
Abstract
Tea plant (Camellia sinensis) is a typical fluoride (F) hyperaccumulator enriching most F in old leaves. There is association between the risk of fluorosis and excessive consumption of teas prepared using the old leaves. It is meaningful to develop methods for controlling F levels in tea leaves. We generated a comprehensive RNA-seq dataset from tea plants grown at various F levels for different durations by hydroponics, aiming at providing information on mechanism of F metabolism in tea plant. Besides raw reads of the RNA-seq dataset, we present assembled unigenes and aligned unigenes with annotations versus the Gene Ontology (GO) databases, Kyoto Encyclopaedia of Genes and Genomes (KEGG) databases, and Nonredundant (Nr) protein databases with low e-values. 69,488 unigenes were obtained in total, in which 40,894 were given Nr annotations.
Design Type(s) | transcription profiling by high throughput sequencing design • factorial design • sequence assembly objective |
Measurement Type(s) | transcription profiling assay |
Technology Type(s) | RNA sequencing |
Factor Type(s) | Fluoride Measurement • experimental condition • Exposure Duration |
Sample Characteristic(s) | Camellia sinensis • leaf |
Machine-accessible metadata file describing the reported data (ISA-Tab format)
Similar content being viewed by others

Background & Summary
Tea plants accumulate abundant fluoride (F) from environments without toxicity1–3. The F concentration in mature tea leaves was up to 2800 mg/kg4. In general, tea brewing leaches 24–83% of total F into infusions4. An adult drinking five cups of tea per day would intake 8.0–303% F content of the Polish SAI (Safe and Adequate Daily Intake)5. Long-term consumption of brick teas with excess F would increase the risk of dental and skeletal fluorosis6,7.
Fluoride uptake by tea plant is highly related to Al, Ca and anion channels. Adding Al to the soil or hydroponic solution increased F accumulation in tea plant but had no effect on Al accumulation8. Tea plant might absorb F through a F-Al complex9. Endogenous Ca2+ and CaM played an important role in F accumulation when there was Al3+ in the hydroponic solution10. The addition of Ca2+ decreased the passages in cell wall or membrane, resulting in reduction of F uptake11. F uptake by tea plants was related to anion channel, because anionic channel inhibitor NPPB or DIDS could reduce F absorption12.
For non-model organisms with limited information of genome, ribonucleic acid sequencing (RNA-Seq) is an efficient approach to transcriptome profiling13–15. In this study, RNA-Seq was tested on tea leaf samples from hydroponic tea plants grown at three levels of F for two growing duration, with two biological replicates and control. Based on 10 cDNA libraries, a de novo assembled transcriptome was generated using all F treatments and control samples. After de novo assembly, total 69,488 unigenes were obtained with N50 of 869 bp. The unigene dataset can help explore the potential genes towards F metabolism in tea plant.
Methods
Experiment design
Two-year-old tea cuttings of Camellia sinensis cv. ‘JK2’ were cultured by hydroponic method in a climate chamber at 25±2 °C, 70±10% relative humidity and 12 h light/12 h dark16. The nutrient solution was renewed weekly with formula as below: 100.05 mg/L NH4NO3, 34.68 mg/L KH2PO4, 1.64 mg/L K2HPO4, 2.15 mg/L CaSO4·2H2O, 49.00 mg/L MgSO4·7H2O, 33.32 mg/L Al2(SO4)3·10H2O, 0.28 mg/L FeSO4·7H2O, 14.21 mg/L Na2SiO3·9H2O, 5.00 mg/L H3BO3, 3.00 mg/L MnSO4, 0.44 mg/L ZnSO4·7H2O, 0.16 mg/L CuSO4·5H2O, 0.16 mg/L Na2MoO4·2H2O, and the pH of the solution was adjusted to 4.8–5.2 by 0.1 mol/L HCl or 0.1 mol/L NaOH16. After eight weeks acclimation in the nutrient solution, the tea plants were treated with three levels of F (0, 5, 20 mg/L). Third leaf from apical bud with biological replicates were sampled for F content analysis and RNA extraction on the day before F treatment using 0 mg/L group as control, and 5 mg/L, 20 mg/L combined with 1st, 3rd day as treatment groups. The labels of F treatment were as below, sampling before F treatment from 0 mg/L F group was used as control (tabbed as Ftea-CK), 5 mg/L F for 1 day (tabbed as Ftea-S5-1), 5 mg/L F for 3 days (tabbed as Ftea-S5-3), 20 mg/L F for 1 day (tabbed as Ftea-S20-1), 20 mg/L F for 3 days (tabbed as Ftea-S20-3). The experiment design and the sampling standard were illustrated in Fig. 1.

Sampling on 0 day under 0 mg/L F treatment was used as control (tabbed as Ftea-CK), and sampling on 1 and 3 days under 0 mg/L F treatment were used for F determination; Ftea-S5-1: 5 mg/L F treatment for 1 day; Ftea-S5-3: 5 mg/L F treatment for 3 days; Ftea-S20-1: 20 mg/L F treatment for 1 day; Ftea-S20-3: 20 mg/L F treatment for 3 days. F levels in all samples were measured and the de novo assembled transcriptome was based on Ftea-CK, Ftea-S5-1, Ftea-S5-3, Ftea-S20-1, and Ftea-S20-3 with biological replicates.
Fluoride determination
F contents in tea samples were determined using F ion selective electrode (Shanghai Ruosull Technology Co., Ltd., Shanghai China) mainly following the method described by Stevens et al.17. To inactivate enzymes in tea leaves, we added a pretreatment of F determination by microwaving tea leaves for 60 s. Then the leaves were dried at 120 °C for 30 min and at 75 °C for 3 h to 48 h until the weights of the leaves remained unchanged. Dry samples (0.15 g) were accurately measured into conical flasks with 20 mL boiling water for 30 min and shook up per 10 min. Then the solutions were transferred into 50 mL volumetric flask with TISAB solution (3 mol/L sodium acetate: 0.75 mol/L sodium citrate=1:1 v/v) and metered the solution to 50 mL with ddH2O. Each solution was measured by F ion selective electrode until the change in mV was less than 0.2 mV/min. The standard curve was constructed by NaF (AR grade, dried at 105 °C for 2 h). All F concentrations of samples were calculated by direct calibration from the standard curve. The reclaim rate of the added F was 95.0–99.3%, with coefficient of variation 2.1%. The results of F contents in different tea samples were listed in Table 1.
RNA extraction
Total RNA was extracted using an RNAprep pure plant kit special for plants with high content of polysaccharide or polyphenols (TIANGEN Biotech Co., Ltd., Beijing, China). The quality and quantity of extracted RNA were measured by agarose gel electrophoresis and Nanodrop 2000 (Quawell Technology, Inc., San Jose, USA). The extracted RNA samples were stored at −80 °C.
Library construction and transcriptome sequencing
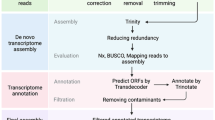
Three μg total RNA of each sample was used for cDNA library construction using TruSeq Stranded mRNA LT Sample Prep Kit (Illumina, San Diego, CA, USA). The mRNA was extracted from total RNA by oligo (dT)-attached magnetic beads. A cDNA library was generated before Next-Generation Sequencing (NGS) in five steps: (1) The mRNA was fragmented using divalent cations under elevated temperature in an Illumina proprietary fragmentation buffer, with mRNA fragment length ranging from 200 to 300 bp; (2) First-strand cDNA was synthesized by random oligonucleotides and SuperScript II using the mRNA fragments as template. (3) Second-strand cDNA was synthesized in a mixture of buffer, dNTPs, RNase H, and DNA polymerase I, in which thymine (T) was replaced by uracil (U) so as to generate strand-specific library. (4) DNA fragments with ligated adaptor molecules on both ends were selectively enriched using Illumina PCR Primer Cocktail in a 15 cycle PCR reaction. After PCR amplification of the cDNA library, the libraries between 300–400 bp were chosen for next step. (5) Products were purified (AMPure XP system) and quantified using the Agilent high sensitivity DNA assay on a Bioanalyzer 2100 system (Agilent). Finally, Illumina NextSeq500 was performed to generate 2×150 bp paired-end (PE) reads. The major process of the study was listed in Fig. 2.

We collected 5 samples with replicates (third leaf from bud) including control and various fluoride treatments. After cDNA construction, Illumina NextSeq500 was used for sequencing in 150 bp paired-end (PE) reads. Trinity was used for clean reads de novo assembly and BlastX was used searching against GO, KEGG and NR databases.
De novo assembly and annotation
Before assembly, the reads with low quality were removed and adapters were filtered with Cutadapt (Version 1.2.1)18. Clean reads were pooled and RNA-Seq de novo assembly was carried out using Trinity, including assembling the reads into contigs by Inchworm, clustering the contigs to generate De Brujin Graph (DBG) by Chrysalis, and obtaining transcripts based on DBG19. The fixed default K-mer value was 25. Details of contigs and unigenes were listed in Table 2. The obtained unigenes were annotated by conducting a local BLASTx search. To classify the functions of contigs, GO annotation was performed using Blast2GO software20, and KEGG orthology and pathway annotations were obtained by KAAS (KEGG Automatic Annotation Server). These methods are expanded versions of descriptions in our related work16.
Data Records
The raw data (Data Citation 1 and Table 3) was deposited in the NCBI Sequence Read Archive. Each accession has two replicates. The assembled unigenes have been deposited at GenBank (Data Citation 2).
Technical Validation
To first control the sequencing quality, we compared total reads and total bases of each sample to ensure the amounts stood the same magnitude. The Q20, base content, GC content and sequence base quality were then determined using FastQC (http://www.bioinformatics.babraham.ac.uk/projects/fastqc). In order to comprehensively cover the transcriptome of Camellia sinensis, ten libraries of control and experimental groups were sequenced and assembled. A total of 270 573 372 raw reads were generated. 268 766 730 clean reads were obtained for de novo assembly after filtration, with clean reads rate being up to 99.33% (Table 4). 219 018 contigs and 69 488 unigenes were obtained (Table 2).
Functional annotations were obtained by sequence based alignments performed by blast search (BLASTx) against the non-redundant protein database (Nr). The Nr species distribution and e-value distribution (Fig. 3) revealed that 55.24% of unigenes with hits had a strong homology with the sequences available in the Nr protein database (e-value < e−45). The distribution of Nr species was revealed that the majority of Camellia sinensis unigenes showed the highest homology with Actinidia chinensis var. chinensis (52.05%), then Vitis vinifera (5.49%), Quercus suber (4.10%), Camellia sinensis (2.69%), Juglans regia (1.47%), Olea europaea var. sylvestris (1.24%), Coffea canephora (1.19%), Nelumbo nucifera (1.02%), Hevea brasiliensis (0.98%), Theobroma cacao (0.94%), Sesamum indicum (0.77%), and others (28.05%). Because of limited Nr annotations of Camellia sinensis, only 2.69% of the unigenes had Nr annotations against Camellia sinensis. However, 93.19% (64 756 out of 69 488) of all unigenes could mapped on newly published tea genome database using tophit software. Unigenes unable to mapped on tea genome database and predicted fusion genes information were offered in appendix (Supplementary Table 1 and Supplementary Table 2). The major distribution of GO annotations was listed in Fig. 4, based on the Blast2GO software analysis. GO database includes three main categories: biological process, cellular component and molecular function. Within the biological process category, “cellular process”, “metabolic process”, and “single-organism process” were the most abundant sub-categories. In cellular component category, the predominant portion of unigenes represented “membrane”, “cell” and “cell part” followed by “membrane part” and “organelle”. Under the molecular function category, “catalytic activity” and “binding” sub-categories were the major proportions of unigenes. The distribution of KEGG pathways annotations was shown in Fig. 5. Sub-categories as “signal transduction”, “translation” and “carbohydrate metabolism” were the most abundant categories in KO hierarchies. Besides, certain category as “transport and catabolism” was highly related to experimental conditions. The summary of databases annotation was listed in Table 5.

Distribution of NR e-value and species.

Bars represent the numbers of unigenes assigned into GO sub-categories of three main categories: biological process, cellular component and molecular function. Within the biological process category, “cellular process”, “metabolic process”, and “single-organism process” were the most abundant sub-categories. In cellular component category, the predominant portion of unigenes represented “membrane”, “cell” and “cell part” followed by “membrane part” and “organelle”. Under the molecular function category, “catalytic activity” and “binding” sub-categories were the major proportions of unigenes.

Bars represent the numbers of unigenes clustered into KEGG Orthology (KO) hierarchies. “Signal transduction”, “translation” and “carbohydrate metabolism” were the most abundant categories in KO hierarchies. Certain category as “transport and catabolism” was highly related to experimental conditions.
Additional information
How to cite this article: Li, Q.S. et al. De novo transcriptome assembly of fluorine accumulator tea plant Camellia sinensis with fluoride treatments. Sci. Data. 5:180194 doi: 10.1038/sdata.2018.194 (2018).
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
References
Ruan, J. Y. & Wong, M. H. Accumulation of fluoride and aluminium related to different varieties of tea plant. Environ Geochem Hlth 23, 53–63 (2001).
Shu, W. S., Zhang, Z. Q., Lan, C. Y. & Wong, M. H. Fluoride and aluminium concentrations of tea plants and tea products from Sichuan Province, PR China. Chemosphere 52, 1475–1482 (2003).
Lu, Y., Guo, W. F. & Yang, X. Q. Fluoride content in tea and its relationship with tea quality. J Agr Food Chem 52, 4472–4476 (2004).
Fung, K. F., Zhang, Z. Q., Wong, J. W. C. & Wong, M. H. Fluoride contents in tea and soil from tea plantations and the release of fluoride into tea liquor during infusion. Environ Pollut 104, 197–205 (1999).
Malinowska, E., Inkielewicz, I., Czarnowski, W. & Szefer, P. Assessment of fluoride concentration and daily intake by human from tea and herbal infusions. Food Chem Toxicol 46, 1055–1061 (2008).
Cao, J. et al. Brick tea fluoride as a main source of adult fluorosis. Food Chem Toxicol 41, 535–542 (2003).
Webbpepl., M. M. & Bradley, W. G. Endemic Fluorosis with Neurological Complications in a Hampshire Man. J Neurol Neurosur Ps 29, 577–583 (1966).
Ruan, J. Y., Ma, L. F., Shi, Y. Z. & Han, W. Y. Uptake of fluoride by tea plant (Camellia sinensis L) and the impact of aluminium. J Sci Food Agr 83, 1342–1348 (2003).
Nagata, T., Hayatsu, M. & Kosuge, N. Aluminium kinetics in the tea plant using 27Al and 19F NMR. Phytochemistry 32, 771–775 (1993).
Zhang, X. C. et al. Ca2+ and CaM are involved in Al3+ pretreatment-promoted fluoride accumulation in tea plants (Camellia sinesis L.). Plant Physiol Bioch 96, 288–295 (2015).
Ruan, J. Y., Ma, L. F., Shi, Y. Z. & Han, W. Y. The impact of pH and calcium on the uptake of fluoride by tea plants (Camellia sinensis L.). Ann Bot-London 93, 97–105 (2004).
Zhang, X. C., Gao, H. J., Zhang, Z. Z. & Wan, X. C. Influences of different ion channel inhibitors on the absorption of fluoride in tea plants (Camellia sinesis L.). Plant Growth Regul 69, 99–106 (2013).
Jain, M. Next-generation sequencing technologies for gene expression profiling in plants. Brief Funct Genomics 11, 63–70 (2012).
Wang, Z., Gerstein, M. & Snyder, M. RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet 10, 57–63 (2009).
Martin, J. A. & Wang, Z. Next-generation transcriptome assembly. Nat Rev Genet 12, 671–682 (2011).
Li, Q. S. et al. Effect of fluoride treatment on gene expression in tea plant (Camellia sinensis). Sci Rep 7 (2017).
Stevens, D. P., Mclaughlin, M. J. & Alston, A. M. Limitations of Acid Digestion Techniques for the Determination of Fluoride in Plant-Material. Commun Soil Sci Plan 26, 1823–1842 (1995).
Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J 17, 10–12 (2011).
Grabherr, M. G. et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol 29, 644–652 (2011).
Conesa, A. et al. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research Bioinformatics 21, 3674–3676 (2005).
Data Citations
NCBI Sequence Read Archive SRP120449 (2017)
GenBank GGEB00000000.1 (2017)
Acknowledgements
The authors appreciate the National Natural Science Foundation of China for financial support to this work (Project No. 31170643).
Author information
Authors and Affiliations
Contributions
L.Q.S. mainly operated the experiments and prepared the manuscript, including de novo assembly, annotations, alignments and analysis. L.X.M., Q.R.Y., S.E.H. and L.X.M. prepared the samples and basic analysis. L.J.L. interpreted the results. J.H.Y. interpreted the results. Z.X.Q. interpreted the results. L.Y.R. designed the study and prepared the manuscript. All the authors read the final version of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Additional information
Supplementary information accompanies this paper at
ISA-Tab metadata
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/ The Creative Commons Public Domain Dedication waiver http://creativecommons.org/publicdomain/zero/1.0/ applies to the metadata files made available in this article.
About this article

Cite this article
Li, QS., Li, XM., Qiao, RY. et al. De novo transcriptome assembly of fluorine accumulator tea plant Camellia sinensis with fluoride treatments. Sci Data 5, 180194 (2018). https://doi.org/10.1038/sdata.2018.194
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/sdata.2018.194
