
Abstract
Catalytic Asymmetric allylation of aldehydes with functionalized allylic reagents represents an important process in synthetic organic chemistry because the resulting chiral homoallylic alcohols are valuable building blocks in diverse research fields. Despite the obvious advantages of allyl halides as allylation reagent under Barbier-type conditions, catalytic asymmetric version using functionalized allyl halides remains largely underdeveloped. Here, we addressed this issue by employing a chromium-catalysis system. The use of readily available allyl bromides with γ substitutions including trimethylsilyl, fluorinated methyl and phenylthio groups provided an efficient and convenient method to introduce those privileged functionalities into homoallylic alcohols. Good yields, high anti-diastereo- and excellent enantioselectivities were achieved under mild reaction conditions.
Similar content being viewed by others

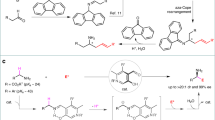
Introduction
Carbonyl allylation with functionalized allyl reagents has been a sustained research topic, as in this transformation, two new functionalities including one alcohol and a terminal C-C double bond are generated1. Thanks to the efforts from many organic chemists, traditional organometallic allylation based on Boron, Silicon and Tin reagents has become a valuable tool to generate C-C bond in a reliable and predictable way (Fig. 1a)2,3,4,5,6,7. Using alcohol as the equal efficient aldehyde surrogate, catalytic transfer hydrogenation protocols have considerably matured in the past decade (Fig. 1b)8,9,10, nonetheless, above mentioned methods possess several minor flaws, such as requiring the pre-generation of organometallic reagents or the usage of precious metals. At the same time, the allylation of carbonyl compounds with allylic halides using metals, typically Mg, In, as mediating reagents under Barbier-type reaction conditions is of great interest11. This strategy offer practical advantages as the nucleophile is generated in situ in the presence of electrophiles, not only circumvents the need of isolating allyl metal species, but also enables an intramolecular version.

(a) Allylation with Silicon, Tin and Boron reagents; (b) Transfer hydrogenation allylation; (c) This work: asymmetric chromium-catalyzed allylation with functionalized allyl bromides.
Chromium mediated Grignard-type addition of carbohalides to aldehyde, the Nozaki-Hiyama-Kishi reaction proved to be one of the most powerful synthetic methods for carbon-carbon bond formation12,13,14,15,16,17,18, its synthetic utilities have been demonstrated in numerous complex natural products total synthesis. Despite the rapid evolution of chiral chromium ligands19, there remains considerable unmet challenges in enantioselective catalysis due to the generally high reactivity of organometallic allylation reagent derived from allyl halides e.g. Grignard reagents. On the other hand, asymmetric chromium catalysis holds great synthetic potential considering its extraordinary affinity towards aldehydes and mild reductive nature, together with the fruitful and ready accessible functionalized allyl halides. In line with our interests in the asymmetric carbonyl allylation with chromium complex, we recently reported enantioselective chromium catalyzed carbonyl 2-(alkoxycarbonyl)allylation leading to synthetic useful α-exo-methylene-γ-butyrolactones20, dearomative coupling of halomethylheteroarenes20, 21, chiral quaternary stereogenic centers formation22. Herein, we would like to report our preliminary results on incorporating synthetically useful and medicinally relevant functionalities including trimethyl silyl, fluorinated methyl and phenylthio groups into homoallylic alcohol (Fig. 1c). The resulting products are not only valuable substances themselves, but also serve as significant building blocks for further derivatizations. The use of readily available γ-functionalized allyl halides, cheap metals and chiral ligands, mild reaction conditions together with easy execution make an attractive approach which would streamline the access to a large variety of related reaction patterns.
Results and Discussion
anti-Diastereo- and Enantioselective Carbonyl (Trimethylsilyl)allylation
Organosilicon represents a privileged functionality in synthetic organic chemistry23,24,25. A variety of named reactions and useful transformations derive from the unique properties of silicon; representative examples including Peterson olefination, Brook rearrangement, Fleming-Tamao oxidation, Prins cyclization and Sakurai allylation. Thus, sustained efforts have been dedicated toward the development of efficient methods for the expedient introduction of silicon into organic molecules. Among various organosilicon compounds, allylsilane is a very important building block leading to diverse useful products including homoallylic alcohols. Chiral β-hydroxy allylsilanes and derivatives have been extensively used by Roush, Panek and others for the synthesis of 1,2- and 1,4-diols in the total synthesis of natural products26,27,28,29,30,31,32. In 2010, Krische reported an iridium catalyzed silylallylation using SEGPHOS as a chiral ligand for the synthesis of α-silyl homoallylic alcohols33. More recently, Barrio and Akiyama reported the chiral BrØnsted acid catalyzed carbonyl allylboration with γ-silylboronates34, 35. Given the broad synthetic utilities of α-silyl homoallylic alcohols, alternative methods for efficient and enantioselective synthesis of this important moiety are highly desired.
At the outset of our study, the coupling of 3-phenylpropanal with easily accessible [(1E)-3-bromoprop-1-enyl]trimethylsilane (1a) was chosen as the model reaction (Fig. 2). With proton sponge as the base in the complexation step, ZrCp2Cl2 as the dissociation agent and Mn as the reducing reagent for chromium turnover. We first tested the reaction in the absence of any chiral ligands, to our delight, the desired homoallylic alcohol was generated in good yield as a single diastereomer (Fig. 2, entry 2).

Evaluation of chiral ligands and other reaction parameters. aThe reactions were carried out at 0.2 mmol scale unless noted otherwise; bIsolated yield; cDetermined by chiral HPLC analysis, the absolute configuration was determined by comparison with reported example; d1 equiv of Mn was added for the complex formation; eComplexation took 8 hrs.
We turned our attention to the development of its asymmetric variant by employing carbazole-based bisoxazoline (Nakada catalyst) as the chiral ligands20, 36. L1 (R = iPr) was first examined; after quite a few trials by varying the solvents and additives based on our previous studies, product 2a could be isolated in 75% with 89% ee (Fig. 2, entry 3). Further ligand screening revealed that an introduction of a bulkier substitution proved deleterious for the enantioselectivity, as L2 (R = tBu) resulted in 2a in 66% with only 28% ee (Fig. 2, entry 4). A comparable enantioselectivity was obtained as L3 (R = PhCH2) (Fig. 2, entry 5) and L4 (R = Et) (Fig. 2, entry 6) were employed as ligands. Finally, L5 (R = iBu) gave rise to the 2a in 90% yield with 95% ee (Fig. 2, entry 1).
All other deviations from the optimal conditions led to a decrease of the enantioselectivity and in some case even the yield. Lowering the reaction temperature didn’t benefit the overall efficiency (Fig. 2, entry 10). Solvents screening revealed that THF gave the best result, the reactions running in either CH3CN or DME gave 2a in slightly lower yields and ee (Fig. 2, entry 12 and 13). Cheaper and easy handling CrCl3 could also be directly used, a comparable result (87% yield, 94% ee, Fig. 2, entry 14) was obtained; in this experiment, the complexation step required the addition of one equivalent of Mn metal.Entrya
It was found that ligand loadings could be lowered to 7 mol% with slight erosion of enantioselectivity (Fig. 2, entry 15). LiCl exhibited an enhancing effect on the coupling rate and enantioselectivity, which is likely to facilitate the formation of allyl species and its transmetallation to the chiral chromium complex (Fig. 2, entry 16). Both TMSCl37,38,39 and ZrCp2Cl2 40 have been previously used as dissociating reagents, and they have various impacts on the reaction. However, in this case, TMSCl had slightly less efficiency (Fig. 2, entry 17). Notably, the reaction scale could be increased to 1 mmol with maintenance of the efficiency (Fig. 2, entry 18).
With these optimized conditions in hand (Fig. 2, entry 1), the generality of this transformation was established using a broad range of aldehydes shown in Fig. 3. Excellent anti - selectivity ( > 98:2) was observed for all the

Substrate scope studies. aAll reactions carried out at 0.2 mmol scale under the standard conditions, ligand L5 was used unless otherwise noted, generally over 50/1 anti-diastereoselectivity was observed; bThe absolute configurations of 2e, 2l, 2m, 2p, and 2r were assigned by comparison with reported examples, others were by analogy; cTHF as solvent; dCH3CN as solvent.
cases and generally high enantioselectivity (90–98% ee) was obtained. Coupling of 1a with representative aliphatic aldehydes including cyclohexyl carboxyaldehyde, heptaldehyde proceeded smoothly; the corresponding homoallylic alcohols 2b, 2c were isolated in high yields (93% and 82%) with excellent enantiomeric excess (98% and 92% ee). Substrate with a chloro group reacted well, giving 2d in 92% yield with 97% ee. Aldehydes bearing a terminal C-C double bond or a cyclohexene moiety both participated in the (trimethylsilyl)allylation efficiently, furnishing the corresponding products 2e and 2f in good yields (94% and 90%) with excellent enantiomeric excess (97% and 90%). Heteroatoms with proper protecting groups such as TBDPS for oxygen atom or phthalimide for nitrogen atom are compatible under current reaction conditions, no erosion of enantiomeric excess of the corresponding products 2g and 2h were detected (97% ee and 98% ee, respectively). The current reaction condition tolerates a sensitive ketal moiety, protected triols 2i was obtained in moderate yield (80%) and good ee (>97%). A naturally occurring aldehyde (−)-citronellal bearing a chiral methyl group β to the carbonyl group exhibited good reactivity and selectivity profile, the corresponding 2j was obtained in 91% yield with 95% de. Reaction of racemic 3,5,5-trimethylhexanal provided 2k in 95% yield with 95% ee for each diastereomer.
Attempts to expand this protocol to aryl aldehydes turned out to be successful. Benzaldehyde reacted well, giving 2l in moderate 56% yield with 93% ee. The effect of benzene substituents was examined next. The phenyl group could be freely halogenated with Cl, Br and F without compromise of the reaction efficiency in terms of yields and ee, as products 2m to 2p were isolated in moderate to good yields (56%-64%) with decent ee (ranging from 91% to 94%). Electron-withdrawing CF3 group and weak electron-donating Me group can both be introduced into the system, leading to products 2q to 2s in useful level of yields (>50%) and excellent ees (>90%). Moreover, heterocycle such as thiophene was compatible under current conditions, giving the 2t in 66% yield with 92% ee. To our delight, another important class of aldehydes, α, β-unsaturated aldehydes are suitable substrates for this chemistry as well, it was found that even higher enantiomeric excesses 97% ee for 2u and 98% ee for 2v were obtained from cinnamaldehyde and 4-fluorocinnamaldehyde.
Notably, for several aliphatic aldehydes (2c, 2f, 2g, 2h), most aromatic and α, β-unsaturated aldehydes, CH3CN was a better solvent than THF in terms of bigger ee value, generally over 20% difference was observed.
anti-Diastereo- and Enantioselective Carbonyl (fluoronated methyl) allylation
The introduction of fluorine atoms into organic molecules often leads to dramatic changes in their properties such as solubility, metabolic stability, and bioavailability41. Additionally, fluoroalkyl groups, especially the trifluoromethyl, difluoromethyl groups are strongly electron-withdrawing and highly hydrophobic. Because of these desirable properties, fluoroalkylated compounds are widely used in materials science, argochemistry and medicinal chemistry42, 43. Crotylation of carbonyl compounds constitutes one significant transformation in synthetic organic chemistry, as the resulting β-methyl homoallylic alcohols serve as indispensable segment in numerous polyketide natural products and advanced intermediates leading to molecules with bio- or medicinal significance. Thus, enantioselective introduction of fluorinated methyl group into the homoallylic system have been an important subject. However, to the best of our knowledge, only a few methods have been reported44,45,46. In 2010, Krische reported an iridium catalyzed (trifluromethyl) allylation using SEGPHOS as a chiral ligand under the transfer hydrogenation conditions47. Despite the above mentioned elegant strategies for introduction of trifluoromethyl group, the demand for alternative efficient methods, and the lack of practical protocol for introducing difluoromethyl and monofluoromethyl groups prompted us to explore chromium-catalyzed asymmetric carbonyl allylation with (fluorinated methyl) allyl halides. γ-trifluoromethylallyl halides are simple and abundant chemicals and easy to prepare. Directly use of them as carbonyl allylation reagents to generate α-trifluoromethyl homoallylic alcohols in racemic manner have been investigated in indium catalysis48,49,50,51. However, neither asymmetric version nor (difluoro- or monofluoromethyl) allylation has been reported. We began with investigating the cross-coupling between γ-trifluoromethylallyl bromide and dihydro cinnamaldehyde. After examination of a considerable variety of reaction parameters, we were pleased to find the anticipated trifluomethylated homoallylic alcohol could be obtained in 90% isolated yield with 95% ee as a single diastereomer. Ligands screening revealed that L1 was the optimal ligand (Figs 3a and 4).

Substrate scope studies. a,b aAll reactions carried out at 0.2 mmol scale under the standard conditions, ligand L1 was used unless otherwise noted, generally over 50/1 anti-diastereoselectivity was observed; bThe absolute configurations of 3a, 3g, and 3h were assigned by comparison with reported examples and X-Ray single crystal analysis of 6, others were by analogy; cTHF as solvent; dDME as solvent.
This highly enantioselective synthesis of 3a can also be expanded to reactions with a variety of aldehydes, and high enantioselectivity (93–98% ee) was obtained (Fig. 4). Heptaldehyde participated in this reaction efficiently; the corresponding 3b was isolated in moderate yield with excellent enantiomeric excess (93% ee). A terminal chloro group was tolerated under current reaction condition; desired 3c was obtained in 82% yield with 95% ee. Reaction of (−)-citronellal bearing a chiral methyl group β to the carbonyl group proceeded well to provide 3d in 70% yield with 96% ee. Substrate with a sensitive ketal moiety was also amenable to the (trifluoro methyl)allylation, giving good overall yield with excellent ee for both diastereoisomers. A range of α, β-unsaturated aldehydes including unsubstitued, para-methoxy, ortho-methoxy and para-fluoro cinnaldehyde reacted well to provide the products (3h–3k) in useful level of yields with excellent ee. However, our attempts to apply the aryl aldehydes to the coupling reaction failed. After assay the full scope of (trifluoromethyl) allylation, we turned our attention to the unprecedented (difluoromethyl) allylation. To our delight, the couplings between γ-difluoromethylallyl bromide with four aliphatic aldehydes including dihydro cinnamaldehyde, hexanal, 5-chloropetanal and (−)-citronellal proceeded well to afford the desired difluoromethylated homoallylic alcohols in moderate to good yields with excellent enantiomeric excess (92–98% ee). However, further scope studies revealed that aryl and α, β-unsaturated aldehydes are not good substrates under current reaction conditions. We then tested the challenging (monofluoromethyl) allylation due to the potential competing (monobromomethyl) allylation. As anticipated, the reactions proceeded sluggishly with most of the aldehydes tested. Nevertheless, we are pleased to find that two α, β-unsaturated aldehydes are suitable substrates under current reaction conditions. The corresponding 3p and 3q were obtained in moderate yields in excellent ee (93% each).
Notably, for (difluoro- or monofluoromethyl) allylation, DME was a better solvent than THF in terms of bigger ee value, generally over 10% difference was observed.
anti-Diastereo- and Enantioselective Carbonyl (phenylthio) allylation
Sulfur-derived functional groups are ubiquitous in synthetic organic chemistry, pharmaceutical industry, material science and food chemistry, which were evidenced by the fact that over 326 FDA approved drugs containing sulfur functionalities52,53,54,55,56,57,58. Among them, thioether is especially popular and can be found in a broad range of pharmaceuticals and natural products59, 60. Although there is a vast array of methods have been developed to incorporating a sulfur into a specific position in a molecule, the catalytic asymmetric construction of a sulfide-bearing carbon centers is still rare61, 62. Thus, the development of an efficient and convenient synthetic method, using readily available building blocks would be of meaningful importance in both the synthetic organic chemistry and pharmaceuticals advancement.
In our attempt to introduce a phenylthio unit into the homoallylic system, model reaction between (E)-(3-bromoprop-1-en-1-yl)(phenyl) sulfane and 3-phenylpropanal was selected to perform in the presence of L1-CrCl2 complex under previously established conditions. To our delight, the desired α-benzylthio homoallylic alcohol was isolated in moderate yield with good dr and excellent ee. After a few reaction optimization trials, the product 4a was obtained in 95% isolated yield with 12:1 dr and 93% ee (Figs 4a and 5). Notably, the diastereoselectivity dropped to 2:1 in the absence of chiral ligand. Having obtained the optimized reaction conditions, the issues with respect to the functional group tolerance were thus addressed, and the results are summarized in the Fig. 5. Reaction of linear hexanal gave similar results in terms of yield and selectivity (4b). Substrates with synthetically useful functional groups such as Cl and terminal double bond (4c and 4d) participated in this reaction efficiently, to deliver the products in decent yield with good dr and excellent ee. We were pleased to find that heteroatoms including O, N, S with proper protecting groups are well tolerated in this reaction, which offers the opportunity for further synthetic elaborations. Beside aliphatic aldehydes, α, β-unsaturated aldehydes are amenable substrates under current reaction conditions. An equal level of yield and enantioselectivity with even higher diastereoselectivity was observed from the reaction of (E)-hex-2-enal. An aryl conjugated enal proved to be beneficial, an improved diastereoselectivity and enantioselectivity were achieved for Cinnamaldehyde. Furthermore, substrates bearing substituents such as para-Cl, para-Br, meta-F, ortho-MeO and ortho-Me on the aryl ring of cinnamaldehyde also were engaged well in this reaction to furnish the desired products in good yields with synthetically useful level of diastereoselectivity and excellent enantioselecitivity.

Substrate scope studies. aAll reactions carried out at 0.1 mmol scale under the standard conditions, ligand L1was used unless otherwise noted; bThe absolute configurations of 4e were assigned by comparison with reported examples, others were by analogy. cat 0 °C for 24 h.
To demonstrate the potential of this protocol in the synthesis of relatively complex molecules, an aldehyde derived from natural occurring lithocholic acid was subjected to the reactions with three allyl bromides, the desired homoallylic alcohols were obtained in good yields with excellent de. The synthetic utility of the resulting homoallylic alcohols were further illustrated in two short transformations of allylsilane (Fig. 6). Following a reported procedure, treatment of 2m with Selectfluor under buffered reaction conditions afforded product 8 in good yield, with almost complete preservation of the optical purity34. Finally, 2a underwent Prins cyclization with dihydrocinnamaldehyde in the presence of TMSOTf to furnish the synthetically useful dihydropyran 9 in good yield and excellent diastereoselectivity with high optical purity.

Synthetic utilities.
Conclusions
In summary, an efficient synthesis of chiral β-functionalized homoallylic alcohols has been achieved through chromium catalyzed asymmetric allylation of aldehydes with three types of γ-substituted allyl bromides. This protocol features readily available allylation reagents, convenient operation and mild reaction condition, broad functional group tolerance and high levels of diastereoselectivity and excellent enantioselectivity. The synthetic value of this methodology was demonstrated in two short transformations. We positively believe this method will find applications in the enantioselective synthesis of related pharmaceutical compounds and biomolecules.
Methods
(For details of the synthetic procedures, see Supplementary Methods pages 3–7).
General Procedure: To a mixture of anhydrous chromium(II) chloride (1.2 mg, 0.01 mmol, 5.0 mmol%), 1,8-bis((S)-4-((R)-sec-butyl)-4,5-dihydrooxazol-2-yl)-9H-carbazole (L-5, 9.0 mg, 0.022 mmol, 10.8 mmol%) and Proton sponge (6.0 mg, 0.028 mmol, 14.0 mmol%) was added THF (1.0 ml) under an nitrogen atmosphere. The mixture was stirred vigorously at room temperature for 3 hours before it was transferred into a vessel charged with Zr(Cp)2Cl2 (60.0 mg, 0.2 mmol, 1.0 eq.), LiCl (8.4 mg, 0.2 mmol, 1.0 eq.) and Manganese powder (22.0 mg, 0.4 mmol, 2.0 eq.). Then (E)-(3-bromoprop-1-en-1-yl)trimethylsilane (77 mg, 0.4 mmol, 2.0 eq.) and aldehyde (0.2 mmol, 1.0 eq.) were added in succession. The resulting suspension was left stirred at room temperature overnight. After the full consumption of aldehyde, the reaction mixture was diluted with undried EA and the resulting suspension was filtered over a pad of silica gel using EA as eluent. Volatiles were evaporated in vacuo. The residue was purified by chromatography to afford the product.
(3R,4S)-1-phenyl-4-(trimethylsilyl)hex-5-en-3-ol (2a)
1H NMR (400 MHz, CDCl3): δ 7.25–7.17 (m, 2H), 7.16–7.05 (m, 3H), 5.73 (dt, J = 17.1, 10.4 Hz, 1H), 4.98 (dd, J = 10.4, 1.6 Hz,1H), 4.88 (d, J = 17.1 Hz,1H), 3.75 (dd, J = 12.7, 5.2 Hz, 1H), 2.72–2.53 (m, 2H), 1.79–1.68 (m, 2H), 1.65 (dd, J = 10.6, 5.4 Hz, 1H), 1.54 (br, 1H), −0.02 (s, 9H); 13C NMR (101 MHz, CDCl3):δ 142.1, 135.7, 128.4, 128.3, 125.7, 115.2, 71.0, 42.6, 39.0, 32.3, −2.0; IR (neat) cm−1 \(\tilde{{\rm{v}}}\): 3456,3027,1247,899,839,749, 698;HRMS (EI(+), 70 eV): C15H24OSi [M-H]+: calcd. 247.1596, found.247.1513; [α]D 20 = +23.2 (c = 1.00, CH2Cl2); HPLC (Chiralcel OD-H column, hexanes:i-PrOH = 95:5, 0.5 mL/min, 210 nm), tminor = 9.8 min, tmajor = 12.1 min, 95% ee.
(3S,4R)-3-(trimethylsilyl)dec-1-en-4-ol (2b)
1H NMR (400 MHz, CDCl3): δ 5.79 (dt, J = 17.1, 10.4 Hz, 1H), 5.03 (dd, J = 10.2, 2.1 Hz, 1H), 4.92 (dd, J = 17.1, 2.0 Hz, 1H), 3.79 (dt, J = 7.5, 5.1 Hz, 1H), 1.67 (dd, J = 10.6, 5.3 Hz, 1H), 1.52–1.36 (m, 3H), 1.35–1.20 (m, 8H), 0.88 (t, J = 6.7 Hz, 3H), 0.04 (s, 9H); 13C NMR (101 MHz, CDCl3):δ 135.9, 115.0, 71.6, 42.6, 37.3, 31.8, 29.3, 25.8, 22.6, 14.1, −2.0. IR (neat) cm−1 \(\tilde{{\rm{v}}}\): 3696, 2962,2856, 1261, 1093, 1021, 800, 690; HRMS (EI(+), 70 eV): C13H28OSi [M-H]+: calcd. 227.1909, found 227.1826; [α]D 20 = +2.8 (c = 2.00, CH2Cl2); Enantiomeric excess was determined by HPLC analysis of the 3,5-nitrobenzoate derivative of the product (Chiralcel OD-H column, hexanes:i-PrOH = 98:2, 0.4 mL/min, 210 nm), tmajor = 13.9 min, tminor = 15.3 min, 96% ee.
(1R,2S)-1-cyclohexyl-2-(trimethylsilyl)but-3-en-1-ol (2c)
1H NMR (400 MHz, CDCl3): δ 5.82 (dt, J = 17.1, 10.5 Hz, 1H), 5.00 (dd, J = 10.2, 2.2 Hz, 1H), 4.89 (dd, J = 17.1, 2.0 Hz, 1H), 3.43 (dd, J = 7.2, 4.5 Hz, 1H),1.91–1.82 (m, 2H), 1.79–1.58 (m, 4H), 1.42–1.32 (m, 1H), 1.30–1.05 (m, 4H), 0.95 (dtd, J = 15.8, 12.3, 2.6 Hz, 2H), 0.05 (d, J = 11.7 Hz, 9H); 13C NMR (101 MHz, CDCl3):δ 135.6, 114.4,76.2, 42.4, 39.3, 29.3, 28.5, 26.4, 26.2, 25.9, −2.1; IR (neat) cm−1 \(\tilde{{\rm{v}}}\): 3492, 2926,2854, 1450, 1248, 1037, 895, 839, 693; HRMS (EI(+), 70 eV): C13H26OSi[M-OH]+: calcd. 209.1753, found: 209.1720;[α]D 20 = −6.1 (c = 0.20, CH2Cl2); Enantiomeric excess was determined by HPLC analysis of the 3,5-nitrobenzoate derivative of the product (Chiralcel OD-H column, hexanes:i-PrOH = 99:1, 0.3 mL/min, 210 nm), tmajor = 23.0 min, tminor = 25.2 min, 92% ee.
(3S,4R)-8-chloro-3-(trimethylsilyl)oct-1-en-4-ol (2d)
1H NMR (400 MHz, CDCl3): δ 5.78 (dt, J = 17.1, 10.4 Hz, 1H), 5.04 (dd, J = 10.2, 2.0 Hz, 1H), 4.93 (dd, J = 17.1, 1.8 Hz, 1H), 3.79 (d, J = 4.6 Hz, 1H), 3.53 (t, J = 6.7 Hz, 2H), 1.85–1.73 (m, 2H), 1.71–1.60 (m, 2H), 1.59–1.40 (m, 4H), 0.04 (s, 9H).13C NMR (101 MHz, CDCl3): δ 135.6, 115.3, 71.3, 45.0, 42.6, 36.4, 32.5, 23.2, −2.0. IR (neat) cm−1 \(\tilde{{\rm{v}}}\): 3463, 2959, 1625, 1449, 1412, 1256, 1089, 1014, 838, 796, 692; HRMS (EI(+), 70 eV): C11H23ClOSi [M-H]+: calcd. 233.1207, found 233.1124; [α]D 20 = +1.5 (c = 1.50, CH2Cl2); Enantiomeric excess was determined by HPLC analysis of the 3,5-nitrobenzoate derivative of the product (Chiralcel OD-H column, hexanes:i-PrOH = 95:5, 0.5 mL/min, 210 nm), tmajor = 14.7 min, tminor = 19.5 min, 97% ee.
(3S,4R)-3-(trimethylsilyl)octa-1,7-dien-4-ol (2e)
1H NMR (400 MHz, CDCl3): δ 5.93–5.69 (m, 2H), 5.16–4.82 (m, 4H), 3.82 (dd, J = 12.6, 5.3 Hz, 1H), 2.24–2.06 (m, 2H), 1.67 (dd, J = 10.6, 5.2 Hz, 1H), 1.64–1.49 (m, 3H), 0.04 (s, 9H); 13C NMR (101 MHz, CDCl3):δ 138.5, 135.8, 115.1, 114.7, 71.0, 42.7, 36.3, 30.3, −2.0; IR (neat) cm−1 \(\tilde{{\rm{v}}}\): 3359, 2922, 2852, 1734, 1658, 1279, 1253, 1087, 801, 700; HRMS (EI(+), 70 eV): C11H22OSi[M-H]+: calcd. 197.1440, found197.1368; [α]D 20 = +5.3 (c = 0.20, CH2Cl2); Enantiomeric excess was determined by HPLC analysis of the 3,5-nitrobenzoate derivative of the product (Chiralcel OD-H column, hexanes:i-PrOH = 98:2, 0.4 mL/min, 210 nm), tmajor = 17.0 min, tminor = 20.2 min, 97% ee.
(1R,2S)-1-(cyclohex-3-en-1-yl)-2-(trimethylsilyl)but-3-en-1-ol (2 f)
1H NMR (400 MHz, CDCl3): δ 5.84 (dt, J = 17.7, 10.4 Hz, 1H), 5.74–5.57 (m, 2H), 5.01 (d, J = 10.2 Hz, 1H), 4.91 (d, J = 17.1 Hz, 1H), 3.53 (d, J = 34.4 Hz, 1H), 2.21–1.79 (m, 5H), 1.77–1.38 (m, 4H), 0.05 (s, 9H).13C NMR (101 MHz, CDCl3):δ 135.4, 135.2, 127.0, 126.2, 126.1, 114.6, 75.6, 75.6, 39.6, 39.3, 38.6, 38.5,28.0, 27.4, 25.2, 25.1,24.5, −2.1, −2.2; IR (neat) cm−1 \(\tilde{{\rm{v}}}\): 2963, 1261, 1091, 1020, 866, 799, 700; HRMS (EI(+), 70 eV): C13H24OSi [M-H]+: calcd. 223.1596, found 223.1514; [α]D 20 = −1.2 (c = 0.40, CH2Cl2). Enantiomeric excess was determined by HPLC analysis of the 3,5-nitrobenzoate derivative of the product (Chiralcel OD-H column, hexanes:i-PrOH = 99.5:0.5, 0.4 mL/min, 210 nm), tmajor = 26.8 min, 27.8 min, tminor = 29.5 min, 31.3 min, 90% de.
(3R,4S)-1-((tert-butyldiphenylsilyl)oxy)-4-(trimethylsilyl)hex-5-en-3-ol (2g)
1H NMR (400 MHz, CDCl3): δ 7.71 (dd, J = 7.6, 1.4 Hz, 4H), 7.50–7.38 (m, 6H), 5.93 (dt, J = 17.1, 10.4 Hz, 1H), 5.01 (dd, J = 10.3, 2.3 Hz, 1H), 4.89 (ddd, J = 17.1, 2.2, 0.6 Hz, 1H), 4.17 (ddd, J = 9.3, 4.1, 2.9 Hz, 1H), 3.92–3.78 (m, 2H), 2.75 (s, 1H), 1.90–1.78 (m, 1H), 1.64 (dd, J = 10.6, 4.4 Hz, 1H), 1.62–1.55 (m, 1H), 1.08 (s, 9H), 0.09 (s, 9H).13C NMR (101 MHz, CDCl3): δ 136.2, 135.6, 135.5, 133.2, 133.1, 129.8, 129.7, 127.7, 114.0, 70.9, 63.2, 43.2, 39.0, 26.8, 19.0, −2.1; IR (neat) cm−1 \(\tilde{{\rm{v}}}\): 3522, 3071, 2955, 1624, 1469, 1426, 1390, 1248, 1081, 899, 837, 738, 702; HRMS (EI(+), 70 eV): C25H38O2Si2[M+H]+: calcd. 427.2410, found 427.2484; [α]D 20 = +0.8 (c = 1.00, CH2Cl2); HPLC (Chiralcel OD-H column, hexanes:i-PrOH = 99:1, 0.4 mL/min, 210 nm), tmajor = 9.5 min, tminor = 10.1 min, 97% ee.
2-((3R,4S)−3-hydroxy-4-(trimethylsilyl)hex-5-en-1-yl)isoindoline-1,3-dione (2h)
1H NMR (400 MHz, CDCl3): δ 7.85 (dd, J = 5.4, 3.1 Hz, 2H), 7.72 (dd, J = 5.4, 3.0 Hz, 2H), 5.82 (dt, J = 17.1, 10.4 Hz, 1H), 5.01 (dd, J = 10.2, 2.0 Hz, 1H), 4.89 (dd, J = 17.1, 1.8 Hz, 1H), 3.91–3.72 (m, 3H), 2.64 (d, J = 4.5 Hz, 1H), 1.84 (ddd, J = 15.0, 10.2, 5.2 Hz, 1H), 1.78–1.68 (m, 1H), 1.62 (dd, J = 10.6, 4.0 Hz, 1H), 0.00 (d, J = 6.1 Hz, 9H); 13C NMR (101 MHz, CDCl3): δ 174.6, 168.9, 135.5, 134.0, 132.0, 123.3, 115.0, 68.3, 42.5, 36.1, 35.1, −2.2; IR (neat) cm−1 \(\tilde{{\rm{v}}}\): 3503, 2957,1707, 1621,1251, 1049, 838, 794, 719; HRMS (EI(+), 70 eV): C17H23NO3Si[M-H]+: calcd. 316.1447, found 316.1364;[α]D 20 = −2.9 (c = 1.00, CH2Cl2). HPLC (Chiralcel OD-H column, hexanes:i-PrOH = 98:2, 0.4 mL/min, 210 nm), tmajor = 27.5 min, tminor = 29.8 min, 98% ee.
(2R,3S)-1-(2,2-dimethyl-1,3-dioxolan-4-yl)-3-(trimethylsilyl)pent-4-en-2-ol (2i)
Minor: 1H NMR (400 MHz, CDCl3): δ 5.92 (dt, J = 17.2, 10.4 Hz, 1H), 4.98 (dd, J = 10.2, 2.1 Hz, 1H), 4.86 (dd, J = 17.2, 2.2 Hz, 1H), 4.25 (ddd, J = 10.1, 8.4, 3.3 Hz, 1H), 4.07 (dd, J = 8.0, 6.0 Hz, 2H), 3.54 (t, J = 7.7 Hz, 1H), 3.05 (br, 1H), 1.75 (dt, J = 14.2, 9.8 Hz, 1H), 1.61–1.51 (m, 2H), 1.41 (s, 3H), 1.36 (s, 3H), 0.05 (s, 9H); 13C NMR (101 MHz, CDCl3): δ 135.8, 114.0, 109.4, 76.1, 71.1, 69.8, 43.3, 40.6, 26.9, 25.8, −2.2. Major: 1H NMR (400 MHz, CDCl3): δ 5.80 (dt, J = 17.1, 10.4 Hz, 1H), 5.04 (dd, J = 10.3, 2.0 Hz, 1H), 4.94 (dd, J = 17.1, 2.0 Hz, 1H), 4.39–4.28 (m, 1H), 4.08 (dd, J = 8.1, 6.1 Hz, 2H), 3.58 (t, J = 7.8 Hz, 1H), 2.14 (br, 1H), 1.73–1.69 (m, 1H), 1.69–1.60 (m, 2H), 1.41 (s, 3H), 1.36 (s, 3H), 0.05 (s, 9H); 13C NMR (101 MHz, CDCl3): δ 136.0, 115.3, 108.6, 73.8, 69.5, 68.3, 43.8, 40.5, 26.9, 25.6, −1.9. IR (neat) cm−1 \(\tilde{{\rm{v}}}\): 3500, 3074, 2934,1625, 1457, 1375, 1247, 1061, 838, 693;HRMS (EI(+), 70 eV): C13H26O3Si[M-H]+: calcd. 257.1651, found 257.1569; [α]D 20 = +9.3 (c = 2.60, CH2Cl2). Enantiomeric excess was determined by HPLC analysis of the 3,5-nitrobenzoate derivative of the product (Chiralcel OD-H column, hexanes:i-PrOH = 80:20, 1 mL/min, 210 nm), tminor = 4.8 min, 5.9 min, tmajor = 5.2 min, 10.7 min, >97% de.
(3S,4R,6S)-6,10-dimethyl-3-(trimethylsilyl)undeca-1,9-dien-4-ol (2j)
1H NMR (400 MHz, CDCl3): δ 5.78 (dt, J = 17.1, 10.5 Hz, 1H), 5.10 (t, J = 7.1 Hz, 1H), 5.04 (dd, J = 10.3, 2.0 Hz, 1H), 4.93 (dd, J = 17.0, 1.5 Hz, 1H), 3.95–3.85 (m, 1H), 2.04–1.93 (m, 2H), 1.72–1.57 (m, 8H), 1.50–1.46 (m, 1H), 1.35–1.27 (m, 2H), 1.23–1.13 (m, 2H), 0.90 (d, J = 6.6 Hz, 3H), 0.04 (s, 9H); 13C NMR (101 MHz, CDCl3):δ 136.2,131.2, 124.8, 115.1, 69.0, 44.8, 43.7, 37.9, 29.0, 25.7, 25.4, 19.0, 17.6, −1.9; IR (neat) cm−1 \(\tilde{{\rm{v}}}\): 2963,1261, 1093, 1020, 866, 799, 700; HRMS (EI(+), 70 eV): C16H32OSi [M-H]+: calcd. 267.2222, found 267.2139; [α]D 20 = +9.4 (c = 0.40, CH2Cl2); Enantiomeric excess was determined by HPLC analysis of the 3,5-nitrobenzoate derivative of the product (Chiralcel OD-H column, hexanes:i-PrOH = 99:1, 0.3 mL/min, 210 nm), tmajor = 19.2 min, tminor = 22.0 min, 97% de.
(3S,4R)-6,8,8-trimethyl-3-(trimethylsilyl)non-1-en-4-ol (2k)
1H NMR (400 MHz, CDCl3): δ 5.87–5.72 (m, 1H), 5.04 (d, J = 10.2 Hz, 1H), 4.93 (d, J = 17.0 Hz, 1H), 3.87 (m, 1H), 1.78–1.56 (m, 2H), 1.55–1.34 (m, 2H), 1.33–1.15 (m, 2H), 1.06 (m, 1H), 0.95–0.91 (m, 3H), 0.90–0.84 (m, 9H), 0.04(s, 9H); 13C NMR (101 MHz, CDCl3):δ 136.2, 135.6, 115.1,115.0, 69.6, 69.2, 52.0, 51.2, 47.3, 47.2, 43.7, 42.2, 31.2, 31.0, 30.1, 30.0, 26.0, 25.8, 23.4, 21.9, −1.8, −2.0; IR (neat) cm−1 \(\tilde{{\rm{v}}}\): 2954, 1626, 1471, 1366, 1249, 1023, 898, 839, 691; HRMS (EI(+), 70 eV): C15H32OSi[M-OH]+: calcd. 239.2222, found 239.2190; [α]D 20 = +10.6 (c = 3.50, CH2Cl2); Enantiomeric excess was determined by HPLC analysis of the 3,5-nitrobenzoate derivative of the product (Chiralcel OD-H column, hexanes:i-PrOH = 99.5:0.5, 0.4 mL/min, 230 nm), tmajor = 14.9 min, tminor = 18.0 min, 95% de.
(1R,2S)-1-phenyl-2-(trimethylsilyl)but-3-en-1-ol (2l)
1H NMR (400 MHz, CDCl3) δ 7.36–7.24 (m, 5H), 5.86 (dt, J = 17.1, 10.3 Hz, 1H), 5.11 (dd, J = 10.3, 1.9 Hz, 1H), 5.03 (ddd, J = 17.1, 1.8, 0.7 Hz, 1H), 4.80 (d, J = 8.4 Hz, 1H), 2.22 (d, J = 1.6 Hz 1H), 2.08 (dd, J = 10.2, 8.6 Hz, 1H), −0.20 (s, 9H); 13C NMR (101 MHz, CDCl3):δ 143.6, 136.6, 128.4, 127.8, 126.9, 116.0,74.5, 45.6, −2.4; IR (neat) cm−1 \(\tilde{{\rm{v}}}\): 3458, 2952, 1626, 1248, 907,764, 699;HRMS (EI(+), 70 eV): C13H20OSi[M-OH]+: calcd. 203.1283., found 203.1249; [α]D 20 = −16.3 (c = 1.00, CH2Cl2); HPLC (Chiralcel OD-H column,hexanes:i-PrOH = 95:5, 1 mL/min, 210 nm), tmajor = 7.0 min, tminor = 12.1 min, 93% ee. The reported value[7] for the (1S,2R)-enantiomer (95% ee) is [α]D 25 = +47.0 (c = 1.0; CHCl3).
(1R,2S)-1-(4-bromophenyl)-2-(trimethylsilyl)but-3-en-1-ol (2m)
1H NMR (400 MHz, CDCl3): δ 7.45 (d, J = 8.2 Hz, 2H), 7.21 (d, J = 8.4 Hz, 2H), 5.82 (dt, J = 17.1, 10.3 Hz, 1H), 5.09 (dd, J = 10.3, 1.7 Hz, 1H), 4.98 (ddd, J = 17.1, 1.7 Hz, 0.8Hz,1H), 4.77 (d, J = 8.0 Hz, 1H), 2.23 (br, 1H), 1.99 (dd, J = 10.3, 8.1 Hz, 1H), −0.16 (s, 9H); 13C NMR (101 MHz, CDCl3):δ 142.8, 135.9, 131.4, 128.5, 121.4, 116.3,73.8, 45.5, −2.4; IR (neat) cm−1 \(\tilde{{\rm{v}}}\): 3435, 2957,1626, 1486, 1409, 1250, 1088,1009, 909, 837, 693.08; HRMS (EI(+), 70 eV): C13H19BrOSi [M-OH]+: calcd. 281.0389, found 281.0356; [α]D 20 = −19.6 (c = 1.10, CH2Cl2); HPLC (Chiralcel OD-H column, hexanes:i-PrOH = 95;5, 1 mL/min, 210 nm), tminor = 6.2 min, tmajor = 6.9 min, 92% ee. The reported value[7] for the (1S,2R)-enantiomer (94% ee) is [α]D 25 = +10.3 (c = 1.0; CHCl3).
(1R,2S)-1-(2-fluorophenyl)−2-(trimethylsilyl)but-3-en-1-ol (2n)
1H NMR (400 MHz, CDCl3): δ 7.45–7.39 (m, 1H), 7.25–7.20 (m, 1H), 7.16–7.10 (m, 1H), 7.04–6.97 (m, 1H), 5.85 (dt, J = 17.2, 10.4 Hz, 1H), 5.17 (dd, J = 7.9, 2.9 Hz, 1H), 5.08 (dd, J = 10.3, 1.9 Hz, 1H), 4.98 (dd, J = 17.0, 1.8 Hz, 1H), 2.18–2.10 (m, 2H), −0.12 (s, 9H).13C NMR (101 MHz, CDCl3): δ159.8 (d, J C-F = 246.4 Hz), 136.0, 130.9 (d, J C-F = 13.0 Hz), 128.9 (d, J C-F = 8.4 Hz), 128.4 (d, J C-F = 4.5 Hz), 124.0 (d, J C-F = 3.4 Hz), 116.0, 115.3 (d, J C-F = 22.2 Hz), 68.0, 44.2, −2.5. IR (neat) cm−1 \(\tilde{{\rm{v}}}\): 3359, 2925,1626, 1487, 1456,1249, 1055, 911, 841, 758, 693;HRMS (EI(+), 70 eV): C13H19FOSi [M-OH]+: calcd:221.1189, found:221.1157; [α]D 20 = −17.5 (c = 1.00, CH2Cl2). HPLC (Chiralcel OD-H column, hexanes:i-PrOH = 95:5, 0.4 mL/min, 210 nm), tmajor = 13.4 min, tminor = 14.2 min, 90% ee.
(1R,2S)-1-(3-fluorophenyl)-2-(trimethylsilyl)but-3-en-1-ol (2o)
1H NMR (400 MHz, CDCl3): δ 7.33–7.24 (m, 1H), 7.13–7.03 (m, 2H), 6.96 (td, J = 8.4, 1.8 Hz, 1H), 5.82 (dt, J = 17.1, 10.3 Hz, 1H), 5.10 (dd, J = 10.3, 1.7 Hz, 1H), 4.99 (dd, J = 17.1, 1.1 Hz, 1H), 4.81 (d, J = 7.9 Hz, 1H), 2.26 (br, 1H), 2.01 (dd, J = 10.2, 8.0 Hz, 1H), −0.15 (s, 9H). 13C NMR (101 MHz, CDCl3):δ 162.9 (d, J C-F = 247.4 Hz), 146.4 (d, J = 6.7 Hz), 135.9, 129.8 (d, J C-F = 8.2 Hz), 122.4 (d, J C-F = 2.8 Hz), 116.3, 114.6 (d, J C-F = 21.2 Hz), 113.6 (d,J C-F = 22.2 Hz), 73.9, 45.5, −2.4; IR (neat) cm−1 \(\tilde{{\rm{v}}}\): 2956, 2927, 1669, 1592, 1451,1257, 1090, 1029, 841, 801, 696; HRMS (EI(+), 70 eV): C13H19FOSi[M-H]+: calcd. 237.1189, found 237.1105; [α]D 20 = −15.2 (c = 0.70, CH2Cl2); HPLC (Chiralcel OD-H column, hexanes:i-PrOH = 95:5, 1.0 mL/min, 210 nm), tmajor = 6.4 min, tminor = 12.4 min, 92% ee.
(1R,2S)−1-(4-chlorophenyl)-2-(trimethylsilyl)but-3-en-1-ol (2p)
1H NMR (400 MHz, CDCl3): δ 7.34–7.22 (m, 4H), 5.83 (dt, J = 17.1, 10.2 Hz, 1H), 5.10 (d, J = 10.2 Hz, 1H), 4.99 (d, J = 17.2 Hz, 1H), 4.79 (d, J = 8.0 Hz, 1H), 2.22 (br, 1H), 2.00 (dd, J = 10.1, 8.3 Hz, 1H), −0.16 (s, 9H).13C NMR (101 MHz, CDCl3):δ 142.2, 136.0, 133.3, 128.5, 128.2, 116.3,73.8, 45.6, −2.4; IR (neat) cm−1 \(\tilde{{\rm{v}}}\): 3714, 3075, 2953,1625, 1491, 1410, 1248, 1010, 911, 836, 694; HRMS (EI(+), 70 eV): C13H19ClOSi [M-OH]+calcd:237.0894, found:237.0861;[α]D 20 = −12.3 (c = 2.00, CH2Cl2); HPLC (Chiralcel OD-H column, hexanes:i-PrOH = 95:5, 0.4 mL/min, 210 nm), tminor = 14.7 min, tmajor = 15.5 min, 91% ee.The reported value[7] for the (1S,2R)-enantiomer (59% ee) is [α]D 25 = +4.8 (c = 1.0; CHCl3).
Materials
NMR spectra were recorded at room temperature on the following spectrometers: Agilent (400 MHz) and VARIAN (400 MHz). Chemical shifts are given in ppm and coupling constants in Hz. 1H spectra were calibrated in relation to the reference measurement of TMS (0.00 ppm). 13C spectra were calibrated in relation to deuterated solvents, namely CDCl3 (77.16 ppm). The following abbreviations were used for 1H NMR spectra to indicate the signal multiplicity: s (singlet), d (doublet), t (triplet), q (quartet) and m (multiplet) as well as combinations of them. When combinations of multiplicities are given the first character noted refers to the largest coupling constant. High performance liquid chromatography (HPLC) was carried out with Agilent 1260 Infinity on a UV spectrophotometric detector (210 nm, Agilent). For ESI+-spectra and EI-HR (GC-TOF) spectrometer was applied. Infrared Spectroscopy (IR) was processed on an FT-IR spectrometer named Nicolet 380. The method is denoted in brackets. For the most significant bands the wave number \(\tilde{{\rm{v}}}\): (cm−1) is given.
Chemicals were purchased from commercial suppliers. Unless stated otherwise, all the substrates and solvents were purified and dried according to standard methods prior to use. Reactions requiring inert conditions were carried out in glove box.
References
Carreira, E. M. & Kvaerno, L. Classics In Stereoselective Synthesis. Wiley-VCH: Weinheim, Germany, 153–185 (2009).
Yus, M., González-Gómez, J. C. & Foubelo, F. Catalytic enantioselective allylation of carbonyl compounds and imines. Chem. Rev. 111, 7774–7854 (2011).
Hall, D. G. Lewis and brønsted acid catalyzed allylboration of carbonyl compounds: from discovery to mechanism and applications. Synlett. 2007, 1644–1655 (2007).
Marek, I. & Sklute, G. Creation of quaternary stereocenters in carbonyl allylation reactions. Chem. Commun. 1683–1691 (2007).
Denmark, S. E. & Fu, J. Catalytic enantioselective addition of allylic organometallic reagents to aldehydes and ketones. Chem. Rev. 103, 2763–2794 (2003).
Kennedy, J. W. J. & Hall, D. G. Recent advances in the activation of boron and silicon reagents for stereocontrolled allylation reactions. Angew. Chem. Int. Ed. 42, 4732–4739 (2003).
Yamamoto, Y. & Asao, N. Selective reactions using allylic metals. Chem. Rev. 93, 2207–2293 (1993).
Han, S. B., Kim, I. S. & Krische, M. J. Enantioselective iridium-catalyzed carbonyl allylation from the alcohol oxidation level via transfer hydrogenation: minimizing pre-activation for synthetic efficiency. Chem. Commun. 7278–7287 (2009).
Bower, J. F., Kim, I. S., Patman, R. L. & Krische, M. J. Catalytic carbonyl addition through transfer hydrogenation: a departure from preformed organometallic reagents. Angew. Chem. Int. Ed. 48, 34–46 (2009).
Shibahara, F. & Krische, M. J. Formation of C–C bonds via ruthenium-catalyzed transfer hydrogenation: carbonyl addition from the alcohol or aldehyde oxidation level. Chem. Lett. 37, 1102–1107 (2008).
Shen, Z.-L., Wang, S.-Y., Chok, Y.-K., Xu, Y.-H. & Loh, T.-P. Organoindium reagents: the preparation and application in organic. Chem. Rev. 113, 271–401 (2013).
Fürstner, A. Carbon−carbon bond formations involving organochromium(III) reagents. Chem. Rev. 99, 991–1045 (1999).
Avalos, M., Babiano, R., Cintas, P., Jiménez, J. L. & Palacios, J. C. Synthetic variations based on low-valent chromium: new developments. Chem. Soc. Rev. 28, 169–177 (1999).
Okude, Y., Hirano, S., Hiyama, T. & Nozaki, H. Grignard-type carbonyl addition of allyl halides by means of chromous salt. A chemospecific synthesis of homoallyl alcohols. J. Am. Chem. Soc. 99, 3179–3181 (1977).
Takai, K., Kimura, K., Kuroda, T., Hiyama, T. & Nozaki, H. Selective grignard-type carbonyl addition of alkenyl halides mediated by chromium(II) chloride. Tetrahedron Lett. 24, 5281–5284 (1983).
Jin, H., Uenishi, J.-I., Christ, W. J. & Kishi, Y. Catalytic effect of nickel(II) chloride and palladium(II) acetate on chromium(II)-mediated coupling reaction of iodo olefins with aldehydes. J. Am. Chem. Soc. 108, 5644–5646 (1986).
Takai, K. et al. Reactions of alkenylchromium reagents prepared from alkenyl trifluoromethanesulfonates (triflates) with chromium(II) chloride under nickel catalysis. J. Am. Chem. Soc. 108, 6048–6050 (1986).
Bandini, M., Cozzi, P. G., Melchiorre, P. & Umani-Ronchi, A. The First Catalytic Enantioselective Nozaki–Hiyama Reaction. Angew. Chem. Int. Ed. 38, 3357–3359 (1999).
Tian, Q. & Zhang, G. Recent advances in the asymmetric Nozaki–Hiyama–Kishi reaction. Synthesis 48, 4038–4049 (2016).
Chen, W., Yang, Q., Zhou, T., Tian, Q. & Zhang, G. Enantioselective synthesis of α-exo-methylene γ‑butyrolactones via chromium catalysis. Org. Lett. 17, 5236–5239 (2015).
Tian, Q., Bai, J., Chen, B. & Zhang, G. Chromium-catalyzed asymmetric dearomatization addition reactions of halomethyl heteroarenes. Org. Lett. 18, 1828–1831 (2016).
Xiong, Y. & Zhang, G. Enantioselective synthesis of quaternary stereocenters via chromium catalysis. Org. Lett. 18, 5094–5097 (2016).
Chabaud, L., James, P. & Landais, Y. Allylsilanes in organic synthesis − recent developments. Eur. J. Org. Chem. 3173–3199 (2004).
Fleming, I., Barbero, A. & Walter, D. Stereochemical control in organic synthesis using silicon-containing compounds. Chem. Rev. 97, 2063–2192 (1997).
Masse, C. E. & Panek, J. S. Diastereoselective reactions of chiral allyl and allenyl silanes with activated C = X π-Bonds. Chem. Rev. 95, 1293–1316 (1995).
Giuffredi, G. T., Purser, S., Sawicki, M., Thompson, A. L. & Gouverneur, V. Asymmetric de novo synthesis of fluorinated D-glucitol and D-mannitol analogues. Tetrahedron: Asymmetry 20, 910–920 (2009).
Va, P. & Roush, W. R. Total synthesis of amphidinolide E. J. Am. Chem. Soc. 128, 15960–15961 (2006).
Su, Q. & Panek, J. S. Total synthesis of (−)-apicularen A. J. Am. Chem. Soc. 126, 2425–2430 (2004).
Greedy, B., Paris, J. M., Vidal, T. & Gouverneur, V. Regio- and enantioselective synthesis of allylic fluorides by electrophilic fluorodesilylation of allyl silanes. Angew. Chem. Int. Ed. 42, 3291–3294 (2003).
Huang, H. & Panek, J. S. Stereoselective synthesis of functonalized dihydropyrans via a formal [4 + 2]-annulation of chiral crotylsilanes. J. Am. Chem. Soc. 122, 9836–9837 (2000).
Panek, J. S. & Yang, M. Diastereoselective additions of chiral (E)-crotylsilanes to α-alkoxy and β-alkoxy aldehydes. A one-step, silicon-directed tetrahydrofuran synthesis. J. Am. Chem. Soc. 113, 9868–9870 (1991).
Panek, J. S. & Yang, M. Diastereofacial selectivity with optically active.alpha.-substituted.beta.-silyl-(E)-hexenoates. Enantioselective construction of homoallylic ethers via reaction with aryl acetals. J. Am. Chem. Soc. 113, 6594–6600 (1991).
Han, S. B., Gao, X. & Krische, M. J. Iridium-catalyzed anti-diastereo- and enantioselective carbonyl (trimethylsilyl)allylation from the alcohol or aldehyde oxidation level. J. Am. Chem. Soc. 132, 9153–9156 (2010).
Barrio, P., Rodriguez, E., Saito, K., Fustero, S. & Akiyama, T. γ-Silylboronates in the chiral brønsted acid-catalysed allylboration of aldehydes. Chem. Commun. 51, 5246–5249 (2015).
For an alternative approach that makes use of chiral organilithium reagents, see: Binanzer, M., Fang, G. Y. & Aggarwal, V. K. Asymmetric synthesis of allylsilanes by the borylation of lithiated Carbamates: formal total synthesis of (–)-decarestrictine D. Angew. Chem. Int. Ed. 49, 4264–4268 (2010).
Inoue, M., Suzuki, T. & Nakada, M. A new asymmetric tridentate carbazole ligand: its preparation and application to Nozaki–Hiyama allylation. Synlett. 4, 570–572 (2003).
Fürstner, A. & Wuchrer, M. Concise approach to the “higher sugar” core of the nucleoside antibiotic hikizimycin. Chem.-Eur. J. 12, 76 (2006).
Fürstner, A. & Shi, N. A multicomponent redox system accounts for the first Nozaki−Hiyama−Kishi reactions catalytic in chromium. J. Am. Chem. Soc. 118, 2533–2534 (1996).
Fürstner, A. & Shi, N. Nozaki−Hiyama−Kishi reactions catalytic in chromium. J. Am. Chem. Soc. 118, 12349–12357 (1996).
Namba, K. & Kishi, Y. New catalytic cycle for couplings of aldehydes with organochromium reagents. Org. Lett. 6, 5031–5033 (2004).
Bégué, J.-P. & Bonnet-Delpon, D. Bioorganic and Medicinal Chemistry of Fluorine. John Wiley & Sons, Inc., Hoboken, NJ (2008).
Purser, S., Moore, P. R., Swallow, S. & Gouverneur, V. Fluorine in medicinal chemistry. Chem. Soc. Rev. 37, 320–330 (2008).
Müller, K., Faeh, C. & Diederich, F. Fluorine in pharmaceuticals: looking beyond intuition. Science 317, 1881–1886 (2007).
Ma, J.-A. & Cahard, D. Update 1 of: asymmetric fluorination, trifluoromethylation, and perfluoroalkylation reactions. Chem. Rev. 108, PR1–PR43 (2008).
Cahard, D., Xu, X., Couve-Bonnaire, S. & Pannecoucke, X. Fluorine & chirality: how to create a nonracemic stereogenic carbon–fluorine centre? Chem. Soc. Rev. 39, 558–568 (2010).
Nie, J., Guo, H.-C., Cahard, D. & Ma, J.-A. Asymmetric construction of stereogenic carbon centers featuring a trifluoromethyl group from prochiral trifluoromethylated substrates. Chem. Rev. 111, 455–529 (2011).
Gao, X., Zhang, Y. J. & Krische, M. J. Iridium-catalyzed anti-diastereo- and enantioselective carbonyl(α-trifluoromethyl)allylation from the alcohol or aldehyde oxidation level. Angew. Chem. Int. Ed. 50, 4173–4175 (2011).
Chen, Q., Qiu, X.-L. & Qing, F.-L. Indium-mediated diastereoselective allylation of D- and L-glyceraldimines with 4-bromo-1,1,1-trifluoro-2-butene: highly stereoselective synthesis of 4,4,4-trifluoroisoleucines and 4,4,4-trifluorovaline. J. Org. Chem. 71, 3762–3767 (2006).
Loh, T.-P. & Li, X.-R. A highly stereoselective synthesis of β-trifluoromethylated homoallylic alcohols in water. Angew. Chem. Int. Ed. 36, 980–982 (1997).
Loh, T.-P. & Li, X.-R. Tin- and indium-mediated allylation reactions in water: highly stereoselective synthesis of β-trifluoromethylated homoallylic alcohols. Eur. J. Org. Chem. 1893–1899 (1999).
Loh, T.-P. & Li, X.-R. A novel method for stereoseleetive synthesis of β-trifluoromethylated homoallylic alcohols in water. Tetrahedron Lett. 38, 869–872 (1997).
Sulfur Compounds: Advances in Research and Application. Acton, Q., Ed., Scholarly Editions: Atlanta, GA, (2012).
Ilardi, E. A., Vitaku, E. & Njardarson, J. T. Data-mining for sulfur and fluorine: an evaluation of pharmaceuticals to reveal opportunities for drug design and Discovery. J. Med. Chem. 57, 2832–2842 (2014).
Smith, B. R., Eastman, C. M. & Njardarson, J. T. Beyond C, H, O, and N! analysis of the elemental composition of US FDA approved drug architectures. J. Med. Chem. 57, 9764–9773 (2014).
Mishra, A., Ma, C. Q. & Bäuerle, P. Functional oligothiophenes: molecular design for multidimensional nanoarchitectures and their applications. Chem. Rev. 109, 1141–1276 (2009).
Bürckstümmer, H., Weissenstein, A., Bialas, D. & Würthner, F. Synthesis and characterization of optical and redox properties of bithiophene-functionalized diketopyrrolopyrrole chromophores. J. Org. Chem. 76, 2426–2432 (2011).
Takimiya, K., Osaka, I., Mori, T. & Nakano, M. Organic semiconductors based on [1]benzothieno[3,2-b][1]benzothiophene substructure. Acc. Chem. Res. 47, 1493–1502 (2014).
Block, E. The Organosulfur chemistry of the genus allium–implications for the organic chemistry of sulfur. Angew. Chem. Int. Ed. 31, 1135–1178 (1992).
Loughlin, K. R., Generali, J., Eds. Prescription Drugs: Alternative Uses, Alternative Cures: Over 1,500 New Uses for FDA-Approved Drugs. The Philip Leif Group, Inc.: New York, NY, (2006).
Damani, L. A., Ed. Sulphur Containing Drugs and Related Organic Compounds. Ellis Horwood Limited: Chichester, UK (1989).
Beletskaya, I. P. & Ananikov, V. P. Transition-metal-catalyzed C−S, C−Se, and C−Te bond formation via cross-coupling and atom-economic addition reactions. Chem. Rev. 111, 1596–1636 (2011).
Kondo, T. & Mitsudo, T. A. Metal-catalyzed carbon−sulfur bond formation. Chem. Rev. 100, 3205–3220 (2000).
Acknowledgements
We are grateful to NSFC-21421091, XDB20000000, the “Thousand Plan” Youth program, State Key Laboratory of Organometallic Chemistry, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences.
Author information
Authors and Affiliations
Contributions
Guo, R. and Yang, Q. contributed equally. Zhang, G. conceived, designed and directed the project. Guo, R., Yang, Q. and Tian, Q. conducted the experiments. Zhang, G. wrote the manuscript with the assistance of Guo, R. and Tian, Q. All authors participated in data analysis and discussions.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Guo, R., Yang, Q., Tian, Q. et al. Barbier-type anti-Diastereo- and Enantioselective Synthesis of β-Trimethylsilyl, Fluorinated Methyl, Phenylthio Homoallylic Alcohols. Sci Rep 7, 4873 (2017). https://doi.org/10.1038/s41598-017-04986-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-04986-x
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
