, R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 27, 2194–2200 (2011).
Templeton, A. R., Crandall, K. A. & Sing, C. F. A cladistic analysis of phenotypic associations with haplotypes inferred from restriction endonuclease mapping and DNA sequence data. III. Cladogram estimation. Genetics 132, 619–633 (1992).
Wang, Q., Garrity, G. M., Tiedje, J. M. & Cole, J. R. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol 73, 5261–5267 (2007).
Cole, J. R. et al. Ribosomal Database Project: data and tools for high throughput rRNA analysis. Nucleic acids research 42, D633–642 (2014).
Diaz, S., Villavicencio, B., Correia, N., Costa, J. & Haag, K. L. Triatomine bugs, their microbiota and Trypanosoma cruzi: asymmetric responses of bacteria to an infected blood meal. Parasites & vectors 9, 636 (2016).
Vieira, C. S. et al. Rhodnius prolixus interaction with Trypanosoma rangeli: modulation of the immune system and microbiota population. Parasites & vectors 8, 135 (2015).
Bosseno, M. F. et al. Identification in triatomine vectors of feeding sources and Trypanosoma cruzi variants by heteroduplex assay and a multiplex miniexon polymerase chain reaction. The American journal of tropical medicine and hygiene 74, 303–305 (2006).
O’Connor, O., Bosseno, M. F., Barnabe, C., Douzery, E. J. & Breniere, S. F. Genetic clustering of Trypanosoma cruzi I lineage evidenced by intergenic miniexon gene sequencing. Infection, genetics and evolution 7, 587–593 (2007).
Gomez-Palacio, A., Arboleda, S., Dumonteil, E. & Peterson, A. T. Ecological niche and geographic distribution of the Chagas disease vector, Triatoma dimidiata (Reduviidae: Triatominae): Evidence for niche differentiation among cryptic species. Infection, genetics and evolution 36, 15–22 (2015).
Ramirez-Sierra, M. J. & Dumonteil, E. Infection Rate by Trypanosoma cruzi and Biased Vertebrate Host Selection in the Triatoma dimidiata (Hemiptera: Reduvidae) Species Complex. J Med Entomol 53, 20–25 (2016).
Nouvellet, P., Ramirez-Sierra, M. J., Dumonteil, E. & Gourbiere, S. Effects of genetic factors and infection status on wing morphology of Triatoma dimidiata species complex in the Yucatan pensinsula, Mexico. Inf Genet Evol 11, 1243–1249 (2011).
Zotzmann, S. et al. Bacterial diversity of cosmopolitan Culex pipiens and invasive Aedes japonicus from Germany. Parasitol Res 116, 1899–1906 (2017).
Audsley, M. D., Ye, Y. H. & McGraw, E. A. The microbiome composition of Aedes aegypti is not critical for Wolbachia-mediated inhibition of dengue virus. PLoS neglected tropical diseases 11, e0005426 (2017).
Coon, K. L., Brown, M. R. & Strand, M. R. Mosquitoes host communities of bacteria that are essential for development but vary greatly between local habitats. Molecular ecology 25, 5806–5826 (2016).
Kautz, S., Rubin, B. E., Russell, J. A. & Moreau, C. S. Surveying the microbiome of ants: comparing 454 pyrosequencing with traditional methods to uncover bacterial diversity. Appl Environ Microbiol 79, 525–534 (2013).
Espino, C. I. et al. Detection of Wolbachia bacteria in multiple organs and feces of the triatomine insect Rhodnius pallescens (Hemiptera, Reduviidae). Appl Environ Microbiol 75, 547–550 (2009).
Duguma, D., Hall, M. W., Smartt, C. T. & Neufeld, J. D. Temporal Variations of Microbiota Associated with the Immature Stages of Two Florida Culex Mosquito Vectors. Microbial ecology 74, 979–989 (2017).
Novakova, E. et al. Mosquito Microbiome Dynamics, a Background for Prevalence and Seasonality of West NileVirus. Frontiers in microbiology 8, 526 (2017).
David, M. R., Santos, L. M., Vicente, A. C. & Maciel-de-Freitas, R. Effects of environment, dietary regime and ageing on the dengue vector microbiota: evidence of a core microbiota throughout Aedes aegypti lifespan. Memorias do Instituto Oswaldo Cruz 111, 577–587 (2016).
Farfan-Garcia, A. E. & Angulo-Silva, V. M. Conducta alimentaria de poblaciones de Triatoma dimidiata (Hemiptera: Reduviidae: Triatominae) en una zona endemica y sus implicaciones epidemiologicas. Rev Salud Publica (Bogota) 13, 163–172 (2011).
Christensen, H. A., Sousa, O. E. & de Vasquez, A. M. Host feeding profiles of Triatoma dimidiata in peridomestic habitats of western Panama. The American journal of tropical medicine and hygiene 38, 477–479 (1988).
Monteon, V., Alducin, C., Hernandez, J., Ramos-Ligonio, A. & Lopez, R. High frequency of human blood in Triatoma dimidiata captured inside dwellings in a rural community in the Yucatan peninsula, Mexico, but low antibody seroprevalence and electrocardiographic findings compatible with Chagas disease in humans. The American journal of tropical medicine and hygiene 88, 566–571 (2013).
Rascalou, G., Pontier, D., Menu, F. & Gourbiere, S. Emergence and prevalence of human vector-borne diseases in sink vector populations. PloS one 7, e36858 (2012).
Lopez, A., Crocco, L., Morales, G. & Catala, S. Feeding frequency and nutritional status of peridomestic populations of Triatoma infestans from Argentina. Acta tropica 73, 275–281 (1999).
Nouvellet, P., Cucunuba, Z. M. & Gourbiere, S. Ecology, evolution and control of Chagas disease: a century of neglected modelling and a promising future. Advances in parasitology 87, 135–191 (2015).
Waleckx, E., Gourbière, S. & Dumonteil, E. Intrusive triatomines and the challenge of adapting vector control practices. Memorias do Instituto Oswaldo Cruz 110, 324–338 (2015).
Gourbière, S., Dumonteil, E., Rabinovich, J., Minkoue, R. & Menu, F. Demographic and dispersal constraints for domestic infestation by non-domiciliated Chagas disease vectors in the Yucatan peninsula, Mexico. The American journal of tropical medicine and hygiene 78, 133–139 (2008).
Guzman-Tapia, Y., Ramirez-Sierra, M. J. & Dumonteil, E. Urban infestation by Triatoma dimidiata in the city of Mérida, Yucatan, Mexico. Vector Borne Zoonotic Dis 7, 597–606 (2007).
Payet, V., Ramirez-Sierra, M. J., Rabinovich, J., Menu, F. & Dumonteil, E. Variations in sex-ratio, feeding and fecundity of Triatoma dimidiata between habitats in the Yucatan Peninsula, Mexico. Vector Borne Zoonotic Dis 9, 243–251 (2009).
Dumonteil, E. et al. Assessment of Triatoma dimidiata dispersal in the Yucatan peninsula of Mexico using morphometry and microsatellite markers. The American journal of tropical medicine and hygiene 76, 930–937 (2007).
Barbu, C., Dumonteil, E. & Gourbière, S. Characterization of the dispersal of non-domiciliated Triatoma dimidiata through the selection of spatially explicit models. PLoS neglected tropical diseases 4, e777 (2010).
Castanera, M. B., Lauricella, M. A., Chuit, R. & Gurtler, R. E. Evaluation of dogs as sentinels of the transmission of Trypanosoma cruzi in a rural area of north-western Argentina. Annals of tropical medicine and parasitology 92, 671–683 (1998).
Crisante, G., Rojas, A., Teixeira, M. M. & Anez, N. Infected dogs as a risk factor in the transmission of human Trypanosoma cruzi infection in western Venezuela. Acta tropica 98, 247–254 (2006).
Gurtler, R. E. et al. Domestic dogs and cats as sources of Trypanosoma cruzi infection in rural northwestern Argentina. Parasitology 134, 69–82 (2007).
Zavala-Velázquez, J., Barrera-Perez, M., Rodriguez-Felix, M. E., Guzman-Marin, E. & Ruiz-Piña, H. Infection by Trypanosoma cruzi in mammals in Yucatan, Mexico: a serological and parasitological study. Rev. Inst. Med. Trop. Sao Paulo 38, 289–292 (1996).
Cruz-Chan, J. V. et al. Immunopathology of natural Trypanosoma cruzi infection in dogs. Veterinary parasitology 162, 151–155 (2009).
Gurtler, R. E., Ceballos, L. A., Stariolo, R., Kitron, U. & Reithinger, R. Effects of topical application of fipronil spot-on on dogs against the Chagas disease vector Triatoma infestans. Transactions of the Royal Society of Tropical Medicine and Hygiene 103, 298–304 (2009).
Reithinger, R., Ceballos, L., Stariolo, R., Davies, C. R. & Gurtler, R. E. Chagas disease control: deltamethrin-treated collars reduce Triatoma infestans feeding success on dogs. Transactions of the Royal Society of Tropical Medicine and Hygiene 99, 502–508 (2005).
Amelotti, I., Catala, S. S. & Gorla, D. E. Effects of fipronil on dogs over Triatoma infestans, the main vector of Trypanosoma cruzi, causative agent of Chagas disease. Parasitol Res 111, 1457–1462 (2012).
Basombrio, M. A., Segura, M. A., Mora, M. C. & Gomez, L. Field trial of vaccination against American trypanosomiasis (Chagas’ disease) in dogs. The American journal of tropical medicine and hygiene 49, 143–151 (1993).
Quijano-Hernandez, I. A. et al. Preventive and therapeutic DNA vaccination partially protect dogs against an infectious challenge with Trypanosoma cruzi. Vaccine 31, 2246–2252, https://doi.org/10.1016/j.vaccine.2013.03.005 (2013).
Panti-May, J. A. et al. A survey of zoonotic pathogens carried by house mouse and black rat populations in Yucatan, Mexico. Epidemiol Inf, in press 145, 2287–2295 (2017).
Lopez-Cancino, S. A. et al. Landscape ecology of Trypanosoma cruzi in the southern Yucatan Peninsula. Acta tropica 151, 58–72 (2015).
Jimenez-Coello, M., Acosta-Viana, K. Y., Guzman-Marin, E., Gomez-Rios, A. & Ortega-Pacheco, A. Epidemiological survey of Trypanosoma cruzi infection in domestic owned cats from the tropical southeast of Mexico. Zoonoses and public health 59(Suppl 2), 102–109 (2012).
De Urioste-Stone, S. M. et al. Development of a community-based intervention for the control of Chagas disease based on peridomestic animal management: an eco-bio-social perspective. Transactions of the Royal Society of Tropical Medicine and Hygiene 109, 159–167 (2015).
Jimenez-Coello, M., Acosta-Viana, K. Y., Guzman-Marin, E. & Ortega-Pacheco, A. American trypanosomiasis infection in fattening pigs from the south-east of Mexico. Zoonoses Public Health 59(Suppl 2), 166–169 (2012).
Ruiz-Piña, H. A., Abán-Cauich, A. S., Rosado-Barrera, M. E., Arjona-Torres, A. D. & Mendoza-Camargo, L. A. Natural infection by Trypanosoma cruzi in Didelphis virginiana in Yucatan, Mexico. Proc. Seventh Int. Theriological Congress, Acapulco, Mexico, 286 (1997).
Goncalves Santana, R. A. et al. Trypanosoma cruzi strain TcI is associated with chronic Chagas disease in the Brazilian Amazon. Parasites & vectors 7, 267, https://doi.org/10.1186/1756-3305-7-267 (2014).
Cura, C. I. et al. Multiplex Real-Time PCR Assay Using TaqMan Probes for the Identification of Trypanosoma cruzi DTUs in Biological and Clinical Samples. PLoS neglected tropical diseases 9, e0003765 (2015).
Marcet, P. L. et al. PCR-based screening and lineage identification of Trypanosoma cruzi directly from faecal samples of triatomine bugs from northwestern Argentina. Parasitology 132, 57–65 (2006).
Devillers, H., Lobry, J. R. & Menu, F. An agent-based model for predicting the prevalence of Trypanosoma cruzi I and II in their host and vector populations. J Theor Biol 255, 307–315 (2008).
Tomasini, N., Ragone, P. G., Gourbiere, S., Aparicio, J. P. & Diosque, P. Epidemiological modeling of Trypanosoma cruzi: Low stercorarian transmission and failure of host adaptive immunity explain the frequency of mixed infections in humans. PLoS Comput Biol 13, e1005532 (2017).
Acknowledgements
This work received financial support from the UNDP/World Bank/WHO Special Program for Research and Training in Tropical Diseases (TDR)/International Development Research Center (IDRC), Project #A90276, from Grant #632083 from Tulane University School of Public Health and Tropical Medicine, and from CONACYT (National Council of Science and Technology, Mexico) Basic Science (Project ID: CB2015-258752) and National Problems (Project ID: PN2015-893) Programs.
Author information
Authors and Affiliations
Contributions
E.D., S.G. and E.W. developed the conceptual framework and designed the research. M.J.R.S., S.P.C. and C.T.P. performed the experiments. S.G., E.W. and C.H. contributed analytical tools. E.D., C.H., E.W. and S.G. analyzed the data. All authors contributed to writing the manuscript and approved the final submission.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Dumonteil, E., Ramirez-Sierra, MJ., Pérez-Carrillo, S. et al. Detailed ecological associations of triatomines revealed by metabarcoding and next-generation sequencing: implications for triatomine behavior and Trypanosoma cruzi transmission cycles. Sci Rep 8, 4140 (2018). https://doi.org/10.1038/s41598-018-22455-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-22455-x
This article is cited by
-
Biological, ecological and trophic features of invasive mosquitoes and other hematophagous arthropods: What makes them successful?
Biological Invasions (2024)
-
“Mi Casa, Tu Casa”: the coati nest as a hub of Trypanosoma cruzi transmission in the southern Pantanal biome revealed by molecular blood meal source identification in triatomines
Parasites & Vectors (2023)
-
Humans as blood-feeding sources in sylvatic triatomines of Chile unveiled by next-generation sequencing
Parasites & Vectors (2023)
-
Diagnosis of animal trypanosomoses: proper use of current tools and future prospects
Parasites & Vectors (2022)
-
Parasites and blood-meal hosts of the tsetse fly in Tanzania: a metagenomics study
Parasites & Vectors (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
