
Abstract
Tea plant is a typical fluorine (F) accumulator. F concentration in mature tea leaves is several hundred times higher than that in normal field crops. Long-term consumption of teas with high level F will increase the risks of dental and skeletal fluorosis. The mechanism of F accumulation in tea stands unclear. RNA-Seq and digital gene expression (DGE) techniques were used to investigate the effect of F on the differential expressions of transcriptome in tea plant. The results showed that F content in mature tea leaves was increased with increase in F concentration of cultural solution and duration of F treatment time. Based on comparison with data of GO, COG, KEGG and Nr databases, 144 differentially expressed unigenes with definite annotation were identified. Real-time reverse transcription PCR (qRT-PCR) was used to validate the effect of F on expression of 5 unigenes screened from the 144 unigenes. F treatment induced the expression of defense genes such as receptor-like kinases (RLKs) and U-box domain-containing protein. Based on the present study, F uptake is considered to be related to calcium-transporting ATPase, especially autoinhibited Ca2+ ATPase (ACAs) which was activated by the RLKs and worked as a carrier in uptake of F by tea plant.
Similar content being viewed by others


Introduction
Tea plant is known as a fluorine (F) accumulating plant. Mature tea leaves have much higher F concentration than normal field crops such as rice and wheat1. Some brick teas prepared using mature tea shoots had F content more than 800 mg/kg2. Long-term consumption of brick teas containing high level F would lead to excessive intake of F and increase the risks of dental and skeletal fluorosis3. For many higher plants, F uptake is a passive process through extra-cellular pathways by leaking past the endodermal barrier at the root tips and the uptake is linearly correlated with environmental F concentration4. F is not an essential element to plant and high-level F is phytotoxic to many higher plants because it inhibits respiration and photosynthesis5. Hydroponic experiments showed that many factors affected the F uptake by plant, such as plant species, F concentration, pH of culture medium, duration of F exposure, and interaction between fluoride and other environmental mineral elements such as Al3+ and Ca2+ 4, 6. It was reported that chlorine competitively inhibited F transfer in plant and aquaporins might be involved in the transmembrane transport of F7.
Tea plant enriches a large amount of fluoride in mature leaves without toxicity symptoms. The F accumulation mechanism in tea plant remains to be investigated. Revealing the molecular mechanism of F accumulation in tea plant will be helpful to develop agronomic techniques to controlling F accumulation in tea leaves and to develop screening indicators for breeding new tea cultivars with decreased level of leaf fluoride.
Ribonucleic acid sequencing (RNA-Seq) is a recently developed approach to transcriptome profiling, which provides a more precise measurement of levels of transcripts and their isoforms than previous methods8. Digital gene expression (DGE) follows the RNA-Seq protocol and it can effectively determine the differential expression of genes under experimental conditions9. These high-throughput sequencing and high-performance computing technology are comprehensive and rapid methods for measuring expression levels of various genes in specific tissues at a particular state of an organism. Indeed, DGE can reveal novel genes related to development and pathogenicity. Compared to microarray, DGE is a more powerful tool for searching differentially expressed and low-abundance transcripts. In the present study, we used DGE method to search differentially expressed genes under various F levels. Quantitative real-time RT-PCR (qRT-PCR) was used to verify the relationship of expression levels of the DGE screened genes to F treatment in tea plant.
Results
Effect of cultural solution F concentration on F accumulation in tea leaves
During the testing period, tea leaf F concentration in the control group without F addition remained at a same level. However, F concentration in both 5 mg/L and 20 mg/L F treatments increased with the increase in F level of culture solution and F exposure time (Fig. 1). On the 7th day of F treatment, F concentrations in the both F treatments were more than six times of that of control, suggesting environmental F level and F exposure time have great impact on tea leaf F concentration.

Effect of fluoride treatment on F level in tea leave.
RNA-Seq, DGE and de novo assembly
Trinity, a single k-mer assembler, is a good tool to de novo assemble short-read RNA-Seq raw data10. A transcriptome was obtained by Trinity using mixed tea leaf sample. The transcriptome was approximately 66 giga base pairs (Gbp), 219 thousand contigs with max length 7149 bp, and 117 thousand transcripts with 45.01% GC and 95% Q30. When the low-quality nucleotides were trimmed by Trinity, 84913 unigenes were obtained with N50 of 771 bp, in which 43619 unigenes were given functional annotations and 11774 unigenes were more than 1000 bp in length. The sequence assembly of the tested samples showed that total reads were more than 27 million, with average 53% of GC and 95% of Q30 (Table 1). The unigenes with expression strength being 2-fold higher or 50% lower than the control were defined as differentially expressed unigenes. The number of differentially expressed unigenes was 4805 in all the tested groups, among which 193 markedly differentiated unigenes were screened and 144 of them were given annotations. Further comparisons revealed that the expressions strength of five unigenes out of the 144 annotated unigenes were both F dose-dependent and F exposure time-dependent (Fig. 2). The related information of the 5 genes corresponding to the 5 unigenes was obtained by BLAST search in database of National Center for Biotechnology Information (Table 2). qRT-PCR was used subsequently further to verify the expression patterns of these five genes.

Differentially expressed genes with F dose-dependent and exposure time-dependent. F5D1: 5 mg/L F for 1 day; F5D3: 5 mg/L F for 3 days; F20D1: 20 mg/L F for 1 day; F20D3: 20 mg/L F for 3 days.
Annotation and classification of unigenes
Gene Ontology (GO) is a dynamic vocabulary comprehending the accumulation and variation of gene or protein functions in cell that can be applied to eukaryotes, in which there are three independent ontologies, i.e., molecular function, biological process and cellular component11. To analyze the function of the assembled transcripts, non-redundant sequences were submitted to a BLASTx (E-value ≤ 10−5) for searching against GO database, and major annotations of the differentially expressed unigenes were shown in Fig. 3. To functionally categorize the assembled transcripts, GO terms were assigned to each transcript based on the best BLASTx hit from the nr database. 239 annotations were given to the above 144 F-induced differentially expressed unigenes, among which 85 (35.56%) were related to biological processes, 139 (58.16%) to molecular functions and 15 (6.28%) to cellular components. Based on these annotations, it is suggested that F had greater impact on expression of genes involving in molecular function and biological process than the genes involving in cellular component. The results also revealed that genes involving in cell death and immune system processes were differentially expressed by F exposure, suggesting F imparted stress on the tested tea plants. It was reported that tea plant uptook F via active and passive pathways4. The differential expression of genes involving electron carrier activity and metallochaperone activity might play a role in the active F absorption process.

Major sub-categories of GO annotations.
The database of Clusters of Orthologous Groups of proteins (COGs) is a phylogenetic classification of the proteins encoded in completely sequenced genomes, which is suitable for functional and phylogenetic annotation of newly sequenced genomes12. Among the above 144 F-induced differentially expressed unigenes, 136 were assigned to the appropriate COG clusters, involving in “signal transduction mechanisms”, “defense mechanisms”, “protein turnover”, “chaperones”, “secondary metabolites biosynthesis”, “transport and catabolism”, “cell wall/membrane/envelope biogenesis”, “inorganic ion transport and metabolism”, and “lipid transport and metabolism”, suggesting the F treatment affected the genes relating to signal transduction, defense mechanisms, protein turnover and chaperones. The alteration of genes involving cell wall/membrane/envelope, lipid transport and metabolism implies that tea plant might uptake F through both intracellular pathway and extra-cellular pathway by leaking through endodermal barrier.
Validation of the differentially expressed genes by qRT-PCR
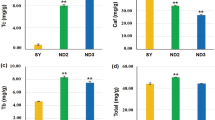
Though DGE is widely utilized in transcriptome study, it is limited to measure the abundance of transcripts because several transcripts might share a same tag as two unrelated genes, paralogs or alternatively spliced isoforms are involved13. The qRT-PCR method is usually used to further verify the DGE results. The verification results of the above five unigenes were listed in Table 2 and it showed that the highest expression strength was observed on third day after F treatment except for that of unigene c41019_g2 (Gene symbol: LOC100256306, Gene ID: 100256306) whose expression was the highest on the first day of F treatment (Fig. 4). On the third day of F treatment, the expression of all the 5 tested unigenes showed dose-dependent effect, which increased with the increase in F concentration. On the 7th day, high-level F treatment (F20D7) had weaker expression than the low level F treatment (F5D7) (Fig. 4). Unigenes c9240_g1 (Gene symbol: LOC110410305, Gene ID: 110410305) and c10652_g1 (Gene symbol: LOC108203676, Gene ID: 108203676) were very weakly expressed in the control without F addition. However, low-level F (5 mg/L) strongly stimulated their expression on the third day of F treatment (F5D3). These results suggest that the optimum sampling time in F-induced test was third day of F-exposure. The decrease in expression strength in treatments of the high level F (20 mg/L) on the seventh day (F20D7) might be a toxic effect of the F stress.

Effect of Fluoride treatment on expression of screened genes.
Discussion
Fluoride treatment triggers defense related genes differentially expressed in tea plant. Receptor-like protein kinases (RLKs) are a superfamily of signal transduction genes encoded by plant genomes, which perceive different signals from the distal cells responding to stresses and diseases14, 15. The RLKs gene superfamily has more than 600 members, which can be divided into 44 sub-families in Arabidopsis 16. The present study revealed that there were 740 unigenes which were given functional annotations relating to RLKs. 134 out of the 740 members were involved in leucine-rich repeat protein kinase such as unigene c46934_g3 (Gene symbol: POPTR_0019s10720g, Gene ID: 7455276), 40 involved in cysteine-rich receptor like protein kinase, and 5 involved in receptor-like protein kinase haiku2. Leucine-rich repeat protein kinase is the largest group of RLKs involving in defense, while the cysteine-rich receptor like protein kinase plays a role in oxidative responses of these receptors in extracellular domains. The gene haiku2 is a mutant allele of gene iku2, which is a leucine-rich repeat kinase gene involving in regulation of seed size17. The qRT-PCR in this study revealed that F showed stimulative effect on expression of RLK gene POPTR_0019s10720g in a dose-dependent manner on third day of F treatment. This can be considered to be a signaling response of tea plant to F treatment. The decrease in the expression of POPTR_0019s10720g on seventh day of F treatment might be a symptom of F stress.
U-box domain-containing proteins (U-box proteins) are a group of ubiquitin-protein ligase E3 that determines the substrate specificity in ubiquitin-26s proteasome pathway (UPP). In Arabidopsis, there are 37 predicted proteins containing U-box, which are classified into 5 subclasses based on structural characteristics and phylogenetic tree18. Though the specific functions of U-box proteins remain to be further investigated, they are largely considered to be involved in biotic and abiotic stress responses19. The present study revealed that there were 124 unigenes involved in U-box proteins whose expression was induced by F treatment, such as unigene c9240_g1 (Gene symbol: LOC110410305, Gene ID: 110410305) (Fig. 4). The LOC110410305 was up-regulated on the third day of F treatment, but was down-regulated on the seventh day of F treatment in F dose-dependent manner. The responses of U-box proteins to F induction on the seventh day of F treatment are considered to be a symptom of F-induced injury of tea plant.
Fluoride uptake in tea plant is closely related to plasma membrane, especially Ca2+ pumps and channels. F uptake by tea root is partially considered to be an active and energy-dependent process involved in transmembrane20. The present study revealed that there were 77 genes related to calcium-transporting ATPase, in which 61 were related to calcium-transporting ATPase as autoinhibited Ca2+ ATPase (ACAs), including 21 ACA9, 6 ACA2, 6 ACA13, 5 ACA1 and others. Ca2+ triggers numerous cell biological processes such as modulating protein kinases, ion channels, and other cellular control proteins. These pathways are involved in plasma membrane-associated and intracellular control channels. The plasma membrane Ca2+ channels include voltage-dependent permeable Ca2+ channel, abscisic acid-activated Ca2+-permeable channel and stretch-activated channel, while the intracellular organelles Ca2+ channel includes insP3-activated Ca2+ channel, cADPR-activated Ca2+ release channel, Ca2+-induced Ca2+ release channel and voltage-dependent Ca2+ channel21. The ACAs in Arabidopsis thaliana can be divided into 4 clusters, based on sequence alignments and intron numbers and/or position22. Isoforms from clusters 1, 2 and 4 have been characterized, in which ACA2 plays a role in relieving salt hypersensitivity23. However, ACA12 and ACA13, as members of cluster 3, are barely investigated. Unlike other ACAs, ACA12 is not stimulated by calmodulin. ACA12 and ACA13 are highly expressed under stress conditions24. The LOC108203676 equivalent to ACA9 in the present study was dramatically upgraded by F induction on first day (Fig. 4), suggesting ACA9 might be involved in responses of F uptake or F stress in tea plant. It is hypothesized that the environmental F triggers the expression of RLKs, which in turn activate Ca2+ ATPase25. The activated Ca2+ ATPase might work as a carrier of fluorine in the F sorption process (Fig. 5).

Hypothetical schematic diagram for fluorine uptake. The RLKs activate Ca2+ ATPase after perceiving F signals, and Ca2+ ATPase helps F enter into cells.
F stimulation accelerates the expression of defense genes such as RLKs and U-box proteins. The RLKs is considered to play important role in the absorption of environmental fluorides. F triggers the expression of RLKs, which in turn activate Ca2+ ATPase. The activated Ca2+ ATPase promotes the absorption and transport of fluoride.
Materials and Methods
Fluorine treatment and leaf sampling
Two-year-old tea cuttings of Camellia sinensis cv. ‘JK2’ were hydroponically cultivated in an air-conditioned chamber. The plants were grown and acclimated for eight weeks in basic nutrient solution (Table 3) at 25 ± 2 °C, 70 ± 10% relative humidity and 12 h light/12 h dark. The renewal of the nutrient solution was carried out weekly. The plants were then divided into 3 groups (10 plants in each group) and treated at three levels of F− (0, 5, 20 mg/L), respectively. The chemical source of F− was NH4F, and the pH of the solution held 4.8–5.2 adjusted by 0.1 M HCl or 0.1 M NaOH. Third leaf from apical bud was sampled with three repetitions for total RNA extraction and F analysis on the day before F treatment, and 1st, 3rd and 7th day of F treatment. The samples for RNA extraction were frozen in liquid nitrogen and then stored at −80 °C till use, and those for F determination were fixed in steam for 1 min and then dried at 80 °C.
Determination of fluorine in tea samples
F content in the tea samples was determined using F ion selective electrode (Shanghai Ruosull Technology Co., Ltd., Shanghai China) following the method described by Stevens et al.26. The reclaim rate of the added F was 95.0–99.3%, with coefficient of variation 2.1%.
RNA-Seq and DGE
Five representative samples were used in the RNA-Seq and DGE analysis and they included the one before F treatment (tabbed as Control) and four samples from various F level and exposure time combination treatments: 5 mg/L F for 1 day (tabbed as F5D1), 5 mg/L F for 3 days (F5D3), 20 mg/L F for 1 day (tabbed as F20D1), 20 mg/L F for 3 days (F20D3), each group has biological replicate. Total RNA was prepared using RNAprep pure Plant Kit (TIANGEN Biotech Co., Ltd., Beijing, China) according to the kit instruction. The amount of total RNA should be more than 10 μg and the concentration of total RNA should be bigger than 300 ng/μL. The mRNA was enriched using Oligo (dT) magnetic beads and fragmented in fragmentation buffer. Agilent 2100 Bioanalyzer (Agilent Technologies, Inc., Santa Clara, USA) was used to check the obtained RNA integrity, and the total RNA concentration was determined using Nanodrop 2000 (Quawell Technology, Inc., San Jose, USA). RNA-Seq and DGE were carried out following Illumina protocol27. The unigenes with expression strength being 2-fold higher or 50% lower than the control were defined as differentially expressed unigenes. Trinity was used to identify SNPs, small insertions or deletions and this set of transcripts was used for downstream statistical analysis. All non-redundant transcripts were searched against the GO, COG, nr database by BLASTALL package with the significant threshold of E-value ≤ 10−5.
Quantitative real-time RT-PCR
Quantitative real-time RT-PCR (qRT-PCR) was used to verify the expression levels of the differentially expressed genes screened by RNA-Seq and DGE profiling. cDNA was synthesized by PrimeScriptTM RT reagent Kit with gDNA Eraser (TaKaRa Biotechnology Co., Ltd., Dalian, China). The qRT-PCR was carried out on the Applied Biosystems StepOnePlus Sequence Detection System (Carlsbad, CA, USA) with SYBR® Premix Ex TaqTM II (TaKaRa Biotechnology Co., Ltd., Dalian, China). The RNA samples used for qRT-PCR were biological replicate from the same experiment. The β-Actin was used as reference gene in the qRT-PCR and the test was carried out in triplicate. The relative gene expression levels were estimated by average threshold cycle which was calculated by \({2}^{-{\rm{\Delta }}{\rm{\Delta }}{C}_{T}}\) 28. The data were expressed as the mean ± SD (standard deviation), and the primer sequences used in the qRT-PCR were listed in Table 4.
References
Lu, Y., Guo, W. F. & Yang, X. Q. Fluoride content in tea and its relationship with tea quality. J Agr Food Chem 52, 4472–4476 (2004).
Fung, K. F., Zhang, Z. Q., Wong, J. W. C. & Wong, M. H. Fluoride contents in tea and soil from tea plantations and the release of fluoride into tea liquor during infusion. Environ Pollut 104, 197–205 (1999).
Webbpepl, M. M. & Bradley, W. G. Endemic Fluorosis with Neurological Complications in a Hampshire Man. J Neurol Neurosur Ps 29, 577-& (1966).
Ruan, J. Y., Ma, L. F., Shi, Y. Z. & Han, W. Y. Uptake of fluoride by tea plant (Camellia sinensis L) and the impact of aluminium. J Sci Food Agr 83, 1342–1348 (2003).
Fornasiero, R. B. Phytotoxic effects of fluorides. Plant Sci 161, 979–985 (2001).
Ruan, J. Y., Ma, L. F., Shi, Y. Z. & Han, W. Y. The impact of pH and calcium on the uptake of fluoride by tea plants (Camellia sinensis L.). Ann Bot-London 93, 97–105 (2004).
Calvo-Polanco, M., Zwiazek, J. J., Jones, M. D. & MacKinnon, M. D. Effects of NaCl on responses of ectomycorrhizal black spruce (Picea mariana), white spruce (Picea glauca) and jack pine (Pinus banksiana) to fluoride. Physiol Plantarum 135, 51–61 (2009).
Wang, Z., Gerstein, M. & Snyder, M. RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet 10, 57–63 (2009).
Audic, S. & Claverie, J. M. The significance of digital gene expression profiles. Genome Res 7, 986–995 (1997).
Zhao, Q. Y. et al. Optimizing de novo transcriptome assembly from short-read RNA-Seq data: a comparative study. Bmc Bioinformatics 12 (2011).
Ashburner, M. et al. Gene Ontology: tool for the unification of biology. Nat Genet 25, 25–29 (2000).
Tatusov, R. L., Galperin, M. Y., Natale, D. A. & Koonin, E. V. The COG database: a tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Res 28, 33–36 (2000).
Wang, X. C. et al. Global transcriptome profiles of Camellia sinensis during cold acclimation. Bmc Genomics 14 (2013).
Gish, L. A. & Clark, S. E. The RLK/Pelle family of kinases. Plant J 66, 117–127 (2011).
Greeff, C., Roux, M., Mundy, J. & Petersen, M. Receptor-like kinase complexes in plant innate immunity. Front Plant Sci 3 (2012).
Shiu, S. H. et al. Comparative analysis of the receptor-like kinase family in Arabidopsis and rice. Plant Cell 16, 1220–1234 (2004).
Luo, M., Dennis, E. S., Berger, F., Peacock, W. J. & Chaudhury, A. MINISEED3 (MINI3), a WRKY family gene, and HAIKU2 (IKU2), a leucine-rich repeat (LRR) KINASE gene, are regulators of seed size in Arabidopsis. P Natl Acad Sci USA 102, 17531–17536 (2005).
Azevedo, C., Santos-Rosa, M. J. & Shirasu, K. The U-box protein family in plants. Trends Plant Sci 6, 354–358 (2001).
Glickman, M. H. & Ciechanover, A. The ubiquitin-proteasome proteolytic pathway: Destruction for the sake of construction. Physiol Rev 82, 373–428 (2002).
Zhang, L., Li, Q., Ma, L. F. & Ruan, J. Y. Characterization of fluoride uptake by roots of tea plants (Camellia sinensis (L.) O. Kuntze. Plant Soil 366, 659–669 (2013).
Schroeder, J. I. & Thuleau, P. Ca2+ Channels in Higher-Plant Cells. Plant Cell 3, 555–559 (1991).
Baxter, I. et al. Genomic comparison of P-type ATPase ion pumps in Arabidopsis and rice. Plant Physiol 132, 618–628 (2003).
Anil, V. S., Rajkumar, P., Kumar, P. & Mathew, M. K. A plant Ca2+ pump, ACA2, relieves salt hypersensitivity in yeast - Modulation of cytosolic calcium signature and activation of adaptive Na+ homeostasis. J Biol Chem 283, 3497–3506 (2008).
Limonta, M. et al. ACA12 is a deregulated isoform of plasma membrane Ca2+ -ATPase of Arabidopsis thaliana. Plant Mol Biol 84, 387–397 (2014).
Huda, K. M. K. et al. Salinity and drought tolerant OsACA6 enhances cold tolerance in transgenic tobacco by interacting with stress-inducible proteins. Plant Physiol Bioch 82, 229–238 (2014).
Stevens, D. P., Mclaughlin, M. J. & Alston, A. M. Limitations of Acid Digestion Techniques for the Determination of Fluoride in Plant-Material. Commun Soil Sci Plan 26, 1823–1842 (1995).
Xia, Z. H. et al. RNA-Seq analysis and de novo transcriptome assembly of Hevea brasiliensis. Plant Mol Biol 77, 299–308 (2011).
Pfaffl, M. W. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res 29, e45 (2001).
Acknowledgements
The authors appreciate the National Natural Science Foundation of China for financial support to this work (Project No. 31170643).
Author information
Authors and Affiliations
Contributions
Qing-Sheng Li: mainly operate the experiments, writing the article; Xiao-Ming Lin: operate the experiments, analysis; Ru-Ying Qiao: operate the experiments; Xin-Qiang Zheng: analysis, interpretation of the results; Jian-Liang Lu: interpretation of the results; Jian-Hui Ye: interpretation of the results; Yue-Rong Liang: design of the study, preparation of the manuscript.
Corresponding authors
Ethics declarations
Competing Interests
This work was financially support by the National Natural Science Foundation of China. All the authors read the final version of the manuscript and the authors declare no conflict of interest in the paper.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Li, QS., Lin, XM., Qiao, RY. et al. Effect of fluoride treatment on gene expression in tea plant (Camellia sinensis). Sci Rep 7, 9847 (2017). https://doi.org/10.1038/s41598-017-08587-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-08587-6
This article is cited by
-
Fluoride-Induced Mitochondrial Dysfunction and Approaches for Its Intervention
Biological Trace Element Research (2024)
-
A comparative assessment of fluoride uptake and toxicity in four major crop seedlings: reduction of α-amylase activity and gibberellic acid
Acta Physiologiae Plantarum (2023)
-
Gene expression analysis in leaf of Camellia sinensis reveals the response to fluoride
Acta Physiologiae Plantarum (2021)
-
Genome-wide analysis of fluoride exporter genes in plants
3 Biotech (2021)
-
Metabolomics Analysis Reveals Major Differential Metabolites and Metabolic Alterations in Tea Plant Leaves (Camellia sinensis L.) Under Different Fluorine Conditions
Journal of Plant Growth Regulation (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
