
Abstract
Asphaltenes constitute the heaviest, most polar and aromatic fraction of petroleum crucial to the formation of highly-stable water-in-crude oil emulsions. The latter occur during crude oil production as well as spills and cause difficulties to efficient remediation practice. It is thought that in nanoaggregate form, asphaltenes create elastic layers around water droplets enhancing stability of the emulsion matrix. Ultrasonic characterisation is a high-resolution non-invasive tool in colloidal analysis shown to successfully identify asphaltene nanoaggregation in toluene. The high sensitivity of acoustic velocity to molecular rearrangements and ease in implementation renders it an attractive method to study asphaltene phase properties. Currently, aggregation is thought to correspond to an intersection of two concentration-ultrasonic velocity regressions. Our measurements indicate a variation in the proximity of nanoaggregation which is not accounted for by present models. We attribute this uncertainty to physico-chemical heterogeneity of the asphaltene fraction driven by variation in molecular size and propose a critical nanoaggregation region. We treated asphaltenes from North and South American crude oils with ruthenium ion catalysed oxidation to characterize their n-alkyl appendages attached to aromatic cores. Principal component analysis was performed to investigate the coupling between asphaltene structures and velocity measurements and their impact on aggregation.
Similar content being viewed by others

Introduction
Water-in-oil emulsions (WOE) are highly stable mixtures occurring during crude oil production and spills1,2,3. Emulsion removal from the water column is problematic due to the inherent high elasticity, viscosity and stability of these mixtures2,3. For efficient removal, WOEs require separation into water and oil phases3,4 which is inhibited by the presence of asphaltenes at the water/oil boundary2,3,4,5.
Asphaltenes are the heaviest, most aromatic and polar constituents of crude oil, soluble in aromatic solvents and precipitating upon the addition of low molecular weight n-alkanes2,6,7,8. Their solubilisation may change depending on solvent9 and the number of aromatic rings10. The asphaltene molecules have a wide distribution of architectures with two broad types, island and archipelago11, recently thought of as two extremes of a structural continuum12,13,14. The island architecture is most common11,15,16 and comprises a single polycyclic aromatic hydrocarbon (PAH) centre and aliphatic appendages11,15. The archipelago architecture is characterised by several (smaller) PAHs connected by an aryl linkage16. Recent studies using atomic force microscopy15,16 presented images of asphaltene coal and petroleum specimens and confirmed their PAH-side chain architecture as well as the high structural variability; less than 10% of specimens were archipelago. Studies on model compounds and asphaltenes using two-step laser mass spectroscopy5,17 and laser-induced acoustic desorption electron impact mass spectroscopy5,18 concluded that island molecules remained stable under different conditions whereas archipelago structures were susceptible to fragmentation, which may be the reason for their low abundance. A typical mass of an island monomer is 750 Da (±250 Da) and archipelago specimens may reach 2000 Da5,19,20,21,22,23. The Yen-Mullins model is most widely used in describing asphaltene aggregation and structure5,11. At concentrations below ca. 50 mg/L, asphaltenes are believed to exist as single molecules or ‘monomers’. As concentrations increase to the critical nanoaggregate concentration (CNAC) of ca. 100 mg/L (±50 mg/L) asphaltenes start forming nanoaggregates. In this form (or at concentrations above CNAC) asphaltenes are believed to stabilise WOEs by creating a ‘skin’ around water droplets4,5,24,25,26,27. At concentrations of 2–5 g/L nanoaggregates begin forming clusters, each consisting of less than ten nanoaggregates5,28. In our investigation the concentrations at which asphaltenes are studied are below cluster formation. Previous works regarding asphaltene aggregation include the Yen model29,30, fractal model31,32 and colloidal model33.
In what follows we investigate the aggregation properties of four asphaltene samples using ultrasonic velocity precision measurements of asphaltene-in-toluene solutions, ruthenium ion catalysed oxidation (RICO) and statistical analysis. We also perform a biodegradation characterisation of asphaltene parent oils. Our velocity characterisation results suggest that asphaltene aggregation occurs over a critical nanoaggregation region (CNR), rather than at a fixed CNAC. We review the theory of surface-active compound (surfactant) aggregation to highlight phenomena explaining asphaltene phase behaviour. In particular, multiple micellarisation can be useful in understanding aggregation of multi-sized surfactant systems. We suggest that the dimensions of CNR are related to asphaltene side-chain distribution which we obtain using ruthenium ion catalysed oxidation (RICO)34,35. This method induces selective oxidation of aromatic rings, releasing the aliphatic chains almost unaltered. In order to discern the CNR boundaries, we use a statistical constrained optimisation method36,37 with linear models, the details of which are presented in Supplementary Information. Our results suggest that the asphaltene concentration range has three regions: (i) linear monomeric, (ii) non-linear critical nanoaggregation (CNR) and (iii) linear aggregated. The Discussion section investigates the relationship between combined asphaltene velocity and structural data using principal component analysis (PCA).
Materials and Methods
Theory of surface-active compound aggregation
In the context of WOE formation and remediation, asphaltenes are significant due to their surface activity and emulsion stabilisation properties2,38,39. Thus, asphaltenes are often likened to surfactants40 and below we outline a number of important properties to compare with asphaltene phase behaviour. Surfactants tend to be located at the interface between two liquid phases as this corresponds to their lowest energy state41. Such interfacial activity agrees with the phenomenon of asphaltene ‘skin’ formation around water droplets4,5,24,25,42,43,44. Molecular dynamic simulations of asphaltenes illustrated that model compounds with highest surface activity (with charged functional groups) were located at the toluene/water interface after 7 ns of simulation (mostly in cluster form), whereas non-charged moieties did not show interfacial activity and mostly remained in bulk toluene45,46. The latter study developed compounds that were structurally similar to the atomic force microscopy (AFM) images from15, thus the results are representative. Simulations of non-charged asphaltene model compounds illustrated that such asphaltenes were a lot more likely to remain in bulk oil rather than travel to the water/oil boundary47. Nanoaggregation (self-association) of asphaltenes occurs upon reaching the CNAC, whereby crucial to asphaltene solubilisation are steric repulsion of alkane substituents and \(\pi -\pi \) stacking of aromatic cores21,40,48. Steric hindrance restricts the number of asphaltenes in a nanoaggregate and a further addition of asphaltenes to the system will lead to a change in aggregate number, as opposed to size10,40. In other words, nanoaggregation kinetics are controlled by asphaltene (poor) solubility49 and strong asphaltene-asphaltene interactions10; in an aqueous environment micellarisation is controlled by solvent-surfactant interactions. Interfacial tension (IFT) is one of the most widespread measurements in analysing surfactant emulsion-stabilisation properties10. Obtaining a plot of surface tension versus the logarithm of surfactant concentration will illustrate a decreasing trend and the CMC. Noteworthy, interfacial tension measurements require that the activity coefficient a s in the Gibbs adsorption equation (estimating interfacial tension) is constant, which is true for many aqueous systems10. It may be of use to understand the challenges in applying IFT to asphaltene systems. Firstly, asphaltene activity coefficient is reported to have contradictory properties, whereby some studies report an approximation of unity using Scatchard-Hildebrand solubility theory with a Flory-Huggins term50, and other suggest that it is not constant10. The surface tension of toluene is two and a half times lower than that of water, and loading high-energy asphaltenes onto toluene surface may increase surface tension51. On the other hand, in Langmuir-Blodgett26 and pendant droplet43,44 experiments, asphaltenes were shown to decrease surface tension. Studies by Rane, et al.43,44 illustrated that in water-heptol systems with 10–50 ppm asphaltene fraction dynamic interfacial tension decreased with no reported asymptotic value, whereas emulsions with 50–200 ppm asphaltene illustrated an asymptotic limit at 20 mN/m. Interestingly, the 50–200 ppm asphaltene concentration is consistent with our estimations of the CNR, and their nuclear magnetic resonance (NMR) data illustrates a linear dependence of asphaltene concentration on NMR signal breaking around a similar 80–200 ppm range. Contradicting previous studies5,26, Rane, et al.43,44,52 used the Langmuir equation of state to suggest that it is asphaltene monomers that stabilise water-in-model oil emulsions rather than nanoaggregates although their NMR analysis suggested that nearly half of molecules present in model oil were nanoaggegates43. Despite the above differences, using micellarisation as a proxy for asphaltene nanoaggregation allows the use of sonic velocity models40,53 detecting aggregation as a function of changes in solution apparent densities and compressibilities of the dispersed phase. The full model derivation may be found in Zielinski et al.53, and is summarised as follows.
Within a uniform liquid, the ultrasonic velocity u is related to density ρ and adiabatic compressibility β of the medium according to the Urick equation54
For multi-phase fluids which are well-dispersed, and ignoring the effects of sound scattering (valid for sufficiently low concentration of scatterers and away from scattering resonances)55, Eq. (1) can be applied with density and compressibility represented by weighted averages of the mixture components. An extension of Eq. (1) allows to detect the onset of surfactant aggregation into micelles, as proposed by Zielinski et al.53. In particular, the sound velocity u is related to apparent molar solution quantities following the relation
where v denotes specific volume, c- weight concentration, tilde- apparent quantities and subscripts refer to solvent (0), monomer (1) and micellar (m) quantities. Also,
The model (2) implies that pre- and post-micellarisation, sonic velocity is related to surfactant concentration as a combination of two linear behaviours whose intersection estimates the CMC. Apparent molar properties are difficult to measure in practice and we presume that an accurate determination of the CMC is found by an intersection of two linear regressions, the optimal selected by informing the coefficient of determination R 2. Zielinski et al.53 verified this model by measuring the speed of sound in solutions of alkyltrimethylammonium bromides and illustrated a very good fit. A study by Andreatta et al.40 used high-Q ultrasonic velocity measurements together with model in53 to study asphaltene nanoaggregation in toluene, whereby regressions could describe variation in pre- and post-CNAC regions well. Upon closer examination of the monomer-aggregate boundary the point of asphaltene nanoaggregation is associated with velocity fluctuations, the reasons for which we aim to investigate in more detail.
Further, we consider the phenomenon of multiple micellarisation whereby a surfactant solution is prone to forming micelles of multiple sizes given the variation in surfactant molecular size56,57,58. The latter occurs in some pure substances and is very common in mixtures of cationic surfactants with a variation in the hydrophobic tail length57,58,59. Critically, as asphaltenes constitute a fraction of petroleum with a distribution of molecular weights and structures we suggest that a phenomenon similar to multiple micellarisation will also be observed. We will attempt to illustrate the latter by performing ultrasonic characterisation of alkyltrimethylammonium bromide surfactants in single and mixed forms followed by asphaltene dispersions in toluene.
Materials
Chemical solvents toluene, acetonitrile, and n-pentane were purchased from Sigma Aldrich and Acros Organics, all ≥99% purity. Dichloromethane (DCM), methanol (MeOH) and petroleum ether were distilled in-house. The surfactants used in verification studies were tetradecyltrimethylammonium bromide (CH3(CH2)13N(CH3)3Br; C14TAB) and dodecyltrimethylammonium bromide (CH3(CH2)11N(CH3)3Br; C12TAB) 99% and 98% purity respectively, obtained from Sigma Aldrich. The two compounds differ by only two methyl groups and their mixtures were used to test sensitivity of the ultrasonic instrument to multiple micellarisation. Milli-Q 18 M Ω deionised water was used to make C14TAB and C12TAB solutions.
Asphaltenes were precipitated from four petroleum samples: E1 with E2 and E3 with E4 are from two different source rocks respectively and all are from different reservoirs. E1 and E2 are from South America and E3 with E4 are from North America. Samples were selected such that there are two biodegradation aliquots from a source rock. Biodegradation analysis was performed on deasphalted petroleum using thin layer chromatography with short column elution to obtain aliphatic and aromatic fractions. The latter were then analysed by gas chromatograohy-mass spectrometry and relative biomarker abundance compared with Wenger et al.60,61 scales.
Asphaltene precipitation
Asphaltene precipitation was performed using a 40-fold excess n-alkane addition19,62. Crude oil (5 g) was mixed with 200 ml of n-pentane, ultrasonicated for 2 h and left to equilibrate overnight. The mixture was then centrifuged for 15 min at 3500 rpm and maltene supernatant decanted. Further, the asphaltene fraction was washed following a cycle of (i) 200 ml n-pentane addition, (ii) ultrasonication for 30 min, (iii) equilibration for 1 h, (iv) centrifugation for 15 min at 3500 rpm and (v) decanting of the supernatant. Asphaltenes were then air-dried overnight and/or dried with nitrogen gas before purification using Soxhlet extraction with toluene62. The obtained asphaltene-toluene solution was evaporated under reduced pressure to 2–5 ml and washed for a final time as described above (i-v) for further use in ultrasonic characterisation and oxidation experiments.
Ruthenium ion catalysed oxidation of asphaltenes
Inference about asphltene architecture was performed using ruthenium ion catalysed oxidation (RICO)34. The reaction products are homologous series of n-alkanoic fatty acid methyl esters (FAMEs) representing n-alkyl appendages that were attached to PAHs and \(\alpha ,\omega \)-di-n-alkanoic fatty acid bis-methyl esters (DFAMEs) representing n-alkenyl bridges between two aromatic units34,35,63. Asphaltenes (50 mg) were mixed with 4 ml DCM, 4 ml acetonitrile, 5 ml 12% aqueous sodium periodate (NaIO4) and 5 mg ruthenium trichloride (RuCl3·xH2O). The mixture was shaken for at least 18 h using an orbital shaker. Dichloromethane and MeOH (15 ml each) were added to the mixture, shaken vigorously and centrifuged for 15 min at 3500 rpm; this cycle was repeated four times. The supernatant fractions obtained from centrifugation were combined in a separating funnel with 5 ml of 4% aqueous sodium hydroxide (NaOH), shaken vigorously and left to separate for 30 min. The aqueous phase was washed with DCM three times. The obtained organic fraction was mixed with 5 ml 13.5% hydrochloric acid (HCl) and washed with DCM further three times to obtain the final organic phase. This was then evaporated under reduced pressure until dryness, mixed with 5 ml 98:2 mixture of MeOH and sulfuric acid (H2SO4) and reflux-heated for 3 h. The obtained esters were mixed with 10 ml deionised water and washed with DCM four times. Finally, the washings were mixed with 4 ml 2% aqueous NaHCO3 and evaporated under reduced pressure with sodium sulphate (Na2SO4) to ca. 5 ml. The products were pipetted out avoiding aqueous traces and blown down with N2 gas to 1 ml for gas-chromatography-flame ionisation detection (GC-FID) and gas chromatography-mass spectrometry (GC-MS).
Maltene analysis
Maltene (deasphalted) petroleum samples were separated into aliphatic/saturate, aromatic and polar fractions using thin layer chromatography (TLC) and short column elution. Glass plates were covered with 0.5 mm silica gel, left to air-dry and activated in an oven at 125 °C for at least 4 h or overnight. The plates were then decontaminated by elution in DCM and activated at 125 °C for 30 min. Maltenes (10 mg) were spotted on a plate with eicosane (C20H42; aliphatic), phenyldodecane (C12H25C6H5; monoaromatic) and anthracene (C14H10; triaromatic) elution standards and eluted in petroleum ether. The separated aliphatic and aromatic fractions were placed in short columns and eluted with petroleum ether and DCM respectively. The final fractions were reduced to 1 ml and analysed by GC-FID and GC-MS with heptadecylcyclohexane (C23H46) and p-terphenyl (C6H5C6H4C6H5) as internal aliphatic and aromatic standards respectively.
Analytical instruments
Maltene and RICO products were analysed using an Agilent 6890 instrument for gas-chromatography-flame ionisation detection (GC-FID) and an Agilent 7975 C instrument for gas chromatography-mass spectrometry (GC-MS).
Iinitial compound screening was performed using the GC-FID equipped with a 30 m HP5-MS column (0.25 mm internal diameter, 0.25 μm polysiloxane stationary phase; J&W Scientific, USA). Helium was used as carrier gas at a flow rate of 1 ml/min. The GC oven was initially held at 50 °C for two minutes and then raised at a rate of 5 °C/min to a final temperature of 310 °C where it was held isothermally for 20 min. The instrument was run in splitless mode, whereby the injector was held at 280 °C and the FID at 300 °C.
Product detection was carried out using Agilent 7890 A GC split/splitless injector at 280 °C linked to an Agilent 7975 C mass spectrometer. The oven was operated at the same temperature mode as GC-FID using a 30 m HP5-MS column (specification as above, J&W Scientific, USA). A mass selective detector was used in selected ion monitoring and full scan modes (m/z 50–700). Compound identification was based on the NIST05 mass spectral library as well as comparison to mass spectra and relative retention times reported in other studies and in-house guides.
Ultrasonic velocity measurements
Ultrasonic velocity measurements were performed at 25 °C using the Resoscan Research System (TF Instruments). Resoscan is a high-resolution instrument allowing a simultaneous measurement of velocity and attenuation of liquid samples. The system includes a two-channel resonator unit with gold and lithium niobate piezocrystals, two 250 μl sample cells and a Peltier thermostat. Temperature control and measurement are performed via two external units enabling a resolution of 0.001 °C and precision of ±0.005 °C. The small sample requirement of 170–250 μl and a high temperature stability enable to measure conformational changes on a molecular scale. In the vicinity of 25 °C the sound velocity changes by around 5 ms−1 °C−1. Therefore, the precision limit of temperature corresponds to a systematic error in velocity of around 0.025 ms−1, which is sufficiently small that it does not affect our results. The fundamental frequency of the instrument is 10 MHz, with range of 7–11.5 MHz, the precise frequency chosen to maximise the quality factor of the acoustic resonant cell.
Asphaltene solutions in toluene were injected using a glass syringe with an aluminium needle to avoid reactivity with the toluene solvent. The instrument was allowed to equilibrate for 3–4 min and left to take around 100 measurements for every solution concentration, from which an average velocity was determined. Aqueous surfactant solutions were injected an auto pipette. Up to 30 measurements were taken per concentration, with an equilibration time of 1 min.
During sample insertion an injector was held vertically parallel to the cell borehole and injection performed during 30–60 s to minimize air entrapment. The obtained data was analysed for extreme outliers, caused by air bubbles and other artefacts. Mean velocity values were plotted versus solute concentration to which piecewise linear regression models were fitted.
Results
Biodegradation of maltenes
Biodegradation of hydrocarbons (examples are shown in Table 1) results in the removal of saturated and aromatic compounds61,64. The level (LVL) of petroleum biodegradation (1–10) was estimated using the Wenger et al.60,61 scales and diagnostic mass spectrometric assignments65. Gas chromatograms used in biodegradation analysis are provided in the Supplementary Information (Fig. S1–S3).
E1 has little signs of biodegradation (Fig. S1(a–d)). The m/z 85 mass chromatogram shows a homologous series of n-alkanes from which compounds (<C10) are absent. Regular isoprenoids, such as 2,6,10,14-tetramethylpentadecane (pristane) and 2,6,10,14-tetramethylhexadecane (phytane), are abundant (m/z 183) as are hopanes and steranes (m/z 191 and 218). Therefore, E1 is classified as LVL 2 biodegraded. The biodegradation extent of E2 (Fig. S1(e-h)) is greater than that of E1, indicated by the removal of n-decane from the n-alkanes. The isoprenoid, hopane and sterane distributions (m/z 183, 191 and 218) are very similar to E1. Therefore, E2 is is classed as LVL 2–3 biodegraded. In contrast, E3 illustrates a complete removal of n-alkanes which puts it into the heavily biodegraded range (Fig. S2(a-f)). Hopanes (m/z 191) are partially removed and 25-norhopanes have been formed. Sterane compounds (m/z 218) are difficult to resolve and benzoalkanes are degraded (m/z 91). However, monoaromatic steroids (m/z 253) are present, therefore E3 is estimated to be LVL 6-7 biodegraded. E4 shows signs of early biodegradation (Fig. S2(g,h) and S3) with the homologous n-alkane series starting from n-C11. Isoprenoids, such as pristane and phytane, are detectable. Hopanes and steranes are abundant and only C29-norhopane has formed. Therefore, E4 is estimated to be LVL 3 biodegraded.
RICO of asphaltenes
The products analysed from RICO are homologous series of n-alkanoic and α, ω-di-n-alkanoic fatty acid bis-methyl esters (FAME, m/z 74; DFAME, m/z 98)34. Partial and total ion chromatograms (TICs) are provided in Supplementary Information. Table 2 includes abundances of FAME and DFAME compounds in RICO products.
In all cases the TICs are dominated by FAME series. The TICs for E1 and E2 (Fig. S4) illustrate left-skewed distributions with a mild even-odd predominance of medium molecular weight compounds. We calculated the ratio of even-odd medium molecular-weight n-alkanoic acids (C12-18 to C11-17) as an indicator of biological activity66 (Table 2), which was 1.711 and 1.33 for E1 and E2 respectively. Note that the high C16 and C18 peaks also drive the even-odd predominance of E1 which is highest out of the four samples. The DFAME compounds at C4-C6 are mixed with the baseline and become increasingly more resolved after C7. In comparison, the compound distributions of E3 and E4 (Fig. S4–S5) are more peaked with varying compound resolution. Even-odd ratios are 1.349 and 1.446 respectively. The DFAME series are very well-resolved for E3 but are quite weak for E4. From Table 2 it is evident that the FAME and DFAME compounds are negatively correlated. As nanoaggregation kinetics of asphaltenes are affected by steric hindrance between side-chains5,11,21 we will test the coupling between asphaltene FAME distributions and nanoaggregation results observed in ultrasonic velocity measurements in the Discussion section.
Resoscan ultrasonic velocity measurements
Aqueous solutions of C12TAB, C14TAB and their mixtures (C12TAB/C14TAB, 1/1 and 2/1 molar) were used to verify the sensitivity of the ultrasonic velocity instrument to micellarisation. Figure 1 illustrates velocity-concentration plots with superimposed linear models whose R 2 values are provided in Table 3. Every concentration measurement in an average of ca. 10 points. Confidence intervals are not shown as the standard deviations are below image resolution, their mean values are shown in Table 3. Plots a,b and the corresponding R 2 values provide good evidence of the linearity between velocity and surfactant concentration, and the CMC values correspond to previous measurements53,67,68. Plots c,d illustrate multiple micellarisation in CTAB mixtures, with a strong indication of the primary critical micelle concentration (CMC1); the secondary critical micelle concentration (CMC2) may be highlighted by taking a (natural) log-transformation as shown in plots e,f. Similar results were found by Ray et al.57 illustrating multiple micellarisation of CTAB surfactants using tensiometric, conductometric and other methods.

Concentration-velocity measurements of CTAB pure and mixed aqueous solutions. Dashed lines represent fitted linear regressions, R 2 values are reported in Table 3. In plots c-f, CMC1 and CMC2 refer to primary and secondary micelle formation respectively. Points marked in black indicate data that were not included in linear regression estimation.
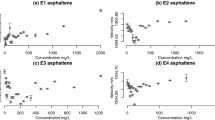
Figure 2 illustrates ultrasonic velocity measurements of four asphaltene samples with linear models superimposed. Every concentration measurement is an average of 60–100 points, the confidence intervals represent the mean ± standard deviation. Piecewise regression models indicated by dashed lines were fitted using a constrained optimisation scheme36,37 which is detailed in the Supplementary Information. When choosing between different regression estimations we deploy this scheme as a guide to indicate where linear behaviour becomes no longer plausible by excessively penalising the R 2 measure given large outliers and changes in regression slope. This should be taken into account when interpreting the penalised R 2 values provided in Table 4, which are expected to be low. In general, the four plots suggest that at either ends of asphaltene concentration range the association with ultrasonic velocity is linear, and the CNR indicated by large outliers. The width of the CNR (Δ CNR) and the velocity difference between the monomeric and aggregated linear models (Δv) are is highly-variable across the samples and are discussed in the next section. Note that the change in the speed of sound is larger than the confidence intervals in the majority of cases. The larger error bars are likely to occur due to trace amounts of dissolved air.

Concentration-velocity measurements of asphaltene-toluene mixtures. Dashed lines illustrate estimated linear models using constrained optimisation, CNR1 and CNR2 refer to the onset and decline of the critical nanoaggregation region respectively.
Discussion
The sample E1 is estimated to have the narrowest Δ CNR of 34.901 mg/L and is also accompanied by the smallest Δv of 0.017 m/s. In contrast, E2 has the largest Δ CNR of 106.776 mg/L and Δv of 0.053 m/s. The total \({R}_{p}^{2}\) values for two samples are similar. The sample E3 has the lowest total \({R}_{p}^{2}\) of 0.4435 although its Δ CNR and Δv are 58.717 and 0.027 m/s which is not the most extreme. E4 has the highest aggregated R 2 and the earliest onset of nanoaggregation at 31.099 mg/L, although Δ CNR is estimated to be twice as great as the monomeric region. The trend below and above the CNR is significant over the measurement error bars for all samples. These trends are similar to those observed in ultrasonic characterisation of Tween 80-toluene mixtures and asphaltene-toluene mixtures by Andreatta, et al.40. In particular, the gradient change is consistent across our four samples, except E1 where both slopes are positive. We are not sure what the reason is for this discrepancy, but a few things can be noted here. The theoretical linear model underlying our measurements that relates the speed of sound to asphaltene concentration40,53 is a function of apparent molar densities and compressibilities. As asphaltenes constitute a petroleum fraction with a range of molecular properties, isolating apparent molar quantities is challenging if not impossible. Understanding the chemico-physical cause of changing gradient in asphaltene ultrasonic velocity measurement would be a very interesting task, however it goes beyond the scope of present investigation.
We performed principal component analysis (PCA)69,70 to assess the combined impact of the above variables on asphaltene nanoaggregation. The variables used in PCA are Δ CNR, Δv, \({R}_{p}^{2}\) and percentage of FAME C11-18 and FAME C>19 out of fatty acid methyl ester products. It is uncertain whether the longer DFAME compounds are PAH linkages as there is no evidence in the literature that inter-aromatic bridges can be over seven carbons long15,16. Therefore, we will exclude this information from PCA. Lower-molecular weight FAMEs (C<7) are highly-volatile and can be partially/completely lost during the RICO procedure34,71, thus will be excluded from the following inference as well. Although the latter is unfortunate, we assume that their contribution is relatively insignificant to hindrance effects as compared to the longer appendages. Table 5 shows the loadings of the principal components (PCs), Minitab software was used for this analysis. Figure 3 illustrates sample division based on PC1 and PC2. Here, principal component 1 (PC1) gives the largest weighting to Δ CNR, Δv and the long aliphatic chains assigning the lowest absolute weight to \({R}_{p}^{2}\). This component can, therefore, be interpreted as the aggregation complexity variable, drawing a positive relation between the size of the CNR and the amount of longer asphaltene side-chains, all of which negatively affect the linear model fit. Observing the distribution in Fig. 3, PC1 indicates that aggregation complexity is lowest for E4 and highest for E2 which is consistent with the aggregation behaviour in Fig. 2. The principal component 2 (PC2) gives the largest weighting to \({R}_{p}^{2}\) whilst making a polar contrast to medium-length asphaltene side-chains. Interestingly, the contrast to long side-chains is half that of medium-sized ones, which implies that the linear model fit is more affected by the abundance of medium-length side-chains or perhaps the size variability. PC2 can be interpreted as the ‘optimal statistical performance’ component. In this domain, E4 and E2 have a very similar performance which is consistent with their fits to linear models (Fig. 2). Together, PC1 and PC2 explain 98% of variation in the data suggesting a link between asphaltene structural and aggregation data.

Division of asphaltene samples based on PC1 and PC2.
Conclusions
In conclusion, we have provided evidence for an asphaltene CNR. Asphaltene samples were selected to represent a range of molecular architectures and biodegradation levels of the parent oils. Their geochemical properties were obtained using maltene analysis, RICO and GC-MS. Ultrasound velocity characterisation was used to detect nanoaggregation in conjunction with the model by Zielinski, et al.53. The analysis of asphaltene solutions in toluene illustrated a variability of aggregation behaviours, subject to asphaltene structural properties. This relationship was confirmed by PCA of asphaltene coupled structural and velocity data. We found that asphaltene structural properties, in particular longer aliphatic appendages and increased variation in size, contribute to a wider aggregation region and a decreased fit to the two-regression model. The results call for a further investigation whereby the origin of longer DFAME compounds is established and their contribution to nanoaggregation also resolved. Ultrasonic velocity results will also be used to construct a probabilistic model of the CNR.
Data availability statement
The datasets generated during and/or analysed during the current study are available from the corresponding author on reasonable request.
References
Bridie, A. J., Wanders, T. H. W., Zegveld, W. & Van Der Heijde, H. B. Formation, prevention and breaking of sea water in crude oil emulsions ‘chocolate mousses’. Mar. Pollut. Bull. 11, 343–348 (1980).
Sjoblom, J. et al. Our current understanding of water-in-crude oil emulsions. recent characterization techniques and high pressure performance. Adv. Colloid Interface Sci. 100-102, 399–473 (2003).
Dicharry, C., Arla, D., Sinquin, A., Gracia, A. & Bouriat, P. Stability of water/crude oil emulsions based on interfacial dilatational rheology. J. Colloid Interface Sci. 297, 785–791 (2006).
Singh, S., McLean, J. D. & Kilpatrick, P. K. Fused ring aromatic solvency in destabilising water-in-asphaltene-heptane-toluene emulsions. J. Dispers. Sci. Technol. 20, 279–293 (1999).
Mullins, O. C. The asphaltenes. Annu. Rev. Anal. Chem. 4, 393–418 (2011).
Sheu, E. Y., Mullins, O. C. & Eds. Asphaltenes: fundamentals and applications (Plenum Publishing, Co, New York, 1995).
Mullins, O. C., Sheu, E. Y., Hammami, A. & Marshall, A. G. Asphaltenes, heavy oils and petroleomics (Springer, New York, 2007).
Sjoblom, J., Simon, S. & Xu, Z. Model molecules mimicking asphaltenes. Adv. Colloid Interface Sci. 218, 1–16 (2015).
Peters, K. E., Walters, C. C. & Moldowan, J. The biomarker guide. Vol. 1 (Cambridge University Press, Cambridge, 2005), 2nd ed. edn.
E., F. S. Micellization. In Mullins, O. C., Sheu, E. Y., Hammani, A. & Marshall, A. G. (eds.) Asphaltenes, heavy oils and petrolemics, 189–203 (Springer Science + Business Media, New York, 2007).
Mullins, O. C. The modified yen model. Energy fuels 24, 2170–2207 (2010).
Durand, E. et al. Effect of chemical composition on asphaltenes aggregation. Energy & Fuels 24, 1051–1062 (2010).
da Silva Oliveira, E. C., Neto, A. C., Junior, V. L., de Castro, E. R. & de Menezes, S. C. Study of brazilian asphaltene aggregation by nuclear magnetic resonance spectroscopy. Fuel 117, 146–151 (2014).
Hosseini-Dastgerdi, Z., Tabatabej-Nejad, S. A. R., Khodapanah, E. & Saharei, E. A comprehensive study on mechanism of formation and techniques to diagnose asphaltene structure; molecular and aggregates: a review. Asia-Pacific J. Chem. Eng. 10, 1–14 (2015).
Schuler, B., Meyer, G., Peña, D., Mullins, O. C. & Gross, L. Unraveling the molecular structures of asphaltenes by atomic force microscopy. J. Am. Chem. Soc. 137, 9870–9876 (2015).
Schuler, B. et al. Heavy oil based mixtures of different origins and treatments studied by atomic force microscopy. Energy & Fuels 31, 6856–6861 (2017).
Sabbah, H., Morrow, A. L., Pomerantz, A. E. & Zare, R. N. Evidence for island structures as the dominant architecture of asphaltenes. Energy & Fuels 25, 1597–1604 (2011).
Borton, D. et al. Molecular structures of asphaltenes based on the dissociation reactions of their ions in mass spectrometry. Energy & Fuels 24, 5548–5559 (2010).
Groenzin, H. & Mullins, O. C. Asphaltene molecular size and structure. The J. Phys. Chem. A 103, 11237–11245 (1999).
Groenzin, H. & Mullins, O. C. Molecular sizes of asphaltenes from different origin. Energy Fuels 14, 677–684 (2000).
Buenrostro-Gonzalez, E., Groenzin, H., Lira-Galeana, C. & Mullins, O. C. The overriding chemical principles that define asphaltenes. Energy Fuels 15, 972–978 (2001).
Groenzin, H. et al. Molecular size of asphaltene solubility fractions. Energy Fuels 17, 498–503 (2003).
Buch, L. et al. Effect of hydrotreatment on asphaltene fractions. Fuel 82, 1075–1084 (2003).
Yarranton, H. W., Hussein, H. & Masliyah, J. H. Water-in-hydrocarbon emulsions stabilized by asphaltenes at low concentrations. J. Colloid Interface Sci. 228, 52–63 (2000).
Jestin, J., Simon, S., Zupancic, L. & Barree, L. Small angle, a neutron scattering study of the adsorbed asphaltene layer in water-in-hydrocarbon emulsions: structural description related to stability. Langmuir 23, 10471–10478 (2007).
Friberg, S. E., AlBawab, A. & Abdoh, A. A. Surface active inverse micelles. Colloid Polym. Sci. 285, 1625–1630 (2007).
Interfacial shear rheology of asphaltenes at oil–water interface and its relation to emulsion stability: Influence of concentration, solvent aromaticity and nonionic surfactan. Colloids Surfaces A: Physicochem. Eng. Aspects 366, 120–128 (2010).
Goual, L. et al. On the formation and properties of asphaltene nanoaggregates and clusters by dc-conductivity and centrifugation. Fuel 90, 2480–2490 (2011).
Yen, T. F., Erdman, J. G. & Pollack, S. S. Investigation of the structure of petroleum asphaltenes by x-ray diffraction. Anal. Chem. 33, 1587–1594 (1961).
Dickie, J. P. & Yen, T. F. Macrostructures of the asphaltic fractions by various instrumental methods. Anal. Chem. 39, 1847–1852 (1967).
Barre, L., Simon, S. & Palermo, T. Solution properties of asphaltenes. Langmuir 24, 3709–3717 (2008).
Simon, S., Jestin, J., Palermo, T. & Barre, L. Relation between solution and interfacial properties of asphaltene aggregates. Energy Fuels 23, 306–313 (2009).
Pfeiffer, J. P. & Saal, R. N. J. Asphaltic bitumen as colloid system. The J. Phys. Chem. A 44, 139–149 (1940).
Peng, P., Fu, J. & Sheng, G. Ruthenium-ions-catalyzed oxidation of an immature asphaltene: structural features and biomarker distribution. Energy Fuels 13, 266–277 (1999).
Snowdon, L., Volkman, J., Zhang, Z., Tao, G. & Liu, P. The organic geochemistry of asphaltenes and occluded biomarkers. Org. Geochem. 91, 3–15 (2016).
Bertsekas, D. P. Constrained optimization and Lagrange multiplier methods (Athena Scientific, Massachussets, 1982).
Smith, A. E. & Coit, D. W. Constraint-handling techniques - penalty functions. In Baeck, T., Fogel, D. & Michalewicz, Z. (eds.) Handbook of Evolutionary Computation (Institute of Physics Publishing and Oxford University Press, Bristol, 1997).
McLean, J. D. & Kilpatrick, P. K. Effects of asphaltene aggregation in model heptane-toluene mixtures on stability of water-in-oil emulsions. J. Colloid Interface Sci. 196, 56–59 (1997).
Grutters, M. et al. Asphaltene induced w/o emulsion: false or true? J. Dispers. Sci. Technol. 28, 357–360 (2007).
Andreatta, G., Bostrom, N. & Mullins, O. C. High-q ultrasonic determination of the critical nanoaggregate concentration of asphaltenes and the critical micelle concentration of standard surfactants. Langmuir 21, 2728–2736 (2005).
Clint, J. H. Adsorption at liquid interfaces. In Surfactant aggregation, 13–32 (Chapman and Hall, New York, 1992).
Khristov, K., Taylor, S. D., Czarnecki, J. & Masliyah, J. Thin liquid film technique - application to water-oil-water bitumen emulsion films. Colloids and Surfaces A 174, 183–196 (2000).
Rane, J. P., Harbottle, D., Pauchard, V., Couzis, A. & Banjaree, S. Adsorption kinetics of asphaltenes at the oil-water interface and nanoaggregation in the bulk. Langmuir 28, 9986–9995 (2012).
Rane, J. P., Pauchard, V., Couzis, A. & Banjaree, S. Interfacial rheology of asphaltenes at oil-water interface and interpretation of the equation of state. Langmuir 29, 4750–4759 (2013).
Kuznicki, T., Masliyah, J. H. & Bhattacharje, S. Molecular dynamics study of model molecules resembling asphaltene-like structures in aqueous organic solvent systems. Energy Fuels 22, 2379–2389 (2008).
Kuznicki, T., Masluyah, J. H. & Bhattacharjee, S. Aggregation and partitioning of model asphaltenes at toluene-water interfaces: molecular dynamics simulations. Energy Fuels 23, 5027–5035 (2009).
Teklebrhan, R. B., Ge, L., Bhattacharjee, S., Xu, Z. & Sjoblom, J. Initial partition and aggregation of uncharged polyaromatic molecules at the oil-water interface: a molecular dynamics simulation study. The J. Phys. Chem. B 118, 1040–1051 (2014).
Zhang, L. Y., Xu, Z. & Masliyah, J. H. Langmuir and langmuir-blodgett films of mixed asphaltene and a demulsifier. Langmuir 19, 9730–9741 (2003).
Buckley, J. S., Wang, J. & Creek, J. L. Solubility of the least-soluble asphaltenes. In Mullins, O. C., Sheu, E. Y., Hammami, A. & Marshall, A. G. (eds.) Asphaltenes, heavy oils and petrolemics, 189–203 (Springer Science + Business Media, New York, 2007).
Yarranton, H. W. & Masliyah, J. H. Molar mass distribution and solubility modeling of asphaltenes. Am. Inst. Chem. Eng. J. 42, 3533–3543 (1996).
Mostowfi, F., Indo, K., Mullins, O. C. & McFarlane, R. Asphaltene nanoaggregates studied by centrifugation. Energy & Fuels 23, 1194–1200 (2009).
Rane, J. P. et al. Applicability of the langmuir equation of state for asphaltene adsorption at the oil-water interface: Coal-derived, petroleum, and synthetic asphaltenes. Energy & Fuels 29, 3584–3590 (2015).
Zielinski, R., Ikeda, S., Nomura, H. & Kato, S. Adiabatic compressibility of alkyltrimethylammonium bromides in aqueous solutions. J. Colloid Interface Sci. 119, 398–408 (1986).
Urick, J. R. A sound velocity method for determining the compressibility of finely divided substances. J.Appl. Phys. 18, 983–987 (1947).
Povey, M. J. W. Ultrasonic techniques for fluids characterisation (Academic Press, California, 1997).
Clint, J. H. Mixed-micelle formation. In Surfactant aggregation, 130–146 (Chapman and Hall, New York, 1992).
Ray, G. B., Chakraborty, I., Ghosh, S., Moulik, S. P. & Palepu, R. Self-aggregation of alkyltrimethylammonium bromides (c10-, c12-, c10-, and c16tab) and their binary mixtures in aqueous medium: a critical and comprehensive assessment of interfacial behavior and bulk properties with preference to two types of micelle formation. Langmuir 21, 10958–10967 (2005).
Georges, J. & Chen, J. W. Micellization study of sodium dodecyl sulfate in water and microemulsion systems by conductivity and counterion-activity measurements. J. Colloid Interface Sci. 113, 143–156 (1986).
Treiner, C. & Makayssi, A. Structural micellar transition for dilute solutions of long chain binary cationic surfactant systems: a conductance investigation. Langmuir 8, 794–800 (1992).
Wenger, L. M., Davis, C. L. & Isaken, G. H. Multiple controls on petroleum biodegradation and impact on oil quality. In SPE Reservoir Evaluation and Engineering, 375–383 (Society of Petroleum Engineers, New Orleans, Louisiana, 2001).
Peters, K. E., Walters, C. C. & Moldowan, J. L. The biomarker guide. Second edition. 2. Biomarkers and Isotopes in Petroleum Systems and Earth History (Cambridge University Press, Cambridge, 2005).
Yu, G. et al. Interfacial behavior and water solubility of various asphaltenes at high temperature. Colloid Interfacial Sci. A 441, 378–388 (2014).
Strausz, O. P., Mojelsky, T. W., Faraji, F. & Lown, E. M. Additional structural details on athabasca asphaltene and their ramifications. Energy Fuels 13, 207–227 (1999).
Milner, C. D., Rogers, M. A. & Evans, C. R. Petroleum transformations in reservoirs. J. Geochem. Explor. 7, 101–153 (1977).
Peters, K. E., Walters, C. C. & Moldowan, J. L. The biomarker guide. Second edition. 1. Biomarkers and isotopes in the environment and human history. (Cambridge University Press, Cambridge, 2005).
Tissot, B. P. & Welte, D. H. Petroleum Formation and Occurrence, 2nd Edition (Springer-Verlag, Berlin, 1984).
Loh, W., Teixeira, L. C. & Lee, L. T. Isothermal calorimetric investigation of the interaction of poly(n-isopropylacrylamide) and ionic surfactants. The J. Phys. Chem. B 108, 3196–3201 (2004).
Sun, D. Z., Wang, S. B., Song, M. Z., Wei, X. L. & Yin, B. L. A microcalorimetric study of host–guest complexes of alpha-cyclodextrin with alkyl trimethyl ammonium bromides in aqueous solutions. J. Solut. Chem. 34, 701–712 (2005).
Mardia, K. V., Kent, J. T. & Bibby, J. M. Multivariate analysis (Academic Press, London, 1979).
Cox, T. An introduction to multivariate data analysis (Oxford University Press, New York, London, 2005).
Ma, A., Shuichang, Z. & Zhang, D. Ruthenium-ion-catalyzed oxidation of asphaltenes of heavy oils in lunnan and tahe oilfields in tarim basin, nw china. Org. Geochem. 39, 1502–1511 (2008).
Brincat, D. & Abbott, G. D. Some aspects of the molecular biogeochemistry of massive and laminated rocks from naples beach section (santa barbara-ventura basin). In Isaacs, C. M. & RüllkÃtter, J. (eds.) The Monterey formation: from rocks to molecules, 140–149 (Colombia University Press, New York, 2001).
Abbott, G. D., Lewis, C. & Maxwell, J. R. Laboratory models for aromatization and isomerization of hydrocarbons in sedimentary basins. Nat. 318, 651–653 (1985).
Abbott, G. D., Lewis, C. A. & Maxwell, J. R. The kinetics of specific organic reactions in the zone of catalysis. Philos. Transactions Royal Soc. Lond. A 315, 107–122 (1985).
Acknowledgements
We thank Dr Melvin Holmes, laboratory technicians David Earley, Bernard Bowler and Dr Paul Donohoe for their kind help, and Dr Jian Qing Shi for assistance in statistical inference. This work is funded by the Natural Environment Research Council Centre for Doctoral Training for Oil and Gas (grant 1522796) and Newcastle University through British Geological Survey Funding Initiative (BUFI). The maintenance of the Resoscan Research System and ultrasound spectrometry instrumentation is supportend through Engineering and Physical Sciences Research Council (grant EP/M026310/1). The funders had no impact on the study design, data collection/analysis, the publication or preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
G.D.A. and M.J.W.P. conceived the project. N.G.P., M.J.W.P. and G.D.A. conceived the ultrasonic characterisation experiments. N.P. provided expertise for ultrasonic characterisation experiments. G.D.A. and A.S. conceived the RICO and biodegradation experiments. G.D.A. provided expertise for GC-MS analysis. A.S. performed the experiments, analysed the results and wrote the manuscript. N.G.P and G.D.A. made the main contribution to the critique of the manuscript. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Svalova, A., Parker, N.G., Povey, M.J.W. et al. Determination of Asphaltene Critical Nanoaggregate Concentration Region Using Ultrasound Velocity Measurements. Sci Rep 7, 16125 (2017). https://doi.org/10.1038/s41598-017-16294-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-16294-5
This article is cited by
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
