
Abstract
Based on our initial genome-wide association study (GWAS) on esophageal squamous cell carcinoma (ESCC) in Han Chinese, we conducted a follow-up study to examine the single nucleotide polymorphisms (SNPs) associated with family history (FH) of upper gastrointestinal cancer (UGI) cancer in cases with ESCC. We evaluated the association between SNPs and FH of UGI cancer among ESCC cases in a stage-1 case-only analysis of the National Cancer Institute (NCI, 541 cases with FH and 1399 without FH) and Henan GWAS (493 cases with FH and 869 without FH) data (discovery phase). The top SNPs (or their surrogates) from discovery were advanced to a stage-2 evaluation in additional Henan subjects (2801 cases with FH and 3136 without FH, replication phase). A total of 19 SNPs were associated with FH of UGI cancer in ESCC cases with P < 10−5 in the stage-1 meta-analysis of NCI and Henan GWAS data. In stage-2, the association for rs79747906 (located at 18p11.31, P = 5.79 × 10−6 in discovery) was replicated (P = 0.006), with a pooled-OR of 1.59 (95%CI: 1.11-2.28). We identified potential genetic variants associated with FH of UGI cancer. Our findings may provide important insights into new low-penetrance susceptibility regions involved in the susceptibility of families with multiple UGI cancer cases.
Similar content being viewed by others

Introduction
Esophageal cancer (EC) represents the sixth most frequent cause of cancer-related deaths worldwide1, 2. Over half of all EC-related deaths occur in China where esophageal squamous cell carcinoma (ESCC) is the predominant histologic subtype, particularly in high-risk populations3, 4. Both genetic and environmental risk factors are believed to play roles in the development of ESCC. In Western populations, smoking and heavy alcohol intake are established dominant risk factors for ESCC5. However, smoking and alcohol are not major contributing factors in the high-risk populations in north central China6, 7, where causes underlying the carcinogenesis of ESCC remain poorly defined.
We and others have conducted several genome-wide association studies (GWAS) and identified a number of genetic loci linked to risk of ESCC8,9,10,11,12,13,14,15, but these loci account for only a small fraction of the genetic susceptibility for ESCC risk. Previous studies in the high-risk populations in north central China have demonstrated consistent associations between family history (FH) of cancer, particularly that of upper gastrointestinal (UGI) cancer, and risk of ESCC16,17,18. This strong tendency toward familial aggregation suggests the potential usefulness of looking at genetic predisposition for UGI cancer by examining family-history-related genetic loci. Based on our initial GWAS for ESCC in high-risk populations of Han Chinese ethnicity9, 10, we conducted additional analyses of the association of single nucleotide polymorphisms (SNPs) with FH of UGI cancer in ESCC cases. First, SNPs consistently associated with FH of UGI cancer in a case-only analysis of two GWAS were identified (discovery stage)9, 10. Second, top SNPs associated with FH of UGI cancer in the GWAS (or their surrogate SNPs) were evaluated in a second stage replication in additional cases.
Results
Meta-analysis of GWAS for the discovery stage
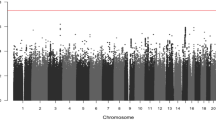
A total of 19 SNPs were associated with FH of UGI cancer in ESCC cases with a P < 0.05 in both the National Cancer Institute (NCI) and Henan GWAS data in the discovery stage (Supplementary Table 1) and a combined P < 10−5 in the meta-analysis (Tables 1 and 2). None reached genome-wide significance. The smallest P value was observed for rs140792366 (P = 7.65 × 10−7), which is in the gene GRIK4 (located at 11q23.3).
SNP Replication analysis in Henan Sample
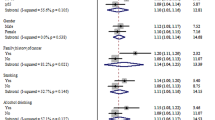
Of these 19 SNPs, rs57921607 and its surrogate failed design and were dropped from further consideration. Ten original and 8 surrogate SNPs were genotyped in the replication stage (Table 2 and Supplementary Table 1). Among the 18 SNPs tested in replication, we did not see an alternative (minor) allele for rs141703242 (surrogate of rs186503151) or rs184911713 (surrogate of rs187481103). Further, we found very few minor alleles for rs140792366. Association analyses were, therefore, limited to the remaining 15 SNPs. The candidate SNP with strongest associations with FH in the discovery (rs140792366) had much lower MAF in the stage 2 and was not replicated. Rs79747906 in DLGAP1 (located at 18p11.31, odds ratio [OR] = 1.91 and P = 5.79 × 10−6 in the meta-analysis of discovery set) had the smallest p-value in the replication analysis with FH defined for relatives in three generations and was significant at a nominal significance level (OR = 1.23, P = 0.006) (Tables 1 and 2). Based on the random-effect model (P for heterogeneity = 0.02), the pooled OR (95% CI) for the discovery and replication set was 1.59 (1.11–2.28) (P = 0.01). The association for rs79747906 was also evident in the secondary model of replication analysis, for first degree relatives only (OR = 1.24, P = 0.008) (Table 1 and Supplementary Table 2). The pooled OR (95% CI) for the second replication model and the discovery set was 1.59 (1.12–2.26) (P = 0.009).
We also found a suggestive association for rs12461816 (located at 19p13.12) and FH in the secondary replication model (OR = 1.39, P = 8.91 × 10−6 in the discovery set, and OR = 1.09, P = 0.08 in replication; the pooled OR = 1.27, P = 0.03) (Table 2 and Supplementary Table 2). However, the primary analysis did not support a significant association for this SNP (OR = 1.04 and P = 0.37) (Table 2).
In silico and cis-eQTL functional annotation
We annotated the two SNPs (rs79747906 and rs12461816) that showed suggestive association with FH in the replication. The functional analysis revealed that rs79747906 C allele may have a weak CTCF binding function. SNP rs12461816 has a potential weak polycomb-repressed state of transcription in stomach smooth muscle and the T allele of rs12461816 abolishes a methylated CpG in normal esophagus and gastric tissues (Table 3 and Supplementary Figures 1 and 2).
In the expression quantitative trait loci (eQTL) analyses, rs12461816 T allele was significantly associated with increased cis expression of AC004791.2 in normal esophageal muscularis (P = 6.00 × 10−7) and mucosa (P = 7.30 × 10−4), normal stomach mucosa (P = 9.50 × 10−7), and whole blood (P = 2.5 × 10−5). It was also associated with increased expression of CYP4F24P (P = 4.50 × 10−4) in the gastroesophageal junction, decreased expression of CYP4F11 (P = 8.50 × 10−4) in esophageal mucosa, and showed nominal associations with several other genes (Table 3 and Supplementary Table 4). For rs79747906, the C allele was suggestively associated with altered expression of several genes such as LPIN2, although none of these associations were significant after adjustment for multiple comparisons (Supplementary Tables 3 and 4).
Discussion
We conducted analyses based on our initial GWAS for ESCC, examining the SNPs associated with FH of UGI cancer in Han Chinese. We found 19 SNPs consistently associated with FH of UGI cancer in two GWAS combined. Of these, rs79747906 (18p11.31) was replicated in analyses of additional cases. To our knowledge, no previous studies have reported this SNP as associated with FH of UGI cancer or UGI cancer risk.
Rs79747906 is located in the intergenic region close to DLGAP1, SNPs of which have been associated with obsessive-compulsive disorder19. Based on Roadmap Epigenomics data rs79747906 does not appear to map to a regulatory region in normal UGI tissues or blood, but the C allele, which was positively associated with FH, overlapped with a weak CTCF binding function in ENCODE cells including Human Esophageal Epithelial Cells (HEEpiC) (Table 3 and SI Figure 1). However, we did not observe potential regulatory function for any SNPs in LD with rs79747906 (r2 ≥ 0.40, http://analysistools.nci.nih.gov/LDlink/) in normal esophageal tissue (data not shown). We also found that the rs79747906 C allele may alter several DNA binding motifs of homeobox transcriptional factors and proteins, including CDP, Nanog, PAX4, and SMAD2 (ENCODE and HaploRegv4) and scores high as a regulatory SNP for embryonic stem and progenitor cells of other tissues (RegulomeDB and HaploRegv4). Collectively, these findings suggest that the genetic region containing rs79747906 has the potential to change chromatin architecture in esophageal epithelia and/or in esophageal stem cells via protein binding. Our eQTL analysis of rs79747906 C allele showed only suggestive and tissue-specific evidence of a potential cis-eQTL effect on transcription, such as increased expression of LPIN2 (more distal protein coding gene) in blood and RP13-270P17.3 (a long intergenic non-coding RNA [LincRNA] gene) in stomach mucosa. It is worth noting that metabolic changes have been reported for UGI cancer patients previously20, 21. LPIN2 protein is known for its role in metabolism, with LPIN2 SNPs associated with metabolic traits22, which may be important for UGI cancer. Our study provides evidence for future examination of LPIN2 and RP13-270P17.3 and risk of UGI cancer.
The T allele of rs12461816 (19p13.12) was associated with FH and can abolish a methylated CpG in normal esophagus and stomach tissues. In keeping with a methylated and condensed DNA status, rs12461816 is also located to a potential polycomb-repressed DNA region in stomach smooth muscle, suggesting a possible transcriptional repression function for this region regulated by epigenetics. Notably, rs12461816 T allele was strongly associated with increased expression of AC004791.2. The expression of multiple other genes with important functions may be altered by rs12461816 variant too, such as CYP4F11, which encodes a cytochrome P450 enzyme (and is important for arachidonic acid or fatty acid metabolism).
UGI cancer is a major public health concern worldwide, among the most frequent causes of cancer-related deaths1, 2, highlighting the urgent need to elucidate the mechanisms underlying carcinogenesis. We identified potential genetic variants associated with FH of UGI cancer in ESCC cases. Our findings may provide important insights into new low-penetrance susceptibility regions not only for ESCC but possibly for UGI cancer overall, contributing to the understanding of ESCC and UGI cancer pathogenesis. Further investigation of the underlying genetic susceptibility may reveal new pathways predisposing to UGI cancer and potentially identify new therapeutic targets for drug discovery, aiding in the prevention and management of these common tumors.
Methods
Study population
Discovery stage sample
The discovery stage was based on the NCI GWAS and Henan GWAS. Participants for the NCI GWAS were drawn from the Shanxi UGI Cancer Genetics Project with participants residing in the western Taihang Mountain area; and the Nutrition Intervention Trials (NITs), with participants from the southern Taihang Mountain area. The Shanxi study was conducted between 1997 and 2007 and included case-control and case-only study components. Newly-diagnosed, histologically-confirmed ESCC and gastric cancer cases were identified and blood samples collected at enrollment for all cases7. The NITs were initiated in Linxian in 1985 and tested the effect of multiple vitamin and mineral combinations taken for up to six years on the incidence and mortality of EC and gastric cardia cancer4. Following a blood survey conducted in 1999–2000, all newly-diagnosed, histologically-confirmed ESCC cases documented during the follow-up through December 31, 2007, were included in the current analysis. Both the Shanxi and NIT studies were approved by their respective Institutional Review Boards and written informed consent was obtained from all subjects prior to participation. The NCI Special Studies Institutional Review Board approved both the Shanxi and NIT studies as well as the overall GWAS.
Participants for the Henan GWAS were collected from an ongoing hospital-based ESCC case-control study from northern China. The cases for the current study were restricted to the ‘genetically matched’ subset pool that was obtained from within Henan province. The study was approved by each institutional and hospital ethical committee and conducted according to the principles of the Declaration of Helsinki.
Our study was restricted to ESCC cases only as all traditional controls in the Henan GWAS were selected originally to have a negative family history, thus precluding an examination of FH differences using controls.
Replication stage sample
Replication of the top SNPs in the discovery stage was based on additional ESCC cases from Henan. Participants for the replication stage were part of the same ongoing hospital-based ESCC case-control study as for the Henan GWAS, but case ascertainment occurred subsequent to the Henan GWAS study. Similarly, the study population for the replication stage was restricted to ESCC cases only.
Family history of UGI cancer
For all studies from the discovery stage and replication stage, information on the diagnosis of UGI cancer, including age at diagnosis, gender, and tumor type, within family members for each study participant was collected through questionnaire by interview.
In the discovery stage, FH was defined according to available information to maximize the number of subjects, as available data on FH varied slightly for the three studies in the discovery stage. For NCI/Shanxi study and Henan study, ESCC cases with FH of UGI cancer (FH+) were primarily defined as those having one or more relative(s) diagnosed with UGI cancer within three generations. ESCC cases without FH of UGI cancer (FH−) were defined as those who did not have any relatives diagnosed with esophageal or gastric cancer within three generations. For the smaller NCI study (NIT), definition of FH was limited to first-degree relatives, with FH+ defined as one or more first-degree relatives diagnosed with UGI cancer. In sum, the discovery of the SNPs associated with FH of UGI cancer in cases of ESCC was based on a total of 541 cases with FH and 1399 without FH from the two NCI GWAS, including 343 cases with FH and 1076 without FH in NCI/Shanxi study, and, 198 cases with FH and 323 without FH in NIT, and 493 cases with FH and 869 without FH from the Henan GWAS.
For the replication phase sample, we considered two different definitions of a positive FH of UGI cancer: For the primary analysis, we used the same FH definition used for the majority of the discovery stage (NCI/Shanxi and Henan), in which we defined FH+ as having one or more relatives within three generations diagnosed with UGI cancer. Based on this definition, we have 2801 cases with FH and 3136 without FH. We also examined a more conservative definition of FH (limited to having one or more relatives with UGI cancer within first-degree relatives) to make sure that results did not differ based on FH definition; this secondary analysis included 1937 cases with FH and 3136 without FH.
Genotyping and quality control
GWAS data for the discovery stage
The details of the analytic preprocessing for the NCI GWAS and Henan GWAS were described previously9, 10. Additional quality control procedures were implemented in a joint analysis that included these two GWAS13, 15. Specifically, SNPs with a call rate < 95%, a Hardy-Weinberg proportion test P < 0.000001 or a MAF < 1% were excluded before the subsequent imputation analysis that was conducted separately for each of the two GWAS scans. Details of the imputation analysis have been previously published15.
SNP Replication in Henan Sample
A total of 19 top SNPs associated with FH of UGI cancer in the discovery stage (P < 0.05 in each individual GWAS and < 10−5 in the meta-analysis) were further examined in the 5937 additional Henan subjects. We tested the original SNPs (10 of 19) or their surrogates (8 of 19) if the original SNP failed assay design. The surrogate SNPs were selected by searching within 200 kb on either side of each targeted SNP. We used the genotype data from 1000 Genomes project JPT + CHB population to estimate the pair-wise LD and selected the three best LD surrogates based on r2 values for each targeted SNP. If the best one (that is, the SNP with the highest r2 with the targeted SNP) failed the assay design, we nominated the second SNP. If the second one also failed the assay design, we then nominated the third SNP as the surrogate. The r2 between the surrogate SNP and the original SNP is included in the footnotes of Table 2 and Supplementary Table 2. One SNP was dropped because both the original SNP and its only available surrogate failed assay design.
The genotyping of the 18 replication SNPs was performed as follows: a segment of DNA which surrounded the SNPs (100 bp) was amplified through PCR by using HotStarTaq (Qiagen). After purification by shrimp alkaline phosphatase and exonuclease I (Epicentre), the PCR products were tested by a primer extension assay by using the SNaPshot Multiplex kit (ABI). An ABI 3130xl capillary electrophoresis DNA instrument with Gene Mapper 4.0 software (Applied Biosystems, Foster City, CA) was used to analyze the resulting primer extension products.
Statistical analysis
Meta-analysis of GWAS for the discovery stage
Details of the statistical analysis methods for the NCI GWAS and Henan GWAS were included in the primary reports9, 10. We conducted meta-analyses to combine the β-estimates and standard errors from each GWAS. We tested the between-study heterogeneity and estimated the overall association from the fixed-effects model (weighted proportionately to the inverse of the study-specific variance). We identified eigenvectors for each GWAS and included the significant eigenvectors (P < 0.05) to control for population stratification in each individual GWAS.
Replication analysis
For 18 SNPs (including 10 original SNPs and 8 surrogates), we examined the association between each SNP and FH by comparing FH+ ESCC cases to FH- cases. The P-values and ORs for the SNPs (per one minor allele) were calculated from unconditional logistic regression models using trend tests adjusted for age, and sex. The definition of FH+ in the primary and secondary models is detailed in the Study Population section. For SNPs with suggestive associations, the pooled ORs were calculated based on random effect models for meta-analysis, as significant heterogeneity was found between the discovery and the replication dataset.
In silico and cis-eQTL functional annotation
To explore whether SNPs associated with FH after replication stage might have potential regulatory functions, we used custom tracks on the UCSC Genome browser (http://genome.ucsc.edu) to screen Roadmap Epigenomics (http://www.roadmapepigenomics.org/) in esophageal and stomach tissues and blood, as well as ENCODE data for each implicated SNP region for evidence of regulatory relevance,23, 24 such as overlap with chromatin marks, CpG-site methylation, and transcription factor binding motifs25. We also used the online tools HaploRegv4 (http://www.broadinstitute.org/mammals/haploreg/haploreg.php) and RegulomeDB (http://regulome.stanford.edu) as complementary analyses to confirm the location of each SNP in relation to annotated protein-coding genes and/or non-coding RNA genes.
To identify SNPs associated with RNA expression, we used publically available data (Genotype-Tissue Expression Project [GTEx], http://www.gtexportal.org/home/) to perform eQTL analyses in relevant normal tissues, including normal esophageal mucosa (n = 241), esophageal muscularis (n = 218), gastroesophageal junction (n = 127), normal stomach mucosa (n = 170), and whole blood (n = 338). We assessed the impact of associated SNPs on coding and non-coding genes in cis or located within 1MB of the signal and known to be expressed at the mRNA in the target tissue from the GTEx Project. For each tissue type, we performed cis-eQTL analysis for each gene-SNP pair. Linear regression was conducted for the association between each SNP and log and quantile normalized RNA-sequencing expression values from each tissue, adjusting for three genotyping principal components, 15 peer factors, and sex (http://www.gtexportal.org/home/documentationPage). P-values were adjusted for multiple comparisons using Bonferroni correction (P = 0.05/total number of genes tested per risk locus).
References
Ferlay, J. et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. International journal of cancer. Journal international du cancer 136, E359–386, doi:10.1002/ijc.29210 (2015).
Torre, L. A. et al. Global cancer statistics, 2012. CA: a cancer journal for clinicians 65, 87–108, doi:10.3322/caac.21262 (2015).
Li, J. Y. et al. Atlas of cancer mortality in the People’s Republic of China. An aid for cancer control and research. International journal of epidemiology 10, 127–133 (1981).
Li, B. et al. Linxian nutrition intervention trials. Design, methods, participant characteristics, and compliance. Ann Epidemiol 3, 577–585 (1993).
Freedman, N. D. et al. A prospective study of tobacco, alcohol, and the risk of esophageal and gastric cancer subtypes. Am J Epidemiol 165, 1424–1433 (2007).
Tran, G. D. et al. Prospective study of risk factors for esophageal and gastric cancers in the Linxian general population trial cohort in China. International journal of cancer. Journal international du cancer 113, 456–463 (2005).
Gao, Y. et al. Risk factors for esophageal and gastric cancers in Shanxi Province, China: a case-control study. Cancer Epidemiol 35, e91–99 (2011).
Cui, R. et al. Functional variants in ADH1B and ALDH2 coupled with alcohol and smoking synergistically enhance esophageal cancer risk. Gastroenterology 137, 1768–1775 (2009).
Abnet, C. C. et al. A shared susceptibility locus in PLCE1 at 10q23 for gastric adenocarcinoma and esophageal squamous cell carcinoma. Nature genetics 42, 764–767 (2010).
Wang, L. D. et al. Genome-wide association study of esophageal squamous cell carcinoma in Chinese subjects identifies susceptibility loci at PLCE1 and C20orf54. Nature genetics 42, 759–763 (2010).
Tanaka, F. et al. Strong interaction between the effects of alcohol consumption and smoking on oesophageal squamous cell carcinoma among individuals with ADH1B and/or ALDH2 risk alleles. Gut 59, 1457–1464 (2010).
Wu, C. et al. Genome-wide association study identifies three new susceptibility loci for esophageal squamous-cell carcinoma in Chinese populations. Nature genetics 43, 679–684 (2011).
Abnet, C. C. et al. Genotypic variants at 2q33 and risk of esophageal squamous cell carcinoma in China: a meta-analysis of genome-wide association studies. Hum Mol Genet 21, 2132–2141 (2012).
Wu, C. et al. Genome-wide association analyses of esophageal squamous cell carcinoma in Chinese identify multiple susceptibility loci and gene-environment interactions. Nature genetics 44, 1090–1097 (2012).
Wu, C. et al. Joint analysis of three genome-wide association studies of esophageal squamous cell carcinoma in Chinese populations. Nature genetics 46, 1001–1006 (2014).
Hu, N., Dawsey, S. M., Wu, M. & Taylor, P. R. Family history of oesophageal cancer in Shanxi Province, China. European journal of cancer 27, 1336 (1991).
Hu, N. et al. Familial aggregation of oesophageal cancer in Yangcheng County, Shanxi Province, China. International journal of epidemiology 21, 877–882 (1992).
Guo, W. et al. A nested case-control study of oesophageal and stomach cancers in the Linxian nutrition intervention trial. International journal of epidemiology 23, 444–450 (1994).
Stewart, S. E. et al. Genome-wide association study of obsessive-compulsive disorder. Molecular psychiatry 18, 788–798 (2013).
Abbassi-Ghadi, N. et al. Metabolomic profiling of oesophago-gastric cancer: a systematic review. European journal of cancer 49, 3625–3637 (2013).
Wang, L. et al. 1H-NMR based metabonomic profiling of human esophageal cancer tissue. Molecular cancer 12, 25 (2013).
Reue, K. The lipin family: mutations and metabolism. Current opinion in lipidology 20, 165–170 (2009).
Rosenbloom, K. R. et al. ENCODE data in the UCSC Genome Browser: year 5 update. Nucleic Acids Res 41, D56–63 (2013).
Meyer, L. R. et al. The UCSC Genome Browser database: extensions and updates 2013. Nucleic Acids Res 41, D64–69 (2013).
Kheradpour, P. & Kellis, M. Systematic discovery and characterization of regulatory motifs in ENCODE TF binding experiments. Nucleic acids research 42, 2976–2987 (2014).
Acknowledgements
This research was supported by the Intramural Research Program of Division of Cancer Epidemiology and Genetics, National Cancer Institute, National Institutes of Health.
Author information
Authors and Affiliations
Contributions
A.M.G. and L.D.W. had full access to all the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis. Study concept and design: P.R.T., L.D.W., A.M.G. Acquisition, analysis, or interpretation of data: W.Q.L, Z.W., P.L.H., P.R.T., L.D.W, A.M.G. Draft of the manuscript: W.Q.L., A.M.G. Critical revision of the manuscript for important intellectual content: X.S., W.Q.L., N.H., X.K.Z., Z.W., P.L.H., T.J., G.Q.K., H.S., C.W., L.W., L.S., Z.M.F., H.M., T.J.Z., L.F.J., S.J.H., W.L.H., M.J.W., P.Y.Z., S.L., X.M.L., F.Y.Z., L.B., T.D., Y.L.Q., J.H.F., X.Y.H., C.G., M.A.T., S.M.D., N.D.F., S.J.C., C.C.A., P.R.T., L.D.W., A.M.G. Statistical analysis: Z.W., W.Q.L. Obtained funding: A.M.G., L.D.W. Study supervision: W.Q.L., A.M.G., L.D.W.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Song, X., Li, WQ., Hu, N. et al. GWAS follow-up study of esophageal squamous cell carcinoma identifies potential genetic loci associated with family history of upper gastrointestinal cancer. Sci Rep 7, 4642 (2017). https://doi.org/10.1038/s41598-017-04822-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-04822-2
This article is cited by
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
