
Abstract
A series of 3-aryl propionic esters and their analogues were designed and evaluated for acaricidal activity in vitro against Psoroptes cuniculi, a mange mite. The structure–activity relationship (SAR) was also discussed. The results showed that 6 compounds possessed the excellent activity (LC50 = 0.17–0.24 mM, LT50 = 1.5–2.9 h), superior to ivermectin (LC50 = 0.28 mM, LT50 = 8.9 h) (P < 0.05), a standard drug. Furthermore, 7 compounds showed the good activity (LC50 = 0.25–0.37 mM, LT50 < 3.9 h), slightly lower or close to that of ivermectin. One compound displayed super-fast acaricidal property, far superior to ivermectin. SAR analysis found that the ester group is vital for the activity and the small steric hindrance adjacent to the ester group is advantageous for the high activity. The <C4 linear alcohol esters can give the higher activity. The substituents on the 3-phenyl ring or replacement of the 3-phenyl with heterocyclic aryl generally decreases the activity. The position of the ester group in the ester chain also influences the activity, where the 3-phenyl propionate and the benzoate had the highest and lowest activity, respectively. Thus, 3-arylpropionates emerged as new and promising high-efficient acaricide candidates.
Similar content being viewed by others
Introduction
Acariasis is a skin disease caused by mites, which are widely parasitic on the body surfaces or epidermises of animals or human. Psoroptes cuniculi is an animal ear mite living in the ear canal of animals, and can infect rabbits, goats, horses, buffalo, sheep and so on1. More importantly, Psoroptic acariasis is a highly contagious disease. The infestation can cause intense pruritus, inflammation, serous exudations, the formation of crusts and scabs, reduction of weight gain, and even death2. Therefore, psoroptic acariasis has been a global disease causing serious economic losses for the animal industry3.
Therapy and control of both human scabies and animal mange mainly depend on the use of drugs and chemicals4. Among them, organochlorine, organophosphates, pyrethrins5, ivermectin and abamectin6 have been more commonly used drugs. However, continuous use of these drugs has presented some serious problems such as drug-resistance7,8, toxicity and environmental damage9. Therefore, it is urgent to develop new effective and safe acaricidal agents for treatment and control of animal acariasis.
Cinnamic acids are a class of natural phenylpropanoid compounds widely distributed in plants in the form of free acid and ester10. In the past decades, cinnamic acid derivatives have been attracting much attention due to their common occurrence in plants10, low toxicity11 and various important biological activities12, such as anticancer13, antimicrobial14, antioxidative15,16, anti-inflammatory17, anti-Mycobactrium tuberculosis18, anti-HIV19, antidiabetic20, anticholesterolemic21, hepatoprotective22, inducing neural progenitor cell proliferation23 and leishmanicidal activities24. In addition, we recently also found cinnamic acid esters had excellent acarcidal activity against P. cuniculi25,26 and strong inhibition activity against plant pathogenic fungi27. Thus, cinnamic acid is one promising molecular scaffold for the development of new drugs.
As one dihydrogen derivative of cinnamic acid, 3-phenylpropanoic acid is also one natural compound28. Its esters extensively exist in some plants29,30, rice wine31 and propolis32 as odor-active components. However, unlike cinnamic acid esters, few of study has been found on the bioactivity of 3-phenylpropanoic acid esters until now.
Especially interesting for us is that as the derivate of ethyl cinnamate, ethyl 3-phenylpropanoate showed greater acaricidal potential than ethyl cinnamate25. This result strongly suggests that it is necessary to take a systematical investigate on acaricidal activity of 3-phenylpropanoic acid esters in order to discover more potent acaricidal agents. As our continuing study, herein, a series of 3-aryl propionic acid esters and their analogues were reported for design, syntheses, acaricidal activity against P. cuniculi and preliminary structure-activity relationship (SAR).
Results and Discussion
Design of compounds
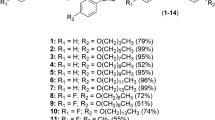
In order to explore SAR and find more potent compounds, based on the principle of structural similarity and the consideration of molecular diversity, four series of the target compounds were designed by using ethyl 3-phenylpropinate (2) as a lead compound. The first series is a group of 3-phenylpropionic acid esters (1–31) derived from various alcohols or phenols to explore the effect of various alkyl groups of the alcoholic moiety on the activity. The second series is a group of methyl 3-arylpropanoates (32–42) with various substituents on the benzene ring in order to explore the effect of the substitution pattern of the benzene ring on the activity. The substituents include electron-donating groups like Me, OMe and OH and electron-withdrawing groups like halogen atoms and NO2. The substitution position involves the o-, m- or p-site. The third series is a class of heteroaryl analogues (43–49) of ethyl 3-phenylpropionate in order to know the effect of different aromatic rings on the activity. The last group is comprised of the other compounds (50–58) with the aim of exploring the effect of the ester group itself and its location on the ester chain on the activity. The structures of the compounds are showed in Table 1. Since the bio-target and action mechanism of 3-phenylpropionate are yet unknown, we aimed mainly to gain insights into structural motifs that influence the biological activity of 3-phenylpropionic acid ester.
Synthesis of compounds
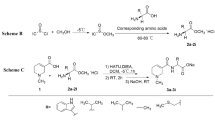
As shown in Fig. 1, commercial available aryl fatty acids as starting materials were treated by thionyl chloride to provide the corresponding acyl chlorides. The latter reacted with the corresponding alcohols or phenols to obtain compounds 1‒42 and 50‒53 in 70–97% yields. 3-Phenylpropanoyl chloride reacted with diethylamine to afford compound 55 in 82% yield. Compounds 43‒49 were obtained by selective reduction of the carbon carbon double bond of the corresponding ethyl (E)-3-heteroarylpropionates with NaBH4 in the presence of CuCl in 63–90% yields. Compound 56 was prepared by selective reduction of the carbon carbon double bond of (E)-1-phenylhex-1-en-3-one with NaBH4 in the presence of Pd/C and CH3CO2H in 87% yield. Compound 57 was obtained by Williamson ether synthesis from 3-phenylpropan-1-ol and ethyl iodide in 53% yield.

Synthesis of compounds 1‒53 and 55‒57. Reagents and conditions. (a) SOCl2, 75 °C, 2 h; (b) R′OH, DCM, 0 °C, 1 h, 70–97% yield; (c) Et2NH/water, 0 °C, 1 h, 82% yield; (d) NaBH4-CuCl/EtOH, rt, 1 h, 63–90% yield; (e) NaBH4, Pd/C, CH3COOH, toluene, 0 °C, 2 h, 87% yield; (f) NaH, THF, 0 °C, 0.5 h; then CH3CH2I, 0 °C, 12 h, 53%.
The known compounds were confirmed by comparison of the NMR data and those reported in literature. The new compounds (13, 29 and 30) were identified by 1H NMR, 13C NMR and HRMS analysis. The purity of all the compounds was determined to be >98% by 1H NMR analysis.
Acaricidal activity
The synthesized compounds along with commercial available 3-phenylpropionic acid (58) and N-ethyl-3-phenylpropanamide (54) were screened for acaricidal activity in vitro against P. cuniculi according to our previous method25,26. Ivermectin, an acaricidal drug standard, was used as a reference control. The results listed in Table 1 showed that all the compounds showed some activity at 0.5 or 0.25 mg/mL. Among them, twenty-five compounds (1–6, 10, 11, 18, 24, 27, 32, 37–43, 46–48, 51–53) displayed the good to excellent activity with mite mortality rates of >66% at 0.25 mg/mL, higher or equal to that of ivermectin (68.3%) (p ≤ 0.05), and twenty-one compounds (1–5, 10, 11, 27, 32, 37–43, 46, 47, 51–53) showed the mortality rates of ≥96%.
According to the same method described above, the compounds with the higher initial activity in Table 1 were further determined for median lethal concentrations (LC50) and median lethal times (LT50) in order to get insight into their acaricidal potency. Ivermectin was used as a reference drug. The mortality rates caused by treatment of various concentrations of the compounds for 24 h or various treatment times of each the compound at 4.5 mM are shown in Figs 2 and 3, respectively. Figures 2 and 3 showed that the activities of all the compounds were concentration- and time-independent, but the change trend (or steepness) of the various curves is different, indicating that the mites had different susceptibility to the concentration and treatment time of the various compounds. It was found from the steepness of the various curves that the mites were more sensitive to the compounds than ivermectin in two aspects of concentration effect and treatment-time effect. Meanwhile, most of the test compounds showed the higher activity in most of the test concentrations or treatment times than ivermectin (Figs 2 and 3). As far as concentration effect was concerned, compounds 1 showed the highest activity followed by compounds 3 and 41 (Fig. 2) while for treatment-time effect, compound 27 displayed the highest activity followed by 24, 51, 1, 2, 3, 46 and 47 (Fig. 3). It was worth noting that the result of compound 27 in Fig. 3A was obtained at 2.0 mM but not 4.5 mM for the other compounds. Interestingly, two compounds 6 and 10, 1 and 41 or 37 and 38 showed the very similar concentration-mortality curve (Fig. 2A,B). Similar cases were also observed for the time-mortality curves of both 1 and 2, 32 and 37, 38 and 39 or 46 and 47 (Fig. 3).

Effects of test concentrations of the compounds on the acaricidal activity against P. cuniculi (24 h).

Effects of treatment times (B) of the compounds (4.5 mM) on the acaricidal activity against P. cuniculi. (a) compound 27 was tested at 2.0 mM (Fig. 3A). The other compounds were tested at 4.5 mM.
The results above were further confirmed by LC50 and LT50 values of the various compounds (Table 2). Seven compounds (1‒4, 41 and 53) showed smaller LC50 values of 0.17‒0.25 mM than ivermectin (LC50 = 0.28 mM) (P < 0.05). Compounds 1, 3 and 41 showed the highest activity (LC50 = ca. 0.17, P < 0.05), and their relative activities reach more than 1.5-fold of ivermectin. Compounds 2, 4, 40 and 53 showed the second highest activity (LC50 = ca. 0.24, P < 0.05). Additionally, compounds 11, 27, 42, 51 and 52 also showed the good activity (LC50 0.29‒0.32 mM), slightly lower or close to that of ivermectin. It is worth noting that the molecular weight of ivermectin (MW 875) is much higher than that of the test compounds (MWs 164–271). Therefore, when LC50 values are expressed in mass concentration, all the compounds possessed the smaller LC50 values of 27.3–184.0 μg/mL than ivermectin (LC50 247.4 µg/mL) (Table 2). On the other hand, all the compounds in Table 2 also showed faster acaricidal action with LT50 values of <8.4 h than ivermectin (LT50 = 8.9 h) at the same concentration of 4.5 mM. Especially, compound 27 showed super-fast acaricidal action (LT50 < 0.7 h, at 2.0 mM), far superior to the other compounds or ivermectin. Even if at 1.0 mM, its relative activity (LT50 = 3.6 h) still reached up to 2.5-fold that of ivermectin at 4.5 mM.
Discussions—Structure-activity relationship
By comparison of the activity and structures of the various compounds in Tables 1 and 2 coupling with Figs 2 and 3, we found some clear and important SARs for 3-aryllpropoionic acid esters. (1) For the alcohol esters, the type and steric hindrance of the alcoholic alkyls (R) significantly influence the LC50 values of the compounds. Generally, the activity of the compounds decreases with elongation of the linear R (1–9, Tables 1 and 2). Compared with the ≥C5 alkyl groups (5–9, LC50>100 µg/mL), the ≤C4 alkyl (1–4, <50 µg/mL) can dramatically improve the activity. Compared with the secondary or tertiary alcohol esters, the primary alcohol esters can give the higher activity (3 vs 10; 4 vs 12; 5 vs 13). A similar case was also found for the iso-butyl ester (11) (a primary alcohol ester) and the iso-propyl ester (10) (a secondary alcohol ester) (Table 2) although the former possesses a larger spatial volum than the latter. The results above suggest that the steric hindrance around the α-C of the alcoholic moiety or near the ester group can significantly impact the activity. The small steric hindrance adjacent to the ester group is helpful for the high activity. The opinion above can be aslo supported by the fact that the n-hexyl ester (6) is more active than the cyclohexyl ester (14). Interestingly, the SARs above is very similar to that of cinnamic acid esters26.
On the other hand, the alcoholic alkyls also have the similar influence trend for the LT50 values of the compounds. The methyl, ethyl and n-propyl esters (1‒3) showed the very close-by LT50 values, superior to that of the n-butyl, n-amyl, n-hexyl esters (4‒6). The iso-butyl ester (11) also showed the smaller LT50 values than the iso-propyl ester (10). Interestingly, compounds 1‒3 displayed very similar time-effect curves in Fig. 3A.
2) The introduction of substituents to the 3-phenyl ring of the methyl propionate (1) leads to decrease of the activity (32‒40, 42 vs 1) except 3ʹ-NO2 (41). A similar case was also observed for cinnamates25. Since the substituents not only include various groups with different electron effect like OH, OMe, Me, halogen atoms or NO2 but also involve hydrogen-bond acceptors (NO2, OMe) and hydrogen-bond donors (OH), we speculated that the effect of substituents on the 3-phenyl ring on the activity should be mainly steric effect but not electron effect or hydrogen-bond effect.
3) Compared with the n-hexyl ester (6) or the cyclohexyl ester (14), the phenyl ester (15) showed the lower mite mortality (Table 1). Some similarly, the benzyl ester (28) was less active than the n-heptyl ester (7). The results above suggest that the fatty or alicyclic alcohol esters have the higher activity than the phenol or aromatic alcohol esters. However, it is worth noting that for the phenolic esters, the substituents on the phenolic ring can significantly impact the activity (15–31). Compared with the phenyl ester (15), the introduction of methyl, chlorine atom or nitro to the phenyl ring leads to significant improvement of the activity (16–18, 22–24, 26, 27 vs 15) (Table 1). A similar case was also found in cinnamic acid esters26. Remarkably, among the compounds in Table 2, the 4-nitrophenyl ester (27) and 4-chlorophenyl ester (24) gave the lowest and second lowest LT50 values, respectively. The results above showed that the presence of p-cholor or p-nitro on the phenolic ring can endow the phenolic esters with the fast-acting property to the mites.
4) Replacement of the phenyl of ethyl 3-phenylpropionate (2) by heterocyclic aryls like pyridyl, thienyl or furyl leads to slight decrease of the activity (43–49 vs 2). This result is similar but completely the same as that of the heterocyclic analogues of cinnamates, in which the pyridin-2-yl or furan-2-yl analogues of ethyl cinnamate were more active than ethyl cinnamate26.
5) Compounds 3 and 50–53 have the same ester chain length consisting of seven atoms. Their structural difference only lies in the position of the ester group on the chain. From the activities of these compounds, it was found that the position of the ester group on the ester chain is able to influence the activity also. Among them, the propionate (3) has the highest activity although the acetate, butyrate and pentanoate isomers (51–53) also showed the good activity. Unusually, the benzoate (50) showed the much lower activity than the other isomers (3, 51–53). This result is probably attributed to the π–π conjugation effect existing between the ester carbonyl group and the benzene ring in 50. Similarly, the lower activity of ethyl cinnamate than its dihydro-derivate (2) may be also related with the same reason25.
6) Compounds 54–57 may be considered as analogues or derivatives of ethyl 3-phenypropionate (2). These compounds have the same chain length consisting of six atoms. Obviously, the ester (2) were much more active than its corresponding amides (54, 55), ketone (56), ether (57) or acid (58) (Table 1). These results show that the ester group is vital to the activity. On the other hand, the ketone 56 being more active than its corresponding ether 57 suggests that the carbonyl of the ester group should be more important than its saturated oxygen for the activity. The results above are the same as that of cinnamates26. Based on the results above, we conjectured that the activity of the esters very likely involves an acyl transfer mechanism, in which the oxygen atom of the ester carbonyl probably acts as a hydrogen bond receptor or nucleophile when the compounds interact with their biological receptor. If so, the lower electron density on the carbonyl carbon should be helpful for the activity. The opinion above can be supported by the fact that the ester (2) was more active than the amides (54, 55) to some extent. At present, the study on the acaricidal bio-target and mechanism of these esters has been in our plan.
In conclusion, a series of 3-arylpropionate derivatives were designed and evaluated for acaricidal activity in vitro against Psoroptes cuniculi. Twenty-four compounds were found to possess the lower LT50 values than ivermectin, a standard drug, and of which seven compounds also showed the higher LC50 activity at the same time than ivermectin. Besides having the excellent LC50 activity, compound 27 also showed a super-fast acaricidal property, far superior to ivermection. Compared with ivermectin, the present compounds displayed some obviously attractive advantages, such as natural compounds or natural compound framework, the higher activity, the quick-acting property, simple structure, easy preparation and low-cost. Thus, these compounds possess great potential for development of novel acaricidal agents. Additionally, some important SARs were derived. The ester group was found to be crucial for the activity. The type of the alcoholic alkyls and its steric hindrance around the alcoholic α-C or near the carbonyl group are two main influence factors on the activity. The fatty alcohol esters were generally more active than the phenol esters or aromatic alcohol esters. The distance between the ester group and the phenyl ring slightly affects the activity except the benzoate with the lower activity. The introduction of substituents to the 3-phenyl ring or replacement of the 3-phenyl by heterocyclic aryls leads to decrease of the activity. Further research is very necessary on the acaricidal mechanism, therapeutic effect and toxicity of the compounds. At present, these works are partly underway in our lab.
Methods
Chemicals
3-Phenylpropanoic acid (58), N-ethyl-3-phenylpropanamide (54) and ivermectin (≥91% 22,23-dihydroavermectin B1 consisting of 95% avermectin B1a and 5% avermectin B1b) was purchased from Sigma-Aldrich Trading Co., Ltd., Shanghai, China. Other chemicals used in the present study were purchased from J&K Chemical Ltd., Beijing, China, and used without further purification.
Instruments
Melting points (m.p.) were determined on an XT-4 micro-melting point apparatus and uncorrected. 1H- and 13C-NMR spectra were recorded on a Bruker AVANCE III (Bruker, Karlsruhe, Germany) operating at 500 and 125 MHz, respectively and using tetramethylsilane (TMS) as an internal standard. Chemical shifts (δ values) and coupling constants (J values) are given in ppm and Hz, respectively. Mass spectra (HR-MS) were carried out with a Micromass Auto Spec-3000 instrument.
Synthesis
Synthesis of compounds 1–42 and 50–53
The general procedure is as follows. The mixture of aryl- or heteroaryl-substituted fatty acid (10 mmol) and thionyl chloride (41 mmol, 3 mL) was refluxed for 2 h. The excess thionyl chloride was removed under reduced pressure to provide the corresponding acyl chloride. The acyl chloride without further purification was dissolved in 6 mL dry DCM, and cooled to 0 °C. The corresponding alcohol or phenol (10 mmol) was added, and stirred at 0 °C for 1 h. The mixture was diluted by DCM (50 mL), successively washed with water and 5% Na2CO3 solution, and dried over anhydrous sodium sulfate. After removal of the solvent under reduced pressure, the residue was subjected to silica gel column chromatography using petroleum ether–ethyl acetate (10:1) to afford the desired compounds.
The following known compounds were obtained as a colorless oil in 70–97% yield: methyl 3-phenylpropanoate (1), ethyl 3-phenylpropanoate (2), propyl 3-phenylpropanoate (3), butyl 3-phenylpropanoate (4), pentyl 3-phenylpropanoate (5), hexyl 3-phenylpropanoate (6), heptyl 3-phenylpropanoate (7), octyl 3-phenylpropanoate (8), nonyl 3-phenylpropanoate (9), iso-propyl 3-phenylpropanoate (10), iso-butyl 3-phenylpropanoate (11), t-butyl 3-phenylpropanoate (12), cyclohexyl 3-phenylpropanoate (14), phenyl 3-phenylpropanoate (15), p-tolyl 3-phenylpropanoate (18), m-methoxyphenyl 3-phenylpropanoate (20), o-chlorophenyl 3-phenylpropanoate (22), benzyl 3-phenylpropanoate (28), methyl 3-(4-fluoromophenyl)propanoate (34), methyl 3-(4-chlorophenyl)propanoate (35), methyl 3-(4-bromophenyl)propanoate (36), 3-(2-methoxyphenyl)propanoate (37), 3-(3-methoxyphenyl)propanoate (38), pentyl benzoate (50), butyl 2-phenylacetate (51), ethyl 4-phenylbutanoate (52), methyl 5-phenylpentanoate (53). The known compounds were obtained as a yellow or pale yellow oil in 72%–85%: m-tolyl 3-phenylpropanoate (17), p-methoxyphenyl 4-phenylbutanoate (21), p-nitrobenzyl 3-phenylpropanoate (31) and methyl 3-(4-methoxyphenyl)propanoate (39).
Known compounds p-chlorophenyl 4-phenylbutanoate (24) in 77% yield, m.p. 55–56 °C, methyl 3-(4-methylphenyl)propanoate (32) in 84% yield, m.p. 35–36 °C and methyl 3-(4-hydroxyphenyl)propanoate (33) in 51% yield, m.p. 39–40 °C were obtained as a white solid; known compound p-nitrophenyl 4-phenylbutanoate (27) in 83% yield, m.p. 96–97 °C, methyl 3-(2-nitrophenyl)propanoate (40) in 76% yield, m.p. 55–56 °C and methyl 3-(4-nitrophenyl)propanoate (42) in 80% yield, m.p. 73–74 °C were obtained as a yellow solid.
The 1H and/or 13C NMR data of the compounds above were showed in Supporting Information and consistent with those in literature.
tert-Pentyl 3-phenylpropanoate (13)
Yield: 70%; a colorless oil; 1H NMR (500 MHz, CDCl3) δ: 7.34–7.31 (m, 2 H, Ar-H), 7.26–7.22 (m, 3 H, Ar-H), 2.96 (t, J = 7.8 Hz, 2 H, H-3), 2.60 (t, J = 7.8 Hz, 2 H, H-2), 1.79 (q, J = 7.5 Hz, 2 H, CH2CH3), 1.44 (s, 6 H, C(CH3)2), 0.88 (t, J = 7.5 Hz, 3 H, CH2CH3); 13C NMR (125 MHz, CDCl3) δ: 172.2 (C=O), 140.8 (Ph, C-1′), 128.4 (C-3′, C-5′), 128.3 (C-2′, C-6′), 126.1 (C-4′), 82.8 (O-C), 37.1 (C-2), 33.5 (CH2CH3), 31.2(C-3), 25.6 (CH3 × 2), 8.2 (CH2CH3). HR-ESI-MS [M+Na]+ calcd for C14H20NaO2+, 243.1356, found 243.1351.
o-Tolyl 3-phenylpropanoate (16)
Yield: 71%; a yellow oil; 1H NMR (500 MHz, CDCl3) δ: 7.41–7.34 (m, 4 H, Ar-H), 7.32–7.24 (m, 3 H, Ar-H), 7.21–7.18 (m, 1 H, Ar-H), 7.00 (d, J = 7.7 Hz, 1 H, H-3″), 3.17 (t, J = 7.7 Hz, 2 H, H-3), 2.99 (t, J = 7.7 Hz, 2 H, H-2), 2.15 (s, 3 H, CH3); 13C NMR (125 MHz, CDCl3) δ: 171.2 (C=O), 149.4 (C-1″), 140.2 (C-1′), 131.2 (C-3″), 130.1 (C-2″), 128.7 (C-3′, C-5′), 128.5 (C-2′, C-6′), 126.9 (C-5″), 126.5 (C-4″), 126.1 (C-4′), 121.9 (C-6″), 35.8 (C-2), 31.1 (C-3), 16.1 (CH3).
2-Methoxyphenyl 3-phenylpropanoate (19)
Yield: 80%; a yellow oil; 1H NMR (500 MHz, CDCl3) δ: 7.38–7.34 (m, 4 H, Ar-H), 7.31–7.25 (m, 2 H, Ar-H), 7.03–6.99 (m, 3 H, Ar-H), 3.85 (s, 3 H, CH3), 3.16 (t, J = 7.7 Hz, 2 H, H-3), 2.98 (t, J = 7.7 Hz, 2 H, H-2); 13C NMR (125 MHz, CDCl3) δ: 171.0 (C=O), 151.2 (C-2″), 140.4 (C-1″), 139.9 (C-1′), 128.6 (C-3′, C-5′), 128.5 (C-2′, C-6′), 126.9 (C-4″), 126.4 (C-4′), 122.8 (C-5″), 120.8 (C-6″), 112.5 (C-3″), 55.9 (OCH3), 35.6 (C-2), 31.0 (C-3).
3-Chlorophenyl 3-phenylpropanoate (23)
Yield: 79%; a brown solid; m.p. 34–35 °C; 1H NMR (500 MHz, CDCl3) δ: 7.41–7.38 (m, 2 H, Ar-H), 7.36–7.30 (m, 4 H, Ar-H), 7.29–7.26 (m, 1 H, Ar-H), 7.11 (t, J = 1.9 Hz, 1 H, H-2″), 7.01–6.94 (m, 1 H, Ar-H), 3.14 (t, J = 7.6 Hz, 2 H, H-3), 2.95 (t, J = 7.7 Hz, 2 H, H-2); 13C NMR (125 MHz, CDCl3) δ: 171.0 (C=O), 151.2 (C-1″), 140.0 (C-1′), 134.7 (C-3″), 130.2 (C-5″), 128.7 (C-3′, C-5′), 128.5 (C-2′, C-6′), 126.6 (C-4′), 126.2 (C-4″), 122.3 (C-2″), 120.0 (C-6″), 36.0 (C-2), 30.9 (C-3).
2-Nitrophenyl 3-phenylpropanoate (25)
Yield: 81%; a pale yellow solid; m.p. 74–75 °C; 1H NMR (500 MHz, CDCl3) δ: 8.14 (dd, J = 8.2, 1.5 Hz, 1 H, H-3″), 7.69 (t × 2, J = 7.9, 1.5 Hz, 1 H, H-4″), 7.44 (t × 2, J = 7.9, 1.2 Hz, 1 H, H-5″), 7.40–7.37 (m, 2 H), 7.34–7.29 (m, 3 H), 7.21 (d, J = 8.1 Hz, 1 H, H-6″), 3.16 (t, J = 7.8 Hz, 2 H, H-3), 3.04 (t, J = 7.8 Hz, 2 H, H-2); 13C NMR (125 MHz, CDCl3) δ: 170.6 (C=O), 144.1 (C-1″), 141.8 (C-2″), 139.9 (C-1′), 134.8 (C-5″), 128.7 (C-3′, C-5′), 128.4 (C-2′, C-6′), 126.7 (C-6″), 126.5 (C-4′), 125.8 (C-3″), 125.3 (C-4″), 35.7 (C-2), 30.5 (C-3).
3-Nitrophenyl 3-phenylpropanoate (26)
Yield: 82%; a yellow solid; m.p. 67–68 °C; 1H NMR (500 MHz, CDCl3) δ: 8.16–8.14 (m, 1 H, H-4″), 7.95 (t, J = 2.2 Hz, 1 H, H-2″), 7.58 (t, J = 8.2 Hz, 1 H, H-6″), 7.42–7.38 (m, 3 H), 7.35–7.30 (m, 3 H), 3.15 (t, J = 7.8 Hz, 2 H, H-3), 3.00 (t, J = 7.8 Hz, 2 H, H-2); 13C NMR (125 MHz, CDCl3) δ: 170.8 (C=O), 151.0 (C-1″), 148.8 (C-3″), 139.7 (C-1′), 130.0 (C-5″), 128.7 (C-3′, C-5′), 128.4 (C-2′, C-6′), 128.1 (C-6″), 126.7 (C-4′), 120.8 (C-4″), 117.4 (C-2″), 35.9 (C-2), 30.9 (C-3).
2-Nitrobenzyl 3-phenylpropanoate (29)
Yield: 80%; a brown oil; 1H NMR (500 MHz, CDCl3) δ: 8.14 (dd, J = 8.2, 1.0 Hz, 1 H, H-3″), 7.62 (t × 2, J = 7.7, 1.1 Hz, 1 H, H-5″), 7.51 (t × 2, J = 7.7, 1.1 Hz, 1 H, H-4″), 7.43 (d, J = 7.7 Hz, 1 H, H-6″), 7.36–7.33 (m, 2 H), 7.28–7.25 (m, 3 H), 5.19 (s, 2 H, OCH2), 3.05 (t, J = 7.8 Hz, 2 H, H-3), 2.81 (t, J = 7.8 Hz, 2 H, H-2); 13C NMR (125 MHz, CDCl3) δ: 172.4 (C=O), 147.7 (C-2″), 143.3 (C-1″, C-6″), 140.1 (C-1′), 128.6 (C-3′, C-5′), 128.34 (C-2′, C-6′), 128.31 (C-4″, C-5″), 126.5 (C-4′), 123.8 (C-3″), 64.8 (OCH2), 35.7 (C-2), 30.9 (C-3). HR-ESI-MS [M+Na]+ calcd for C16H15NNaO4+, 308.0893, found 308.0889.
3-Nitrobenzyl 3-phenylpropanoate (30)
Yield: 78%; a pale yellow oil; 1H NMR (500 MHz, CDCl3) δ: 8.23–8.21 (m, 2 H, H-2″, H-4″), 7.64 (d, J = 7.7 Hz, 1 H, H-6″), 7.58–7.55 (m, 1 H, H-5″), 7.34–7.31 (m, 2 H), 7.26–7.23 (m, 3 H), 5.23 (s, 2 H, CH2), 3.03 (t, J = 7.7 Hz, 2 H, H-3), 2.78 (t, J = 7.7 Hz, 2 H, H-2); 13C NMR (125 MHz, CDCl3) δ: 172.5 (C=O), 148.4 (C-3″), 140.1 (C-1′), 138.1 (C-1″), 134.0 (C-6″), 129.6 (C-5″), 128.6 (C-3′, C-5′), 128.3 (C-2′, C-6′), 126.4 (C-4′), 123.2 (C-4″), 122.9 (C-2″), 64.8 (OCH2), 35.7 (C-2), 30.9 (C-3). HR-ESI-MS [M+Na]+ calcd for C16H15NNaO4+, 308.0893, found 308.0879.
Methyl 3-(3-nitrophenyl)propanoate (41)
Yellow solid in 78% yield, m.p. 45–46 °C. 1H NMR (500 MHz, CDCl3) δ: 8.12–8.11 (m, 2 H, H-2′, H-4′), 7.59 (d, J = 7.6 Hz, 1 H, H-6′), 7.50 (t, J = 8.3 Hz, 1 H, H-5′), 3.72 (s, 3 H, CH3), 3.11 (t, J = 7.6 Hz, 2 H, H-3), 2.72 (t, J = 7.6 Hz, 2 H, H-2); 13C NMR (125 MHz, CDCl3) δ: 172.6 (C=O), 148.4 (C-3′), 142.5 (C-1′), 134.7 (C-6′), 129.4 (C-5′), 123.3 (C-2′), 121.6 (C-4′), 51.8 (OCH3), 35.0 (C-2), 30.5 (C-3).
The general procedure for synthesis of compounds 43–49 is as follows
To the solution of ethyl (E)-3-heteroaryl acrylate (10 mmol) and CuCl (6 mmol) in DCM (50 mL) was slowly added NaBH4 (25 mmol) at room temperature within 20 minutes. After stirring for 0.5 h, the solution was concentrated under reduced pressure. The residue was purified by silica gel column chromatography using petroleum ether–ethyl acetate (3:1 or 10:1) to afford the desired compounds. Compounds 43–49 were obtained as a yellow or pale yellow oil in 63–90%. The 1H and/or 13C NMR data of the compounds were consistent with those in literature.
Synthesis of N,N-diethyl-3-phenylpropanamide (55)
According to the method described above for 1–42, 3-phenylpropanoyl chloride was obtained by reaction of 3-phenyl propionic acid (10 mmol) and thionyl chloride (41 mmol, 3 mL). The acyl chloride was slowly added to the solution of diethylamine (25 mmol) in DCM (50 mL) at 0 °C within 10 minutes. The mixture was stirred at 0 °C for 1 h, and warmed to room temperature. The solution was washed with water, dried over anhydrous sodium sulfate and concentrated. The residue was subjected to column chromatography over silica gel eluting with the mixture of petroleum ether and ethyl acetate (2:1, V:V) to afford N,N-diethyl-3-phenylpropanamide (55) as a yellow oil in 82% yield. The 1H and/or 13C NMR data match those in literature.
Synthesis of 1-phenylhexan-3-one (56)
(E)-1-phenylhex-1-en-3-one (10 mmol), NaBH4 (40 mmol) and Pd/C (12 mmol) was placed in 100 mL round bottomed flask. The flask was evacuated and back filled with argon three times. Toluene of 50 mL was slowly added at 0 °C. After stirring for 5 minutes, acetic acid (20 mmol) was slowly added at 0 °C within 10 minutes. The mixture was stirred for 2 h, filtered and concentrated under reduced pressure. The residue was purified by silica gel column chromatography using petroleum ether–ethyl acetate (10:1) to yield 1-phenylhexan-3-one (56) as a yellow oil in 87% yield. The 1H and/or 13C NMR data match those in literature.
Synthesis of (3-ethoxypropyl)benzene (57)
To the solution of 3-phenylpropan-1-ol (10 mmol) in dry THF (50 mL) was slowly added NaH (12 mmol) at 0 °C. The mixture was vigorously stirred at 0 °C for 0.5 h. Ethyl iodide (15 mmol) was slowly added at 0 °C, and stirred for 12 h. The mixture was quenched with H2O (50.0 mL) and extracted with DCM. The combined organic phase was dried over Na2SO4, filtered and concentrated under reduced pressure. The residue was purified by silica gel column chromatography using petroleum ether–ethyl acetate (15:1) to afford (3-ethoxypropyl)benzene (57) as a yellow oil in 53% yield. The 1H and/or 13C NMR data match those in literature.
Acaricidal Assay
In vitro acaricidal activity of the synthesized compounds was performed according to our previously reported method25,26. In brief, a tested compound was dissolved in a mixed solvent of dimethyl sulfoxide (DMSO), Tween-80 and normal saline (1:1:8, V/V) to prepare a test solution. Psoroptes cuniculi adult mites of both sexes isolated from the scabs and the cerumen of naturally infected rabbits were used as tested objects. The mites were placed in 24-well flat-bottomed cell culture plates (10 adult mites each well) and followed by addition of 0.6 mL of the tested solution into each the well. Five replicates were made for each test. The same mixed solvent without tested compounds as above was used as an untreated control. Ivermectin, an acaricidal drug standard, in the same mixed solvent was used as a reference control.
The plates were placed in separate humidity chambers in saturated humidity conditions at 28 °C. After 24 h, the mites in the plate were observed under a stereomicroscope. When the persistent immobile mites were stimulated with a needle, lack of reaction was considered as the indication of death. Mortality was calculated using the following formula and expressed as means ± standard deviation (S. D.):
where, N is the number of the test mites in each test; Nc is the number of death mites in the untreated test; Nt the number of death mites in one treated test.
According to the same procedure as described above, a series of test concentrations of the compound was prepared by diluting the stock solution with the same mixed solvent and used to assay median lethal concentration value (LC50). Each concentration test was performed in triplicates. The probit value of the average mortality for each tested concentration and the corresponding lg[concentration] value were used to establish toxicity regression equation for concentration–effect by the linear least-square fitting method. The LC50 value of the compound and its confidence interval at 95% probability was calculated from its toxicity regression equation.
The concentration of 4.5 μmol/mL of the compound was used to determine the mortality of the mites at different treatment time. The probit value of the average mortality for each the compound and the corresponding lg[treatment time] value were used to establish toxicity regression equation for time–effect by the linear least-square fitting method. The median lethal time value (LT50) value of the compound and its confidence interval at 95% probability was calculated from its toxicity regression equation. median lethal time value (LT50).
PRISM software ver. 5.0 (GraphPad Software Inc., San Diego, CA,USA) was used to analyze the data and establish toxicity regression equations. Duncan multiple comparison test was performed on the data to evaluate difference significance between the activities of various compounds.
Data
Data will be made available upon request to the corresponding authors.
References
Bates, P. G. Inter- and intra-specific variation within the genus Psoroptes (Acari: Psoroptidae). Vet. Parasitol. 83, 201–217 (1999).
Shang, X.-F. et al. Acaricidal activity of extracts from Adonis coerulea Maxim. against Psoroptes cuniculi in vitro and in vivo. Vet. Parasitol. 195, 136–141 (2013).
Dagleish, M., Ali, Q., Powell, R., Butz, D. & Woodford, M. Fatal Sarcoptes scabiei infection of blue sheep (Pseudois nayaur) in Pakistan. J. Wildl. Dis. 43, 512–517 (2007).
Borges, F. A. et al. Anthelmintic resistance impact on tropical beef cattle productivity: effect on weight gain of weaned calves. Trop. Anim. Health. Prod. 45, 723–727 (2013).
Cetin, H. et al. Acaricidal activity of Satureja thymbra L. essential oil and its major components, carvacrol and γ-terpinene against adult Hyalomma marginatum (Acari: Ixodidae). Vet. Parasitol. 170, 287–290 (2010).
Behera, S. K., Dimri, U., Singh, S. K. & Mohanta, R. K. The curative and antioxidative efficiency of ivermectin and ivermectin+ vitamin E-selenium treatment on canine Sarcoptes scabiei infestation. Vet. Res. Commun. 35, 237–244 (2011).
Synge, B., Bates, P., Clark, A. & Stephen, F. Apparent resistance of P ovis to flumethrin. Vet. Rec. 137, 51–52 (1995).
Clark, A., Stephen, F., Cawley, G., Bellworthy, S. & Groves, B. Resistance of the sheep scab mite Psoroptes ovis to propetamphos. Vet. Rec. 139, 451 (1996).
Coles, T. B. & Dryden, M. W. Insecticide/acaricide resistance in fleas and ticks infesting dogs and cats. Parasite. vector. 7, 113–124 (2014).
Clifford, M. N. Chlorogenic acids and other cinnamates–nature, occurrence, dietary burden, absorption and metabolism. J. Sci. Food Agric. 80, 1033–1043 (2000).
Lafay, S. & Gil-Izquierdo, A. Bioavailability of phenolic acids. Phytochem. Rev. 7, 301–311 (2008).
Sharma, P. Cinnamic acid derivatives: A new chapter of various pharmacological activities. J. Chem. Pharm. Res. 3, 403–423 (2011).
De, P., Baltas, M. & Bedos-Belval, F. Cinnamic acid derivatives as anticancer agents-a review. Curr. Med. Chem. 18, 1672–1703 (2011).
De Vita, D. et al. Activity of caffeic acid derivatives against Candida albicans biofilm. Bioorg. Med. Chem. Lett. 24, 1502–1505 (2014).
Sova, M. Antioxidant and antimicrobial activities of cinnamic acid derivatives. Mini. Rev. Med. Chem. 12, 749–767 (2012).
Lee, I.-K., Han, M.-S., Kim, D.-W. & Yun, B.-S. Phenylpropanoid acid esters from Korean propolis and their antioxidant activities. Bioorg. Med. Chem. Lett. 24, 3503–3505 (2014).
Jayaprakasam, B., Vanisree, M., Zhang, Y., Dewitt, D. L. & Nair, M. G. Impact of alkyl esters of caffeic and ferulic acids on tumor cell proliferation, cyclooxygenase enzyme, and lipid peroxidation. J. Agric. Food. Chem. 54, 5375–5381 (2006).
Guzman, J. D. et al. 2-Hydroxy-substituted cinnamic acids and acetanilides are selective growth inhibitors of Mycobacterium tuberculosis. MedChemComm 5, 47–50 (2014).
Lee, S. U., Shin, C.-G., Lee, C.-K. & Lee, Y. S. Caffeoylglycolic and caffeoylamino acid derivatives, halfmers of L-chicoric acid, as new HIV-1 integrase inhibitors. Eur. J. Med. Chem. 42, 1309–1315 (2007).
Adisakwattana, S., Moonsan, P. & Yibchok-Anun, S. Insulin-releasing properties of a series of cinnamic acid derivatives in vitro and in vivo. J. Agric. Food. Chem. 56, 7838–7844 (2008).
Lee, S. et al. Synthesis of cinnamic acid derivatives and their inhibitory effects on LDL-oxidation, acyl-CoA: cholesterol acyltransferase-1 and-2 activity, and decrease of HDL-particle size. Bioorg. Med. Chem. Lett. 14, 4677–4681 (2004).
Fernández-Martínez, E., Bobadilla, R. A., Morales-Ríos, M. S., Muriel, P. & Pérez-Álvarez, V. M. Trans-3-phenyl-2-propenoic acid (cinnamic acid) derivatives: structure-activity relationship as hepatoprotective agents. Med. Chem. 3, 475–479 (2007).
Yabe, T. et al. Ferulic acid induces neural progenitor cell proliferation in vitro and in vivo. Neuroscience 165, 515–524 (2010).
Otero, E. et al. Synthesis and leishmanicidal activity of cinnamic acid esters: Structure–activity relationship. Med. Chem. Res. 23, 1378–1386 (2014).
Zhang, B. et al. Ethyl Cinnamate Derivatives as Promising High-Efficient Acaricides against Psoroptes cuniculi: Synthesis, Bioactivity and Structure–Activity Relationship. Chem. Pharm. Bull. 63, 255–262 (2015).
Chen, D. et al. Bioactivity and structure–activity relationship of cinnamic acid derivatives and its heteroaromatic ring analogues as potential high-efficient acaricides against Psoroptes cuniculi. Bioorg. Med. Chem. Lett. https://doi.org/10.1016/j.bmcl.2017.08.051 (2017).
Zhou, K. et al. Bioactivity and structure-activity relationship of cinnamic acid esters and their derivatives as potential antifungal agents for plant protection. PLoS One 12, e0176189 (2017).
Liu, Y. X., Li, X., Cai, L. T., Zhang, H. & Shi, J. X. Identification of phenolic acids in tobacco root exudates and their role in gorwth of rhizosphere microorgainisms. Journal of Plant Nutrition and Fertilizer (Zhiwu Yingyang Yu Feiliao Xuebao) 22, 418–428 (2016).
Miranda, E. J., Nogueira, R. I., Pontes, S. M. & Rezende, C. M. Odour-active compounds of banana passa identified by aroma extract dilution analysis. Flavour. Frag. J. 16, 281–285 (2001).
El-Massry, K. F., El-Ghorab, A. H. & Farouk, A. Antioxidant activity and volatile components of Egyptian Artemisia judaica L. Food Chem. 79, 331–336 (2002).
Xiao, Z., Dai, X., Zhu, J. & Yu, H. Classification of Chinese Rice Wine According to Geographic Origin and Wine Age Based on Chemometric Methods and SBSE-TD-GC-MS Analysis of Volatile Compounds. Food Sci. Technol. Res. 21, 371–380 (2015).
Hames-Kocabas, E. E., Demirci, B., Uzel, A. & Demirci, F. Volatile composition of anatolian propolis by headspace-solid-phase microextraction (HS-SPME), antimicrobial activity against food contaminants and antioxidant activity. J. Med. Plants. Res. 7, 2140–2149 (2013).
Acknowledgements
This project was financially supported by the National Natural Science Foundation of China (Nos 31172365, 31572038). This study was also supported by the Agricultural Science and Technology Innovation Project of Shaanxi Province (2016NY-171). We sincerely thank Hongli Zhang (Northwest A&F University) for the NMR determination of the compounds and Professor Zhenghui Guan (Northwest University, China) for the HRMS determination of NMR.
Author information
Authors and Affiliations
Contributions
L.Z., X.Y. and F.M. conceived the research and wrote the manuscript. D.C., Y.T., M.X., X.W. and D.L. performed all experiments. All authors designed experiments, analyzed data, edited and approved the manuscript.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Chen, D., Tian, Y., Xu, M. et al. Design, Bioactivity and structure-activity of 3-Arylpropionate Derivatives as Potential High-Efficient Acaricides against Psoroptes Cuniculi. Sci Rep 8, 1797 (2018). https://doi.org/10.1038/s41598-018-20140-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-20140-7
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
